
Phylogenetic Analysis of Ribonuclease H Domains Suggests a Late, Chimeric Origin of LTR Retrotransposable Elements and Retroviruses
Abstract
We have conducted a phylogenetic analysis of the Ribonuclease HI (RNH) domains present in Eubacteria, Eukarya, all long-term repeat (LTR)-bearing retrotransposons, and several late-branching clades of non-LTR retrotransposons. Analysis of this simple yet highly conserved enzymatic domain from these disparate sources provides surprising insights into the evolution of eukaryotic retrotransposons. First, it indicates that the lineage of elements leading to vertebrate retroviruses acquired a new RNH domain either from non-LTR retrotransposons or from a eukaryotic host genome. The preexisting retroviral RNH domain degenerated to become the tether (connection) domain of the reverse transcriptase (RT)–RNH complex. Second, it indicates that all LTR retrotransposons arose in eukaryotes well after the origin of the non-LTR retrotransposons. Because of the younger age of the LTR retrotransposons, their complex structure, and the absence of any prokaryotic precursors, we propose that the LTR retrotransposons originated as a fusion between a DNA-mediated transposon and a non-LTR retrotransposon. The resulting two-step mechanism of LTR retrotransposition, in which RNA is reverse transcribed away from the chromosomal target site, rather than directly onto the target site, was probably an adaptation to the uncoupling of transcription and translation in eukaryotic cells.
Ribonucleases H (RNH) endonucleolytically cleave the RNA strand of an RNA–DNA hybrid. Because of their unique enzymatic activity, RNH domains are believed to have played an important role in the transition from the RNA world to the DNA world. A remnant of that activity is proposed to be the role of RNH domains in removing RNA primers at the 5′ ends of lagging strand synthesis in DNA replication (e.g., Qiu et al. 1999). RNH enzymes have also been implicated in DNA repair and RNA transcription (Crouch and Toulme 1998).
There appear to be three broadly distributed lineages of RNH enzymes: RNase HI (rnhA gene), HII (rnhB), and HIII (rnhC; Ohtani et al. 1999). Common evolutionary ancestry has been firmly established for rnhB and rnhC, whereas rnhA may represent a case of convergent evolution (see Lai et al. 2000). These three lineages also differ in their phylogenetic distribution among the three kingdoms. Archaea only possess rnhB genes, whereas all Eukarya appear to have bothrnhA and rnhB genes. Eubacteria can possess all three genes, but most encode either rnhA-B orrnhB-C. In cases of eubacterial genomes that have all three genes, (e.g., Bacillus subtilis), one of the encoded proteins might lack enzymatic activity (Ohtani et al. 1999). Among cellularrnhA genes, the gene from Escherichia coli has been most extensively studied, both with respect to cellular function, as well as the structural aspects of its encoded enzymatic activity (Katayanagi et al. 1993; Goedken and Marqusee 2000). The key residues involved in the catalytic mechanism have been identified and found to be the same in all rnhA proteins (Johnson et al. 1986; Davies et al. 1991).
rnhA domains (hereafter referred to as simply RNH) have also been observed as adjunct domains to the RT as part of the pol gene in retroviruses and in other LTR-bearing retrotransposons (for review, seeBoeke and Stoye 1997). In the retroviral and LTR retrotransposon life cycles, RNH performs three related functions: Degradation of the original RNA template, generation of a polypurine tract (the primer for plus-strand DNA synthesis), and final removal of RNA primers from newly synthesized minus and plus strands. RNH domains can be readily aligned between Eubacteria, Eukarya, retroviruses and other LTR-retrotransposons (Johnson et al. 1986; Doolittle et al. 1989). The three-dimensional structures of the HIV-1 and E. coli enzymes are strikingly similar, with the positions of the core catalytic residues virtually invariant (Davies et al. 1991).
RNH domains have also been found in several lineages of non-LTR retrotransposons (Fawcett et al. 1986; Doolittle et al. 1989; Blesa and Martinez-Sebastian 1997). One early study proposed the presence of RNH domains in a wider range of non-LTR element lineages (McClure 1991). However, a recent more comprehensive analysis of all available non-LTR retrotransposons has suggested that only a limited number of lineages possess this domain, and the lineages that do possess it contain many examples in which it has been lost (Malik et al. 1999). The position of the RNH domain of non-LTR retrotransposons carboxy terminal to the RT domain is similar to that of LTR retrotransposons. Because non-LTR retrotransposons reverse transcribe their RNA template directly on to the chromosomal target site (target-primed reverse transcription; (Luan et al. 1993), the cellular RNH activity present in the nucleus may suffice (Malik et al. 1999). In contrast, LTR retrotransposons require RNH activity in RNA-protein particles in the cytoplasm, which may account for the rigid requirement to encode their own RNH domain.
Two of the outstanding questions in the evolution of reverse-transcriptase-bearing elements (retroelements) are: When and from where did retrotransposons arise? Previous phylogenetic analyses based on the RT domain have attempted to address this issue (Xiong and Eickbush 1988,1990; Doolittle et al. 1989; Eickbush 1994; Nakamura et al. 1997). Despite some success with outlining the evolution of retroelements, the LTR retrotransposons have proven especially difficult to place phylogenetically. The RT domain of the LTR retrotransposons are severely truncated compared with all other known examples of RTs; non-LTR retrotransposons and telomerases from Eukarya, and retrons, plasmids, and group-II introns from Eubacteria, making it difficult to unambiguously align the sequences (Malik and Eickbush, in prep.).
In this report we have employed a phylogenetic analysis of RNH domains to address the origin of the LTR retrotransposons. In contrast to the ambiguous RT phylogeny, the RNH phylogeny clearly suggests that the LTR retrotransposons evolved from a late-branching lineage of non-LTR retrotransposons. It also suggests that the lineage of LTR retrotransposons leading to vertebrate retroviruses has “replaced”; its original RNH domain. The present-day tether domain (connection) found in vertebrate retroviruses represents a molecular fossil of the original RNH domain.
RESULTS
LTR-Retrotransposons Lack an Important Catalytic Motif of RNH
We performed a multiple alignment of RNH domains with representative sequences from Eubacteria, Eukarya, non-LTR retrotransposons and seven different lineages of LTR-retrotransposons. The different RNH domains were chosen from published GenBank entries. In addition, we included several previously unreported sequences available in public databases because these sequences expanded the distribution of the BEL and DIRS groups of elements. This multiple alignment is presented in Figure1. Overlaid on to this alignment are the secondary structures from the RNH domains fromE. coli and HIV1 (PDB structures 1RDD and 1RDH respectively; SCOP database, http://scop.berkeley.edu).

Alignment of the Ribonuclease HI (RNH) domains. Representative RNH domains from Eubacteria, Eukarya, non–LTR retrotransposons and each of the seven lineages of LTR retrotransposons were aligned usingCLUSTALX and PSI-BLAST. Highlighted in bold are the residues believed important for the catalytic mechanism of RNH, including the four carboxylate (dark arrows) and the single histidine residue (white arrow) that are numbered according to their position in the Escherichia coli RNH domain. Also overlaid are the secondary structures of E. coli and HIV-1 RNH domains (above and below the alignment, respectively). Note the missing histidine residue in all lineages of the LTR retrotransposons except the vertebrate retroviruses.
The catalytic residues for RNH enzymatic activity, indicated by dark arrows (three aspartatic acid and one glutamatic acid residue), are unvaried across all RNH domains. Also indicated is a histidine residue (white arrow) that is believed to be essential for the enzymatic mechanism of RNH (Oda et al. 1993; Kashiwagi et al. 1996). Surprisingly, although the RNH of vertebrate retroviruses have this histidine, all other LTR retrotransposons appear to lack this residue. This ‘deletion’ has gone unremarked in previous reports as most of these analyses focused on the alignment of RNH domains from E. coli (and other Eubacteria) with those of HIV-1 and other vertebrate retroviruses (Johnson et al. 1986; Doolittle et al. 1989; McClure 1991).
We have presented a simplified topological diagram of the three-dimensional structure of E. coli RNH (Structure 1RDD) in Figure 2A, highlighting the four catalytic residues and the histidine residue believed to play a direct role in the RNH catalytic mechanism. RNH domains have been characterized as α-helix/β-sheet/α-helix with the mixed β-sheet consisting of five strands in the order 3–2–1–4–5 with strand 2 antiparallel to the rest and an α-helix between strands 4 and 5. The proposed active site of rnhA is shown in Figure 2B. An alanine substitution for this histidine residue in E. coli resulted in a large drop in kcat/Km (Kanaya et al. 1990). The deletion of this histidine-bearing subdomain in the LTR retrotransposons would suggest altered (perhaps weaker) enzymatic ability for the LTR retrotransposon-borne RNH domains.
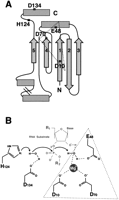
(A) A simplified topological diagram of the Escherichia coli Ribonuclease HI (RNH) domain, indicating the active site residues (see Fig. 1). β-strands are indicated by arrows, and α-helices are shown by boxes. The four carboxylates and single histidine residue are shown. (B) A schematic of the proposed RNH catalytic mechanism is shown (modified with permission from Kanaya et al. 1996). The carboxylate triad typical of other endonucleases with an RNH fold (Yang and Steitz 1995; Rice et al. 1996) is indicated by the dotted triangle.
Vertebrate Retroviruses “Reacquired” Their RNH Domain
One of the most surprising aspects of the RNH sequence alignment in Figure 1 is the suggestion that the vertebrate retroviruses' RNH domain is enzymatically more similar to those of eubacterial and eukaryotic genomes, and non-LTR retrotransposons, than it is to other LTR retrotransposons. We performed a phylogenetic analysis of the RNH domains based on the multiple alignment in Figure 1 to test whether the origin of the RNH domains in vertebrate retroviruses was distinct from other LTR-retrotransposons. Figure 3presents the neighbor-joining tree obtained from this comparison. To test the effect on the phylogeny arising simply from the fact that the subdomain containing H124 is missing from the LTR retrotransposons, we have also excluded this region in a separate analysis. The same phylogeny was obtained as that with the full RNH sequences (data not shown).
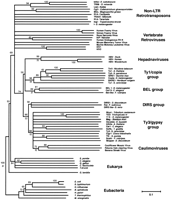
Phylogeny of the Ribonuclease HI (RNH) domains. A Neighbor-Joining (NJ) tree of the various RNH domains was performed based on the alignment of ∼140 amino acid residues in Figure 1. Bootstrap analysis was performed and nodes were collapsed to a 50% consensus. Bootstrap support (percentage from 1000 trials) for the various nodes is shown above the nodes. Maximum parsimony (MP) analysis of the RNH sequences agreed with the NJ analysis but showed lower bootstrap values for most nodes. Bootstrap values from the MP analysis for the major groupings are shown in italics if greater than 50%. The phylogeny is rooted using the eubacterial RNH domains as the outgroup. Note that the phylogenetic position of the vertebrate retroviruses is in conflict with that shown in Figure 4. All retroelement sequences are readily accessible from GenBank and previous reports (Bowen and McDonald 1999;Malik and Eickbush 1999; Malik et al. 2000).
For comparison with the RNH phylogeny, we present in Figure4 a phylogeny of representative LTR retrotransposons based on the RT domain using non-LTR retrotransposons as an outgroup. This phylogeny is in general agreement with those presented earlier with the only significant uncertainty being the relative position of the Ty1/copia and hepadnaviral groups (Xiong and Eickbush 1990; Bowen and McDonald 1999; Malik et al. 2000). Both the RT and RNH phylogeny reveal four distinct lineages of LTR retrotransposons: the Ty1/Copia, BEL, DIRS1 and Ty3/gypsy groups, as well as three classes of viruses: the retroviruses, hepadnaviruses, and caulimoviruses. The phylogenetic relationship within and among these groups is virtually the same using these two sets of data, with one striking exception. In the RNH phylogeny, retroviruses are located distal to the four retrotransposon groups as well as to caulimoviruses and hepadnaviruses, whereas in the RT phylogeny, retroviruses are a sister group to the Ty3/ gypsy elements and caulimoviruses.
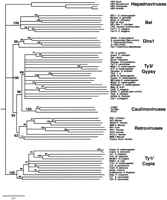
Phylogeny of the long-term repeat (LTR) retrotransposons based on their reverse transcriptase (RT) domains. The phylogram is a 50% consensus tree of the elements' RT domains (∼240 amino-acid residues) based on the neighbor-joining (NJ) method, and is rooted using non-LTR retrotransposon RTs as an outgroup (not shown). Bootstrap values are shown associated with corresponding nodes. This tree is in agreement with prior analyses (Bowen and McDonald 1999; Malik et al. 2000) except for the relative position of the Ty1/copia and hepadnaviral groups and the additional DIRS1-like sequences from sea urchin and two teleosts (accession nos. AZ181274, AL305423, and AF112374, respectively).
Which of these two analyses is an accurate reconstruction of LTR retrotransposon evolution? The RT phylogeny is consistent with the more generally held view of LTR elements. In particular, the integrase (IN) domains of retroviruses is clearly most similar in sequence and domain structure to that of the Ty3/gypsy group of elements (Capy et al. 1996;Malik and Eickbush 1999). Other features of these elements, including the order of the different enzymatic domains of the pol gene are similar between retroviruses and the Ty3/gypsy group. Thus, parsimony suggests that it is the RNH phylogeny that is at odds with the evolution of LTR retrotransposons. This discrepancy could be reconciled if we propose that the ancestral vertebrate retrovirus “replaced” its preexisting RNH domain with another RNH domain from a source outside the LTR retrotransposon group.
Is there any evidence for this ancient replacement of the retroviral RNH domain? In all members of the LTR retrotransposon lineage, the RNH domain is found immediately adjacent to the RT domain. Examination of the relative positions of the RT and RNH domains in retroviruses clearly reveals “paleontological” evidence for an RNH replacement. Retroviruses have an additional domain separating the RT and RNH domains. This additional domain has been referred to as the “tether” or “connection” domain of the retroviral RT–RNH structure (Kohlstaedt et al. 1992). There is little primary sequence similarity to suggest that this retroviral ‘tether’ was the remnant of a previous RNH domain. However, as presented in Figure5C, the three-dimensional structure of the HIV-1 tether region from the HIV-1 RT–RNH crystal structure reveals a remarkable structural similarity between it and the functional RNH domains of HIV-1 (Davies et al. 1991; Kohlstaedt et al. 1992), E. coli (Yang et al. 1990) and Thermus thermophilus (Ishikawa et al. 1993).

Schematic three-dimensional diagrams of the RNH domains fromEscherichia coli (PDB structure 1RDD), Thermus thermophilus (1RIL) and HIV-1 (1RVT) are shown along with the tether domain of HIV-1 (1RVT). β-strands and α-helices are represented by arrows and cylinders, respectively, using theCn3D viewer software (version 3.0). Note that the tether (connection) domain has the same fold (also see Artymiuk et al. 1993) as the enzymatically active ribonuclease HI domains.
The tether domain of HIV-1 has the same organization as the enzymatically functional RNH domains (Fig. 5C), except that it lacks the carboxy-terminal αβα motif and possesses none of the conserved catalytic residues. This similarity of the HIV tether and RNH domains has been previously noted using three-dimensional searching techniques and was suggested to have been the result of an RNH gene duplication event (Artymiuk et al. 1993). These authors found a RMS error of only 1.77 Α over 48 core C-α-atoms on superposition of the proposed equivalent five β strands and single α helix (Fig. 5C). Our phylogenetic analysis suggests that this domain was not the result of a duplication, but was rather the acquisition of a new domain from a source outside the LTR retrotransposons. The “new” RNH domain acquired by vertebrate retroviruses may have been more proficient than the “old” one by virtue of the conserved histidine residue (H124 in Fig. 1) involved in the suggested catalytic mechanism (Fig. 2B).
RNH folds are typical of other endonucleases, including the retroviral and DNA-mediated transposases/INs, the RuvC resolvases as well as exonuclease domains of DNA polymerases (Dyda et al. 1994; Yang and Steitz 1995; Rice et al. 1996). In each of these enzymes, three catalytic carboxylates are similarly arranged (Fig. 2A), whereas RNH and RuvC resolvases have an additional fourth conserved carboxylate (D134). Thus, although an RNH fold by itself is not an absolute indicator that the tether was originally an RNH domain, parsimony argues against the likelihood that the tether was derived from any of the other endonucleases; this would invoke not only the loss of the ancestral RNH domain, but the subsequent acquisition and degeneration of another endonuclease.
Non-LTR Retrotransposons Arose Earlier Than LTR Retrotransposons
The phylogeny in Figure 3 is rooted on the various eubacterial representatives of RNH (the Archaea have no rnhA homolog). Using this rooting, non-LTR retrotransposon and the LTR-retrotransposon RNH domains group together, indicating a common evolutionary origin. The phylogenetic proximity of the retrotransposon lineages to the eukaryotic RNH sequences suggests that the origin of this RNH domain was an early eukaryote. Indeed, the diplomonad Giardia lambliaappears to be the outgroup not only to other eukaryotic but also to all retroelement-encoded RNH domains. Is this (acquisition of an RNH domain from an early eukaryote) yet another example of replacement of a preexisting RNH domains, as hypothesized for retroviruses? Or does this phylogeny reflect the original acquisition of this enzymatic domain by both non-LTR and LTR retrotransposons?
We have addressed this issue previously, in the non-LTR retrotransposon lineage. We and others have postulated that the most likely origin of the non-LTR elements are the group-II introns found in eubacteria and the organelles of fungi and plants (Zimmerly et al. 1995; Cousineau et al. 1998; Malik et al. 1999; Lambowitz et al. 1999). This model is based on both the phylogenetic relationship of their RT domains (Xiong and Eickbush 1990; Malik et al. 1999) and the similarity of their target primed reverse transcription mechanisms used for insertion (Luan et al. 1993; Zimmerly et al. 1995). When the group-II introns are used to root the non-LTR retrotransposon phylogeny, it suggests that the original non-LTR elements were elements which encoded a single open reading frame (ORF) and contained an endonuclease domain with an active site similar to certain restriction enzymes (Malik et al. 1999 Yang et al. 1999). Evolving from these original non-LTR retrotransposons were elements that acquired a gag-like first ORF and replaced the original restriction-like endonuclease with an apurinic-like endonuclease (APE). This nonspecific APE domain enabled the non-LTR elements to insert more widely throughout the genome resulting in the diversification of a number of different lineages. One of these new lineages acquired an RNH domain giving rise to the present day lineages we have termed the I, R1 and Tad clades (see Malik et al. 1999). Thus, our previous analysis of all non-LTR retrotransposon sequences suggests that the phylogeny in Figure 3 reflects the original acquisition of an RNH domain from a eukaryotic host. The phylogeny of the RNH domains is unable to resolve branching order of the three extant non-LTR clades that are derived from this lineage (Malik et al. 1999).
In the case of the LTR retrotransposons (excluding the vertebrate retroviruses), acquisition of the original RNH domain again appears to be monophyletic. This single lineage is not clearly resolved from the multiple extant non-LTR retrotransposon RNH lineages. Because the phylogeny derived from this RNH domain is the same as the RT phylogeny (with the exception of the retroviruses describe above), the entire lineage of LTR retrotransposable elements thus appears to be no older than one of the younger lineages of non-LTR retrotransposons. Consistent with the proposal that the LTR retrotransposons arose later in the eukaryotic lineage than the non-LTR retrotransposons is their phylogenetic distribution. Although non-LTR retrotransposons have been found in the oldest eukaryotes, the diplomonad Giardia lamblia(Arkhipova and Meselson 2000; Burke et al., in prep.) and trypanosomes (Kimmel et al. 1987; Teng et al. 1995), LTR-retrotransposons have not been found in these lineages. This phylogeny thus suggests that the original LTR-retrotransposon RNH domain was acquired from a non-LTR retrotransposon. This event may have been repeated when retroviruses replaced their RNH domain. However, the poor resolution of the non-LTR retrotransposon and vertebrate retroviral lineages does not allow us to rule out alternate possibilities for the source of this acquisition.
What was the structure of the precursor LTR element that acquired this RNH domain? In the following section we present arguments for what we believe is the most likely origin of the LTR retrotransposons, the fusion of a DNA-mediated transposon and a non-LTR retrotransposon.
DISCUSSION
Vertebrate Retroviruses: RNH Connections
In this report we have presented phylogenetic analyses that indicate the vertebrate retroviral lineage has replaced its RNH domain. This event must have occurred early in the evolution of retroviruses because all known retroviral lineages contain this new RNH domain. Our analysis indicates that the retroviruses probably obtained their RNH domain from a non-LTR retrotransposon (Fig. 3). This close relationship of the RNH domain from retroviruses and non-LTR retrotransposons can be observed in the first comparisons of RNH domains in different types of retroelements (Doolittle et al. 1989).
The presence of a connection domain represents the most dramatic difference between retroviral RTs and the RTs of LTR retrotransposons. Because of the advantages of reducing its genome size, we would have expected the preexisting RNH domain to have been rapidly lost after the retroviral lineage gained a new domain. The fact that the connection domain still exists suggests that this “fossilized” RNH domain is performing another important role in the lifecycle of the virus. What is this present-day function? In the case of the HIV protein, the best studied retroviral reverse transcriptase, the active enzyme is a heterodimer composed of a p66 subunit containing an RT, a connection and an RNH domain and a p51 subunit containing only the RT and connection domains. Several studies have remarked on the structural and possible functional role of the connection domain in the formation of this heterodimer (Wang et al. 1994; Divita et al. 1994; Debyser and De Clercq 1996). For example, it has been shown to be crucial in mediating the conformational changes required of the p66/p51 heterodimer for reverse transcription (Bahar et al. 1999) and for RNH activity (Smith et al. 1994). Indeed, contacts by the connection domains make up one-third of the total contacts between the two subunits, and the connection domain in the p51 subunit makes close contact with the tRNA primer annealed to the viral RNA template (Kohlstaedt et al. 1992). Finally, the connection domain may even play a role in the incorporation of protein in the virus particle. Mutations in the connection domain prevent the efficient packaging of HIV viral particles (Mak et al. 1997). It appears likely that, although the RNH domain in other LTR-retrotransposons may carry out both enzymatic and structural roles, the presence of a connection domain in retroviruses has allowed subfunctionalization (Lynch and Force 2000). Thus, although the newly acquired RNH domain is enzymatically active, the connection domain may still carry out its ancestral structural function.
The Chimeric Origin of LTR Retrotransposons
Perhaps the most interesting aspect of the RNH phylogeny described in this report is its implication for the origin of eukaryotic retrotransposons. The RNH domains of the I, R1, and TAD clades of non-LTR elements and the original RNH domain in LTR elements (i.e., before the reacquisition of the RNH domain by retroviruses) appear to have a common origin. These acquisitions appear to have arisen sometime after the origin of eukaryotes. Note that the branch containing these retrotransposon sequences is more closely related to the RNH from the crown group of eukaryotes than is the RNH domain of G. lamblia(Fig. 3). Based on the phylogeny of the RT, APE, and RNH domains of the non-LTR retrotransposons, we had previously concluded that the acquisition of the RNH domain was a monophyletic event occurring late in the evolution of these elements (Malik et al. 1999).
In contrast to the non-LTR retrotransposons, few models have been proposed for the origin of the LTR retrotransposons. First, no prokaryotic elements have been found that could be regarded as likely progenitors of the present-day LTR retrotransposons. Second, the oldest lineage of extant LTR retrotransposons, the Ty1/copia lineage (Xiong and Eickbush 1990; Fig. 4) contains all the components of a complete LTR retrotransposon (a gag-like ORF1, and a polgene with protease, RT, RNH, and IN domains). The only difference between the Ty1/copia group of elements and the other groups of LTR retrotransposons is the position of the IN domain. It is found upstream of the RT/RNH domains in the Ty1/copia group but is downstream from the RT/RNH in the BEL, Ty3/gypsy and retroviral clades. Unlike the gradual addition and replacement of domains in the non-LTR retrotransposons, the only dramatic changes that have occurred since the evolution of LTR retrotransposons were the addition in several lineages of env-like domains (Malik et al. 2000) and the loss of the IN domain in the DIRS group (Cappello et al. 1985).
We propose that the origin of the LTR retrotransposons was the fusion of a DNA-mediated transposon and a non-LTR retrotransposon. Although this model is highly speculative, it is the only simple model that can explain the sudden origin of the two-step mechanism used by LTR retrotransposons in the absence of obvious eubacterial precursors. Based on the similarity of the IN of LTR retrotransposons and the transposases of DNA transposons, Capy et al. (1998) have also recently postulated that one likely origin of the LTR retrotransposons was by a DNA-mediated transposon acquiring RT activity.
Transposition Mechanism
Both DNA-mediated elements and non-LTR retrotransposons have simple, essentially one-step mechanisms of inserting new copies of the element into the genome. DNA transposons encode a transposase, which can directly excise the element from one location for insertion elsewhere. Non-LTR retrotransposons encode a reverse transcriptase, which can synthesize a new DNA copy of the element directly on to the chromosome from an RNA copy by target-primed reverse transcription. LTR retrotransposons, in contrast, use a variation of both of these methods. They use a reverse transcriptase to make a new DNA copy of the element from its RNA transcript, but this copy is made in the cytoplasm separate from the chromosome. Subsequently, they utilize a transposase (IN) to insert this DNA copy into the chromosome by a mechanism similar to that of DNA transposition. Unfortunately, the two most critical enzymatic activities encode by the LTR retrotransposons, RT, and the IN, have not been very useful in tracing the origin of these elements.
IN
The IN domains of retroviruses and LTR retrotransposons have long been known to possess similar structure and enzymatic activity to those of eukaryotic and prokaryotic transposases (for review, see Craig 1995;Mizuuchi 1992). However, this domain does not afford the resolution required to determine the phylogenetic relationship of the LTR element domain to that of the transposon lineage, other than to conclude it is derived from a lineage that contained a D, D35E catalytic site (Fayet et al. 1990; Doak et al. 1994). Indeed this core domain has evolved so quickly, and many subdomains have been added in different lineages, that it is difficult to trace even the phylogeny of the LTR elements themselves using IN sequences (Malik et al. 1999; Capy et al. 1996).
RT
Traditionally, the RT domain has been the favorite phylogenetic tool to trace the evolution of retroelements; it is one of the largest domains and within any group, shows the greatest sequence conservation. However, attempts to trace the origin of the LTR retrotransposons using a RT phylogeny of different retroelements have been beset with artifacts. Previous reports by ourselves and others (Xiong and Eickbush 1988, 1990; Doolittle et al. 1989; Eickbush 1997; Nakamura et al. 1997) have indicated that the LTR retrotransposon RT domains are the most divergent of all elements, even more divergent than telomerase and retron domains. The problem arises from the “pruned” RT domains of LTR retrotransposons, which are only 60% the size of these domains in other retroelements. Some conserved regions of the RT domain appear to be missing in the LTR retrotransposons, whereas other regions appear to have been duplicated. Thus is it difficult to unambiguously align the sequences (Malik and Eickbush, in prep.). Part of the reason for this extensive divergence of the LTR element RT domain may be the different requirements placed on the enzyme. In the case of all other reverse transcriptases (non-LTR, telomerase, group II, and retron) the reverse transcriptase specifically binds its RNA template and primes reverse transcription from the 3′ end of a DNA molecule or the 2‘ hydroxyl residue of an RNA. With the LTR retrotransposons reverse transcription is primed by an annealed primer. Thus the RT of LTR retrotransposons performs what is essentially an extension reaction, not the specific priming reaction carried out by the other RTs.
RNH
Compared to the variation found in RT and IN domains, the size of the RNH domain is very similar between the LTR and non-LTR elements and their eukaryotic and eubacterial sources. There is little ambiguity in the RNH alignments. For example, the alignment shown in Figure 1, although it contains many more types of retroelements, is identical to that derived in the first analysis of such sequences (see Fig. 15 inDoolittle et al. 1989). The RNH phylogeny derived from this sequence comparison clearly suggests that present-day LTR retrotransposons arose later in the eukaryotic lineage than the non-LTR retrotransposons. The most likely source of the RNH domain in LTR retrotransposons is one of the younger lineages of non-LTR retrotransposons. It is ironic that the two enzymes most important to the replication reaction of the LTR retrotransposons (RT, IN) are not very useful in tracing the path of origin of these elements. Meanwhile, RNH (an enzyme that plays a relatively minor role in the process) is more useful in tracing this path because its simple enzymatic function has remained unchanged in both types of retrotransposons as well as in the host genome.
gag-Like ORF
Our model for the chimeric origin of the LTR retrotransposons is also supported by the similarity of the first ORF (gag-like) in the LTR elements and only those non-LTR elements that contain an RNH domain. The gag-like proteins encoded by both elements share one or more cysteine–histidine motifs that are believed to play a role in nucleic acid binding (Jakubczak et al. 1990; Dawson et al. 1997). Although many cellular proteins contain cysteine–histidine-binding motifs, the first ORFs of these retrotransposons share an unusual spacing of residues (C-X2-C-X4-H-X4-C) that is extremely rare in other cellular proteins (Berg and Shi 1996). We would, therefore, argue that the gag genes of LTR retrotransposons and the first ORF of the RNH-containing non-LTR elements share a common ancestry.
Based on these findings, we can summarize the events leading to the proposed chimeric origin of LTR-retrotransposons. We propose that the non-LTR retrotransposons contributed the RT–RNH as well as the first ORF (gag-like) domain to LTR retrotransposons. A DNA transposon contributed the integrase (transposase) as well as the requirement for a short inverted terminal repeat at the ends of the element. To complete the formation of a fully functional LTR retrotransposon required several additional components. The only additional protein domain was a protease domain, which may have been derived from the host’s pepsin gene family (Doolittle et al. 1989). An alternative means to prime reverse transcription was accomplished by the use of an abundant small stable RNA (tRNA) to anneal to the RNA template. Finally, as a means to overcome the problem of replicating the ends of any DNA molecule, the element evolved long direct terminal repeats (LTRs) that promoted jumps between ends.
Why did the fusion of a DNA transposon and a RT- containing element occur in early eukaryotes but not during their long history together in Eubacteria? In bacteria, where transcription and translation are physically linked, an RNA template–RT complex has ready access to new target sites by the simple target-primed reverse transcription mechanism (Fig. 6). In eukaryotes, however, the presence of a nuclear membrane and the export of RNA out of the nucleus for translation in the cytoplasm mean that the non-LTR element is faced with a new problem. Once the protein is translated in the cytoplasm, it must reenter the nucleus either taking its RNA template with it or acquiring a new template in the nucleus. Using the same RNA molecule as template that was used for translation (cis-preference) provides a powerful approach to insuring the duplication of only active elements (see Wei et al. 2001). This necessity of devising a mechanism to take the template back into the nucleus (or obtaining greater stability while waiting for the nuclear envelope to break down) may have provided the selective pressure for the two-step transposition employed by LTR retrotransposons. Reverse transcription of the RNA template in the cytoplasm generates a highly stable DNA copy that can subsequently undergo nuclear import and integration (Fig. 6). This selection pressure to increase template stability in the cytoplasm also provides a rationale for the acquisition of a nucleic-acid-binding chaperone, the ORF1 protein, in later lineages of non-LTR elements that are not present in the original lineages (Malik et al. 1999; Martin and Bushman 2001).
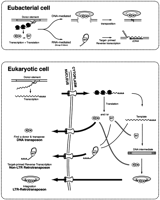
The role of the eukaryotic nucleus in the evolution of long-term repeat (LTR) retrotransposons. In eubacteria, transcription and translation are coupled; thus the encoded transposase (here labeled IN for integrase) of a DNA-mediated element can immediately bind the donor element for transposition. For simplicity, we have only shown the DNA-mediated transposition reaction as cut-and-paste, but in Eubacteria, a replicative form of transposition can also occur (Mizuuchi 1992). In the case of the RNA-mediated reaction, the reverse transcriptase (RT) can immediately bind its own transcript and initiate target-primed reverse transcription. This eubacterial precursor of the eukaryotic retrotransposons is assumed to be a mobile group II intron (Cousineau et al. 1998). The situation differs for mobile elements in eukaryotes where transcription and translation are uncoupled. Synthesis of IN in the cytoplasm means that this enzyme must enter the nucleus and find a donor for transposition. This can result in the transposition of defective copies that only retain the correct terminal repeats. In the case of the non-LTR retrotransposons, the RT must drag its RNA template back into the nucleus (cis preference) or find a new RNA template. Only the former will insure the production of active copies (Wei et al. 2001). This need to stabilize the template for entry back into the nucleus (or to wait for the breakdown of the nuclear membrane during cell division) is postulated to be the selective force that enabled the evolution of the LTR retrotransposons. LTR retrotransposons utilize both RT and IN activities. First, the RNA template is reverse transcribed into a double stranded DNA template. Second, an integrase complex shuttles this complex to a target site for integration either through the nuclear membrane or during nuclear breakdown at cell division.
The two-step LTR retrotransposition mechanism would also have certain advantages over that of the simple DNA transposition reactions. It has been suggested that DNA transposons are not stable for long periods of evolution in a genome both because the transposase made in the cytoplasm has equal probability of binding defective genomic copies for transposition (Kaplan et al. 1985; Hartl et al. 1997) and because the cut-and-paste mechanism of some eukaryotic DNA transposons can not guarantee an increase in copy number. The two-step LTR retrotransposition mechanism overcomes both these problems. Thus, the separation of transcription and translation in early eukaryotes provided the environment for the evolution of a more complex, hybrid mobile element that had advantages over the two classes of elements acquired directly from eubacteria.
METHODS
RNH sequences were obtained from GenBank via PSI-BLASTand TBLASTN (Altschul et al. 1997) searches against the nonredundant database. The sequences were then aligned usingCLUSTALX (Thompson et al. 1997) and manually refined usingPSI-BLAST alignments as a template. RNH structures were obtained from the PDB database (http://www.rcsb.org/pdb). Schematic diagrams of the various PDB files were made using the Cn3Dviewer software version 3.0 (http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml). Phylogenetic analyses were performed according to the neighbor-joining method (Saitou and Nei 1987) using PAUP* (Swofford 1999). Maximum parsimony analysis was also carried out using PAUP* and the heuristic search option with the number of trees retained at each step limited to 10. Although trees obtained by both methods are in strict agreement, the bootstrap support is generally lower using maximum parsimony.
Acknowledgments
We thank Pauline Ng, Bill Burke, and Steve Henikoff for comments on the manuscript. We especially thank Bill Burke for help preparing the figures. We also thank S. Kanaya for permission to use a schematic of RNH's proposed catalytic mechanism. This work was supported by grants from the National Science Foundation to T.H.E. (MCB-9974606), from the National Institutes of Health to Steve Henikoff (GM-29009), and a postdoctoral fellowship to H.S.M. from the Helen Hay Whitney Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL hsmalik{at}fhcrc.org; FAX (206) 667-5889.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.185101.
-
- Received February 2, 2001.
- Accepted April 11, 2001.
- Cold Spring Harbor Laboratory Press








