
A Novel Method of Gene Transcript Profiling in Airway Biopsy Homogenates Reveals Increased Expression of a Na+-K+-Cl− Cotransporter (NKCC1) in Asthmatic Subjects
- Gregory M. Dolganov1,2,5,
- Prescott G. Woodruff2,3,
- Alexander A. Novikov1,
- Yifan Zhang1,
- Ronald E. Ferrando2,
- Richard Szubin4, and
- John V. Fahy2,3
- 1Genelabs Technologies, Inc., Redwood City, California 94063, USA; 2Division of Pulmonary and Critical Care Medicine, Department of Medicine, University of California at San Francisco, San Francisco, California 94143, USA; 3Cardiovascular Research Institute, University of California at San Francisco, San Francisco, California 94143, USA; 4Department of Stomatology, University of California at San Francisco, San Francisco, California 94143, USA
Abstract
Comprehensive and systematic analysis of airway gene expression represents a strategy for addressing the multiple, complex, and largely untested hypotheses that exist for disease mechanisms, including asthma. Here, we report a novel real-time PCR-based method specifically designed for quantification of multiple low-abundance transcripts using as little as 2.5 fg of total RNA per gene. This method of gene expression profiling has the same specificity and sensitivity as RT-PCR and a throughput level comparable to low-density DNA microarray hybridization. In this two-step method, multiplex RT-PCR is successfully combined with individual gene quantification via real-time PCR on generated cDNA product. Using this method, we measured the expression of 75 genes in bronchial biopsies from asthmatic versus healthy subjects and found expected increases in expression levels of Th2 cytokines and their receptors in asthma. Surprisingly, we also found increased gene expression of NKCC1—a Na+-K+-Cl− cotransporter. Using immunohistochemical method, we confirmed increased protein expression for NKCC1 in the asthmatic subject with restricted localization to goblet cells. These data validate the new transcriptional profiling method and implicate NKCC1 in the pathophysiology of mucus hypersecretion in asthma. Potential applications for this method include transcriptional profiling in limited numbers of laser captured cells and validation of DNA microarray data in clinical specimens.
The fundamental mechanisms predisposing an individual to the development of asthma are not understood. Clinical exacerbations and chronicity of bronchial asthma depend on complex mechanisms, some of which appear to be genetically linked (Daniels et al. 1996; Holgate 2000; Sandford and Pare 2000). Many diseases have been linked to specific genetic defects resulting in physiological changes in cells and tissues, which can be correlated with changes in the mRNA levels of many genes. Global gene expression analysis can identify and predict experimentally verifiable phenotypic characteristics that may be of importance to disease progression (Bittner et al. 2000). Measuring gene expression in the airway can help advance understanding of disease mechanisms in asthma and may identify novel drug targets. However, current gene quantification methods including TaqMan and DNA microarrays require relatively large amounts of starting RNA (1–10 μg) and therefore cannot be applied directly to small-sized airway biopsies. In addition, hybridization-based microarray methods have lower sensitivity, specificity, and dynamic range compared with PCR-based approaches, and many genes implicated in asthma are expressed at low levels. RT-PCR has the highest specificity and sensitivity for transcript quantification among all available methods, although low throughput and relatively large amounts of starting RNA required for quantitative RT-PCR have precluded application of this method to small clinical samples, including airway biopsies. To overcome these problems, we developed a novel transcriptional profiling method that allows simultaneous relative quantification of multiple low-abundance transcripts in small biologic samples (Fig. 1). This is a two-step process that incorporates multiplex PCR (typically with a mix of 100–300 gene-specific primer sets) followed by real-time PCR on generated cDNA product with nested TaqMan primers and probes. Using this method, we show the ability to profile expression levels of 75 genes in bronchial biopsies in a study that reveals unexpected differential expression of a Na+-K+-Cl−cotransporter in asthma.
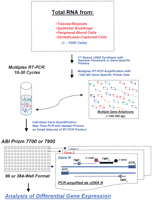
Transcriptional profiling by the two-step RT-PCR method. Total RNA from diseased or healthy tissues or from flow-sorted or laser-captured cells is isolated and reverse-transcribed with random hexamers or gene-specific primers. Then, cDNA product is PCR-amplified with 100–300 gene-specific primer sets (same as in RT reaction). Multiplex amplification is monitored carefully to evade plateau phase of the PCR. All gene-specific primers for multiplex RT-PCR are designed to have Tm = 60°C and produce small cDNA amplicons: 100–250 bp. Gene quantification via real-time PCR is performed on small aliquots of generated cDNA product with nested TaqMan primers in 96- or 384-well plate format using ABI Prizm 7700 or ABI Prizm 7900. RTF and RTR are forward and reverse primers for RT-PCR, TMF and TMR are nested TaqMan primers, and TMP is a TaqMan probe. The probe has a Fluorescein reporter dye at 5′-end (FAM) and a Black Hole Quencher at 3′-end.
RESULTS
Validation of a Novel Transcriptional Profiling Method: Relationship between Cycle Threshold and Gene Copy Number
To simplify statistical analysis, we transformed the cycle threshold values generated in the real-time PCR quantification step into relative gene copy numbers corresponding to gene copies in the aliquot of cDNAs from the RT-PCR step. Absolute copy numbers corresponding to mRNA copies in starting total RNA are not required to identify genes implicated in the disease, although they can be calculated by adding competitive RNA templates to the sample and generating standard curves for genes of interest (Iyer and Struhl 1996). To determine the relationship between cycle threshold (Ct) values in the real-time step and transcript abundance, we generated RT-PCR products for 34 genes of varying abundance in our samples by using reverse transcription forward and reverse primers (RTF/RTR) and cloned them into a dual promoter plasmid vector pCRII-TOPO (Invitrogen, Inc.); the same plasmids could be used to generate corresponding RNAs for quantification of absolute gene copy number. Serial dilutions of plasmids with sequence-verified gene-specific amplicons were generated and used in real-time PCR. The calibration curves for the 34 genes were almost identical with all R-squared values exceeding 94% and many exceeding 99%. Small differences that occurred may have been related to differences in specific fluorescence of corresponding TaqMan probes. Accordingly, we developed a regression equation (Log Copy Number = −0.283 Ct + 11.309) extrapolated from the curves of these 34 genes, and we then linearly transformed PCR threshold cycles for all genes by using this equation.
Validation of a Novel Two-Step Transcription Profiling Method: Optimization of the Multiplex RT-PCR Amplification
Eight aliquots ranging from 10 pg to 175 ng of total RNA from CD4+ lymphocytes of a healthy subject were reverse-transcribed with SuperScript II. Then, each RT reaction was split into five aliquots and PCR amplified with 200 gene-specific primer sets for 0, 15, 20, 25, or 30 cycles, where 0 cycle amplification represents the conventional real-time PCR gene quantification approach. Transferrin receptor was quantified in amplified cDNAs via real-time PCR using 0.025% of the original RT reaction. We found that transferrin receptor could be quantified efficiently using as little as 0.01 ng of starting total RNA, which translates into 5.0 fg of RNA in a single real-time PCR quantification, and that fourfold differences in amount of starting RNA between samples yielded the expected difference of 2 Cts (Fig.2).
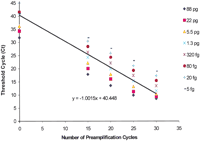
Linearity and sensitivity of two-step RT-PCR for quantification of transferrin receptor gene expression in total RNA from blood CD4+ cells. The number of preamplification cycles in the multiplex step was consistently and inversely related to cycle threshold for transferrin receptor gene in the real-time PCR step. Each real-time PCR contained an equivalent of 0.05% of starting RNA (88 pg to 5.0 fg). Finally, transferrin receptor gene could be quantified using as little as 5.0 fg of starting total RNA. In this instance, the optimal range for multiplex PCR amplification was 10–25 cycles.
Similar data were generated with 11 genes by using five amplification fractions from the RT reaction that contained 2.0 ng of total human tracheal RNA (Fig. 3). As can be seen in Figures 2 and 3, multiplex amplification was linear up to 25 cycles for most abundant messages and up to 30 cycles for less abundant messages, resulting in the overall dynamic range of 2 × 106. Thus, multiplex PCR was efficient and linear at least up to 25 cycles. These data confirm that multiplex PCR did not substantially change the representation of various transcripts after 25 cycles of amplification. The optimal range for a given multiplex PCR amplification was found to be within 10–25 cycles. At 25 cycles of amplification, 0.01–2.0 ng of starting RNA would allow reliable quantification of ∼1000 genes in a dynamic range from 103 to 106, respectively. In contrast, the conventional real-time PCR approach (0 amplification data in Fig. 2) cannot quantify genes with lower expression levels than transferrin receptor, even with 175 ng of starting RNA (88 pg in a real-time PCR).
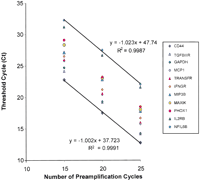
Linearity and sensitivity of two-step RT-PCR for quantification of expression of multiple genes using 2.0 ng of total RNA from human trachea. For each of 11 representative genes, 0.05% of the multiplex PCR product, that is, an equivalent of 1 pg of starting RNA was used in the real-time PCR step. For genes of varying abundance, the number of preamplification cycles in the multiplex step was consistently and inversely related to the corresponding cycle thresholds in the real-time PCR step. At 25 cycles of amplification, 2.0 ng of starting RNA would allow reliable quantification of at least 1000 genes in an optimal cycle threshold range for detection.
In a different experiment (see Fig. 4), we validated multiplex RT-PCR by using only a few CD4+ cells from bronchoalveolar lavage of an asthmatic subject. The cells were counted, and aliquots containing 50, 200, 500, and 2000 cells were processed individually to isolate total RNA. Each sample was reverse-transcribed with 200 gene-specific primer sets and processed as described above for peripheral blood CD4+ cells. We found that multiplex PCR amplification was linear through cycle 25 with the slope for transferrin receptor ∼1.0, and Cts in aliquots of each fraction were proportional to the number of cells used to isolate RNA (data not shown). Amplified cDNA fractions corresponding to 50, 200, 500, and 2000 starting cells then were used to quantify six representative genes of different abundance. We found that these genes could be reliably quantified in amplified 50 cells fractions and their relative expression levels increased proportionally in the aliquots with greater cell numbers (results for 25 cycles of amplification shown in Fig.4). Moreover, in a separate experiment we have individually isolated total RNA from 4, 16, 64, and 128 CD4+ cells, and each sample was reverse-transcribed and preamplified for 25 cycles as described above. Then, similar aliquots of cDNA fractions corresponding to 4, 16, 64, and 128 starting cells were used to quantify GAPDH, interleukin IL-8, and IL-13 (Table1). Currently available methods of RNA isolation may be unreliable when applied to very few cells, and this may introduce variability in measured gene expression levels (Fig. 4) because of statistically unfavorable transcript representation in RT-PCR. Nevertheless, we have found that multiple genes could be reliably quantified by our method even in as few as four cells as evidenced by the standard deviations within an acceptable range for TaqMan measurements (0.52 Ct or below, Table 1).
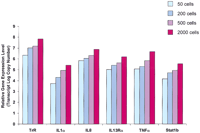
Two-step RT-PCR for quantification of gene expression in varying numbers of CD4+ cells from bronchoalveolar lavage. Using 25 preamplification cycles, the relative expression level of six genes of varying abundance could be accurately measured in as few as 50 starting cells and increased linearly as expected with increasing numbers of cells. (TrR) Transferrin receptor; (IL) interleukin; (TNF) tumor necrosis factor.
Two-Step RT-PCR for Quantification of Gene Expression in Varying Numbers of CD4+ Cells from Bronchoalveolar Lavage
Because the first strand cDNA synthesis is the most critical step for sensitivity of RT-PCR (Henegariu et al. 1997; Freeman et al. 1999), we compared three different reverse transcriptases (see Figs. 5,6) and optimized the conditions of the RT step in our new method. We also compared varying concentrations of random hexamers versus gene-specific primers and optimized the conditions for Superscript II and Sensiscript first strand cDNA synthesis. We found that random priming was better with Superscript II rather than with Sensiscript or Thermoscript, although gene-specific priming with Sensiscript was as efficient as or even better than random priming with Superscript II (see Figs. 5, 6).
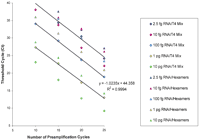
Random hexamers versus T4 Mix priming with Sensiscript. Twenty narograms, 2 ng, 200 pg, 20 pg, and 5 pg of total tracheal RNA (i.e., RNA equivalent to ∼2000, 200, 20, 2, and 0.5 cells) were used in cDNA synthesis with Sensiscript as described in Methods using either random hexamers or T4 Mix with 200 gene-specific primer sets. Aliquots of RT reactions were PCR-amplified with the T4 Mix for 10, 15, 20, and 25 cycles as described in Methods, and 0.05% of each reaction used in real-time PCR to quantify the levels of GAPDH. The number of preamplification cycles in the multiplex step was consistently and inversely related to cycle threshold for GAPDH in the real-time PCR step. Both gene-specific and random hexamers efficiently primed the first strand cDNA with Sensiscript, although gene-specific priming was much more efficient and resulted in consistently lower Cts for GAPDH. Multiplex preamplification with T4 Mix was also efficient and linear and produced slopes of −1.0 (at least for GAPDH).
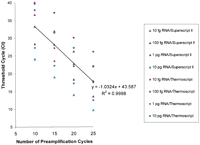
Superscript II versus Thermoscript in cDNA synthesis with random hexamers. Twenty narograms, 2 ng, 200 pg, and 20 pg of total tracheal RNA (i.e., RNA equivalent to ∼2000, 200, 20, and 2 cells) were used in cDNA synthesis with Superscript II or Thermoscript as described in Methods using random hexamers. Aliquots of RT reactions were PCR-amplified with the T4 Mix for 10, 15, 20, and 25 cycles as described in Methods, and 0.05% of each reaction was used in real-time PCR to quantify the levels of GAPDH. The number of preamplification cycles in the multiplex step was consistently and inversely related to cycle threshold for GAPDH in the real-time PCR step. Superscript II was more efficient than Thermoscript in random primed cDNA synthesis and this resulted in lower Cts for GAPDH. Multiplex preamplification with T4 Mix was also efficient and linear and produced slopes of −1.0 (at least for GAPDH).
Application of Transcriptional Profiling Method: Studies in Bronchial Biopsies from Healthy and Asthmatic Subjects
Three bronchial biopsies from each of 13 healthy nonasthmatic subjects and 13 subjects with mild to moderate asthma were frozen in optimally controlled temperature (OCT) compound, cut in 5-μM sections, and stained with Gill's hematoxylin. Clinical characteristics of patients enrolled in this study are described in Methods. Biopsies with intact epithelium but lacking submucosal gland tissue were selected; biopsies from 23 of the 26 patients satisfied these criteria. OCT compound was removed from these biopsies, and RNA was isolated and checked for quality as described in Methods. Four of 23 biopsies yielded partially degraded RNA and were further excluded from this study, leaving samples from the 8 healthy and 11 asthmatic subjects. Variability in RNA quality most likely resulted from the duration of storage of the biopsies (6–18 months at −70°C) or from the handling of individual biopsies before isolating RNA. Duplicate biopsies were available in 3 of the 8 healthy subjects and 4 of the 11 asthmatic subjects.
Two hundred gene-specific primer sets were used in multiplex RT-PCR, including disease gene candidates associated with inflammation and remodeling in asthma such as Th2 cytokines, cytokine receptors, chemokines, transcription factors, proteinases, mucin genes, and ion channels and transporters. These genes also included 13 expressed sequence tags and other genes from the 5q31 locus identified via direct selection (Morgan et al. 1992) or gene prediction within genomic sequences of interest using GENSCAN (Burge and Karlin 1997) in combination with other methods. A list of the disease gene candidates used in present study is available viahttp://asthmagenomics.ucsf.edu/public.
In this study, the real-time PCR quantification step then was performed for 75 of these 200 genes. Mucin gene quantification in these biopsies is reported in a separate study (Ordonez et al. 2001). Because of cellular heterogeneity of airway biopsies, duplicates revealed small differences in their transcriptional profiles. However, too few duplicates were available to formally assess variability between biopsies. Therefore, these data points were averaged to reduce variability and used to evaluate differential gene expression in asthmatic versus healthy subjects.
We found that 19 of the 75 genes were significantly overexpressed in the biopsies from the asthmatic subjects and four were significantly underexpressed. As expected, we found increased expression in asthma of families of genes thought to have important roles in the mechanisms of airway inflammation and remodeling (Table2). For example, of the 17 cytokines measured, five were increased in the asthmatic subjects, including IL-5, which was increased ∼40-fold, and IL-13, which was increased sevenfold (Table 2). IL-4 and IL-9 expression levels were relatively low in the biopsies in both groups and not significantly higher in the asthmatic subjects (Table 2). We found a significant inverse association between expression levels of IL-13 and FEV1% predicted in the asthmatic subjects (rs = −0.65, P = 0.03). In contrast, biopsy gene expression of interferon-γ was decreased in the asthmatic samples and increased with FEV1% predicted (rs = 0.31, P = 0.35). The expression levels of receptors for IL-4, IL-9, and IL-13 were significantly increased in the asthmatic subjects with a notable 30-fold increase in IL-13rα2; the difference in expression of IL-13rα1 was smaller but also significant (Table 2).
Relative Transcript Number of 75 Genes in Endobronchial Biopsy Homogenates of Asthmatic and Healthy Subjects
An unexpected result of our study was the eightfold increase in asthma of gene expression for NKCC1—a Na+-K+-Cl− cotransporter that has not been implicated previously in asthma pathogenesis (Table 2). To further explore this finding, we examined immunhistochemical expression of NKCC1 in paraformaldehyde-fixed, glycomethacrylate-embedded biopsies available from the same groups. Using a monoclonal antibody (T4) specific for NKCC1 and NKCC2, we show increased T4 labeling in the airway epithelium in the asthmatic subjects (Fig. 7). Specifically, we found that T4 labeling localized to the basolateral membrane of goblet cells; no other epithelial cell stained positively (Fig.8). Furthermore, the increased T4 labeling was at least in part because of increased expression of T4 on goblet cells rather than simply increased numbers of goblet cells (surface area of T4 stain per surface area of goblet cells = 0.23 ± 0.16 vs. 0.45 ± 0.13 in healthy and asthmatic subjects, respectively, P = 0.001). We interpret our data as indicating selectively increased protein expression for NKCC1, because NKCC2 represents an absorptive isoform that to date has not been identified outside the kidney (Payne et al. 1995; Haas and Forbush 2000).
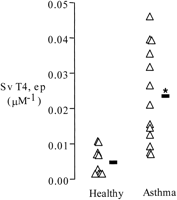
Quantitative measurement of T4 immunostaining in bronchial biopsies from healthy and asthmatic subjects. Open triangles indicate individual data points; boldface dash indicates means; Sv T4, ep is the surface area of T4 immunostaining per volume of epithelium. Mean T4 immunostaining was approximately fourfold higher in the asthmatic subjects (* indicates significantly greater than healthy,P < 0.001).
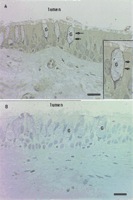
Photomicrograph of T4 immunostaining in a representative section of a bronchial biopsy from an asthmatic subject (A). Also shown is photomicrograph of a representative section stained with negative control (MOPC-31c) (B). The figure shows distinct T4 staining along the basolateral membrane of goblet cells. Arrows point to basalateral immunostaining of goblet cells (G). No other epithelial cells stained positively. The inset in panel Ashows a magnified field of the same specimen. Bar = 20μM.
We also found increased expression for OCTN1—a pH-dependent proton/organic cation transporter—in the biopsies from the asthmatic subjects (Table 2). Like the gene for NKCC1, the gene for OCTN1 resides in the 5q31 locus, and Northern blot analysis has revealed that it is expressed principally in the kidney and in the airway (Tamai et al. 1997). The role of this transporter in the kidney is thought to be in the transport of organic cations (e.g., tetraethyl ammonium), but its physiologic role in the airway is unknown. The expression of OCTN2 was similar in both groups as was the expression of MAXIKP1—the large conductance calcium—and voltage-dependent potassium channel (Table 2).
DISCUSSION
We have developed a novel two-step RT-PCR approach for transcriptional profiling of multiple low-abundance mRNAs that requires significantly less starting RNA than conventional TaqMan approaches. This improved sensitivity allows transcriptional profiling in small biologic samples, such as 1–100 cells. The method relies on final gene quantification via real-time PCR using cDNA product generated by controlled hot start multiplex RT-PCR. In contrast with conventional TaqMan approach, this method requires lower amounts of starting RNA and therefore could be applied to quantify multiple low expressed genes in small clinical samples. Raw data are expressed as the number of PCR cycles needed to reach a detection threshold value (Ct) that is inversely proportional to the exponent of transcript abundance. Use of internal controls allows quantification of mRNAs' abundance in each sample across multiple genes. In our experiments, total RNA from biopsies was converted to double-stranded cDNA with a mixture of 200 gene-specific primer sets, and 75 of these cDNAs were quantified in a subsequent real-time PCR step. The reliability of quantification by this method depends on the initial number of transcripts in the PCR; therefore, statistically it is more favorable to conduct multiplex PCR amplification on a whole rather than on a split sample.
We have shown that both the quality of RNA and the enzymes used in the two-step real-time PCR were critical for reliable transcriptional profiling in small amounts of cells or tissues. Efficiency of RT in our hands fluctuated from 3% to 90%, depending on the type and source of reverse transcriptase, purity and integrity of RNA, concentration and type of primers, and kinetic profiles of the reaction. Because RNA quality is essential for transcriptional profiling, we revised sample handling practices and RNA isolation protocols. Whenever possible, RNA quality first was checked using RNA 6000 LabChip kit (Agilent Technologies, Inc.). Then, all RNA samples were checked for genomic DNA contamination by using −RT controls as described in Methods. Usually the difference between similar aliquots of +RT and −RT reactions was higher than 10 Cts for any given gene; otherwise RNA was excluded from transcriptional profiling. We compared three different RT enzymes in reactions with varying concentrations of random hexamers versus gene-specific primers and found that both Superscript II and Sensiscript can be reliably used in the two-step real-time PCR-based gene quantification. In this method, first strand cDNAs are amplified by multiplex PCR with 100–300 sets of gene-specific primers to produce short amplicons (<250 bp). The number of amplification cycles depends on the efficiency of PCR and the amount of target in the reaction. Therefore, for optimal gene quantification multiplex PCR amplification should be kept out of the plateau phase, and 0.05% aliquots of the original RT reaction should produce 8–12 Cts for GAPDH (or any other housekeeping gene) in subsequent TaqMan quantification. Such optimization usually results in a high dynamic range (up to 109) and accurate gene quantification.
The proposed method has two important advantages over conventional quantitative PCR, TaqMan assays, and gene microrarrays. First, the new method allows simultaneous quantification of hundreds of transcripts using as little as 2.5 fg of total RNA per gene (see Fig. 5), whereas conventional TaqMan assays require substantially larger amounts of total RNA, usually from 10 ng to 1 μg per reaction, depending on the abundance of the genes of interest. In contrast with end point PCR quantification, TaqMan PCR quantification overcomes limitations associated with plateau phase of the PCR. Gene microarrays require even larger amounts of starting RNA (1–10 μg), and individual gene probes in labeled complex cDNA/cRNA mixtures may have unique secondary structures, melting temperatures, and reassociation rates (Southern et al. 1999), which makes hybridization of all gene probes under optimum condition nearly impossible. Second, in contrast with gene microarray methods that have a much smaller dynamic range, lower specificity and sensitivity, real-time PCR has a dynamic range of more than six orders of magnitude and allows simultaneous accurate measurement of low- and high-abundance mRNAs. At this time, no other technique offers the potential for simultaneous rapid, accurate, and reliable quantification of multiple genes in many small biological or clinical samples (1–100 cells).
We applied this new method of transcriptional profiling to the measurement of gene expression in bronchial biopsies from asthmatic subjects and healthy controls. Our data reveal both expected findings that validate our methods and unexpected findings that show the value of our approach for suggesting new hypotheses. As expected, we found increased expression in asthma of many members of the Th2 cytokine family. The genes for this family of cytokines is clustered on 5q31, a chromosomal region consistently associated with asthma in population-based studies of asthma genetics (Postma et al. 1995;Noguchi et al. 1997; Hizawa et al. 1998; Mansur et al. 1998). Our data on Th2 cytokine expression in bronchoalveolar lavages and biopsies confirm current knowledge (Bhathena et al. 2000; Jeffery et al. 2000;Lin et al. 2000) but also extend it because we show new data on expression patterns for Th2 cytokine receptors. In particular, we show increases in gene expression for IL-9 and IL-13 receptors in the asthmatic airway.
An unexpected result of our study was the differential expression in asthma of NKCC1—a gene on 5q31 not previously implicated in asthma. NKCC1 and NKCC2 are bumetanide-sensitive Na+/K+/Cl− cotransporters that transport Na+, K+, and Cl− ions into and out of cells in an electrically neutral manner, in most cases with a stoichiometry of 1Na+ : 1K+ : 2Cl−. These cotransporters acts in concert with other transporters, such as apical Cl− channels and basolateral K+ channels and Na+/K+ pumps, to produce transepithelial Cl− secretion. NKCC1 expression is widespread in various organ tissues (Haas and Forbush 2000). Predictably, mice lacking NKCC1 exhibit decreased chloride secretion in enterocytes (Flagella et al. 1999), but they also have other phenotypic changes including impaired salivation, deafness, ataxia, and infertility (Delpire et al. 1999;Dixon et al. 1999; Flagella et al. 1999). We extended our finding for increased gene expression for NKCC1 in the biopsies from asthmatic subjects by showing increased labeling with a monoclonal antibody (T4) specific for NKCC1 and NKCC2 (Lytle et al. 1995; Crouch et al. 1997). We interpret our data as indicating increased protein expression for NKCC1 only, because NKCC2 represents an absorptive isoform that has not been identified outside the kidney (Payne et al. 1995; Haas and Forbush 2000). We speculate that increased NKCC1 expression in goblet cells may reflect a role for this transporter in the goblet cell hyperplasia characteristic of asthma (Ordonez et al. 2001), because others have shown that overexpression of NKCC1 can induce cell proliferation and phenotypic transformation (Panet et al. 2000; Selvaraj et al. 2000). Although the mechanism of these effects are unknown, it is proposed that NKCC1 may induce changes in intracellular cation concentrations in the early G0/G1 phase, which could induce cell proliferation directly by stimulating signal transduction pathways or indirectly by affecting other membrane transporters (Panet et al. 2000). Also of relevance here is that NKCC1 is inhibited by the loop diuretic furosemide (Panet et al. 2000; Selvaraj et al. 2000), and inhaled furosemide has beneficial effects in asthma. Specifically, pretreatment of asthmatic subjects with inhaled furosemide causes significant attenuations in bronchoconstriction caused by allergen, exercise, and distilled water (Siffredi et al. 1997; Pendino et al. 1998; Rodriguez Vazquez et al. 1998; Tan and Spector 1998; Milgrom and Taussig 1999). Taken together, these data on effects of furosemide in experimental airway challenges and our data for increased NKCC1 expression in asthma indicate NKCC1 as a possible novel therapeutic target for asthma.
We recognize that many factors, including ethnicity, gender, age, and environmental stimuli, may influence the development of asthma. These factors potentially could complicate the detection or validation of altered gene expression in asthmatics versus healthy subjects. However, the present sample sizes are too small to adequately control for the effects of ethnicity, gender, age, or environment. In the future, increasing the number of enrolled subjects and specifically controlling for subject characteristics that influence gene expression may resolve this problem.
In conclusion, we describe a novel method for transcriptional profiling that is applicable to gene profiling in small biologic samples. We provide data validating this method, and we show the utility of the data in measuring gene expression in bronchial biopsies in asthma. In our application of the method in asthmatic subjects, we show increased expression of NKCC1 in goblet cells. Further work is needed to evaluate the role of NKCC1 in goblet cell proliferation or mucus hypersecretion in asthma. We propose that the strategy of transcriptional profiling in airway tissues in asthma is feasible and that this approach could be applied to test existing hypotheses, suggest new hypotheses, and identify novel therapeutic targets.
METHODS
Blood, Bronchoalveolar Lavage, and Biopsy Collection from Human Subjects
CD4+T lymphocyte plays a key role in mediating the inflammatory response in asthma; therefore, we were interested in molecular mechanisms underlying in vivo CD4+ T-cell activation in asthma. For studies on blood CD4+ T cells, blood was obtained from a healthy volunteer. For studies on lung CD4+ T cells, bronchoalveolar lavage was obtained from an asthmatic subject (28-year-old female, FEV1% predicted 97%, PC20 methacholine 1.24 mg/mL) by using methods of lavage (100 mL in right middle lobe) previously described (Fahy et al. 1995). For studies on bronchial biopsies, 13 healthy and 13 asthmatic subjects were studied in a protocol previously described (Ordonez et al. 2001). The asthmatic subjects (four females, mean age 32 ± 4.3) had mild to moderate disease (mean ± SD FEV1 = 83 ± 16% predicted) treated with inhaled short acting β agonists only. The healthy subjects (10 females, mean age 29 ± 6.4 years) were nonsmokers with normal spirometry and no history of allergic disease. All subjects signed consent forms approved by the Committee on Human Research at the University of California, San Francisco.
Isolation of Total RNA from CD4+ Cells
CD4+ cells were isolated from peripheral blood or BAL (bronchoalveolar lavage) by using immunomagnetic enrichment of buffy coats or lavage with MACS CD4-antibody beads (Miltenyi Biotec). Cells were washed and sorted on a magnetic column (RS+ column, 30-nm pore) and used to isolate total RNA with RNeasy mini kit (Qiagen) including DNAse I treatment with RNase-free RQ1 DNAse (Promega Corporation). To 50 μL of RNA in water, 6 μL of 10× DNAse I buffer, 3 μL Superase RNA inhibitor (Ambion), and 1 μL of RQ1DNAse I (1 U/μL) were added, and incubation continued for 20 min at room temperature. Then, 10 μL of stop solution was added, and RNA was cleaned on a second RNeasy column.
Biopsy Processing and RNA Extraction
Three biopsies from each subject were immersed in 20% sucrose at 4°C for 2–4 h and placed in OCT (Sakura Finetek U.S.A. Inc.) before being snap-frozen in liquid nitrogen. Total cellular RNA was obtained by trimming off the OCT compound, sonicating the biopsy in 0.5 mL of RNAzol (Tel-Test, Inc.), and isolating RNA according to manufacturer's protocol.
Two-Step Real-Time PCR-Based Transcriptional Profiling
Gene-specific primers for multiplex RT-PCR and TaqMan were designed using Primer Express software (Perkin Elmer) based on sequencing data from National Center for Biotechnology Information databases and purchased from Biosearch Technologies, Inc. Both sets of the primers were nested, and RT-PCR products were within 250-bp range. All primer sequences and composition of multiplex mixtures used in this study are available from our web site athttp://owl.ucsfmedicalcenter.org/public.
Multiplex RT-PCR Step
Reverse transcription of RNA was performed using three reverse transcriptases, SuperScript II RT, Thermoscript (Life Technologies, Inc.), or Sensiscript (Qiagen), under conditions described by the manufacturers. In all reactions, cDNA was synthesized in 20 μL using 5 pg to 50 ng of human total RNA from CD4+ T cells, endobronchial biopsy homogenates, or whole trachea (Clontech) and either 200 nM random hexamers or 100–300 gene-specific primers sets (MixT4, Mix10, or Mix15) at varying concentrations as indicated, otherwise at 100 nM each (>103-fold excess of primers over template). All reactions contained 1 U of Superase RNase inhibitor (Ambion, Inc.). To control for genomic DNA contamination in total RNA preparations under similar conditions, we conducted +RT and −RT reactions. Optimization of multiplex hot start PCR was performed as described (19–22), using KlenTaq DNA polymerase (cDNA Advantage Mix from Clontech). Each PCR (100 μL) contained 1–20 μL of cDNA material from the RT step and 100–300 gene-specific primer sets, at 20–100 nM each. Before amplification, the reaction was split into five 20-μL aliquots, and each tube heated at 94°C for 2 min to inactivate the anti-Taq antibody followed by 0–25 cycles with 94°C for 30 sec, 55°C for 30 sec, and 70°C for 45 sec.
Real-Time PCR Step
Typically, an equivalent of 2.5 fg to 10 pg of total RNA was used in 25 μL of Universal Master Mix (Perkin Elmer). All forward and reverse TaqMan primers (TMF/TMR) were optimized, and transcript quantifications run in duplicates in parallel with similar aliquots of −RT cDNA controls on ABI Prizm 7700 Sequence Detection System (PE Applied Biosystems, Inc.).
Real-Time PCR Data Analysis
The baseline for each real-time PCR plot was selected above the noise in a window in the linear region of the semi-log plot (three to five cycles or higher). Because of the large dynamic range of gene expression data in a single run, a standardized approach to selection of an individual baseline for each gene was designed. Raw data from ABI Prizm7700 were processed into Excel (Microsoft) spreadsheets using software that automated proper baseline selection and Ct calculation for each of 48 genes on a 96-well plate using both standard curve and dCt methods as described (Fink et al. 1998; Livak 1998).
Conversion of Raw Ct Values to Relative Gene Copy Number
RT-PCR products for most of the genes were generated using RTF/RTR primers and cloned into a dual promoter plasmid vector pCRII-TOPO (Invitrogen, Inc.). All the clones were further verified by PCR and sequencing and used as reference to determine relative gene copy numbers in real-time PCR.
Immunohistochemical Analysis of NKCC1 in Bronchial Biopsies
The NKCC1 IgG monoclonal antibody (clone T4; dilution 1 : 20,000), developed by Drs. Chrisitian Lytle and Bliss Forbush III (Lytle et al. 1995; D'Andrea et al. 1996; Crouch et al. 1997), was obtained from the Department of Biological Sciences, University of Iowa. Biopsies were fixed in 4% paraformaldehyde and embedded in glycomethacrylate (Polysciences, Inc.). Sections from each block were screened to ensure at least two biopsies per subject had two or more high-powered fields of intact epithelium. Twenty-three subjects (13 healthy, 10 asthmatic) met criteria. After blocking with 1.0% horse serum, 2 μM sections were incubated with T4 (1 : 20,000 dilution) for 16–20 h, followed by biotinylated secondary antibody (Vector Labs) for 2 h, and then avidin-peroxidase (Vector Labs) for 2 h. Finally, sections were incubated with DAB chromogen (Zymed Laboratories) for 10 min and counterstained with Gills Hematoxylin #3 (Fisher Scientific) for 30 sec. Mouse purified plasmacytoma IgG1 (1 : 50 dilution, MOPC-31c; Sigma) was used as a negative control. All the reactions were performed at room temperature.
Measurement of T4 antibody immunostaining was performed using a grid of geometric probes applied to video-captured microscopic images of airway epithelium (Olympus Computer Assisted Stereology System). Images were analyzed with the vertical plane perpendicular to the basal lamina (local vertical orientation) at 40× linear magnification. The surface area of T4 stain per volume of epithelium (Sv t4, ep) and surface area of goblet cells per volume of epithelium (Sv goce, ep) were calculated as follows: 2*(sum of test-line intersection with T4 or goblet cell)/(length of test-line per point)* (# of points intersecting the epithelium). The surface area of T4 per surface area of goblet cells (Ss T4 goce) was calculated from (Sv T4, ep)/(Sv goce, ep) (Bolender et al. 1993).
Acknowledgments
We thank Dr. M. Lovett (Washington University, St. Louis) for critical reading of the manuscript and helpful suggestions. This work was supported by Genelabs Technologies, Inc. and a grant from National Institutes of Health (RO1 HL-61662). R.S. has been supported by a grant from the National Institutes of Health (PO1 DE07946).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵5 Corresponding author.
-
E-MAIL gregd{at}itsa.ucsf.edu; FAX (415) 476-5712. Article published on-line before print: Genome Res., 10.1101/gr. 191301.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.191301.
-
- Received April 5, 2001.
- Accepted June 4, 2001.
- Cold Spring Harbor Laboratory Press








