
Evolution of Gene Order in the Genomes of Two Related Yeast Species
- 1Unité de Génétique Moléculaire des Levures, Centre National de la Recherche Scientifique Université Pierre et Marie Curie, Institut Pasteur, 75724 Paris Cedex 15, France; 2Collection de Levures d'Interêt Biotechnologique, Laboratoire de Génétique Moléculaire et Cellulaire, 78850 Thiverval-Grignon, France; 3Laboratoire de Biologie Cellulaire de la Levure, 33077 Bordeaux Cedex, France
Abstract
Changes in gene order between the genomes of two related yeast species, Saccharomyces cerevisiae and Saccharomyces bayanus var. uvarum were studied. From the dataset of a previous low coverage sequencing of the S. bayanus var.uvarum genome, 35 different synteny breakpoints between neighboring genes and two cases of local gene inversion were characterized in detail. The number and the type of the chromosomal rearrangements that have lead to these differences were identified. We show that evolution of gene order in the genomes of these two yeast species is driven mainly by gene duplication onto different chromosomes followed by differential loss of the repeated copies. In addition, local gene inversions also would result from a mechanism of gene duplication, but in an inverted orientation, followed by loss of the original copy. The identification of traces of anciently duplicated genes, called relics, show that the loss of duplicates is more frequently caused by the accumulation of numerous mutations in one of the two copies than by DNA deletion. Surprisingly, gross chromosomal rearrangements such as translocations have only a minor effect on gene order reshuffling as they account for <10% of the synteny breakpoints.
[The sequence data have been submitted to the EMBL Library under accession nos. AJ316068 and AJ316069.]
The comparative genomics approach has proved to be fruitful in retracing chromosome maps evolution between eukaryotic genomes. Nevertheless, most of the studies rely on the relative localization of markers along physical and/or genetic maps (Gale and Devos 1998; O'Brien et al. 1999; Ranz et al. 2001). At this level of resolution, gross chromosomal rearrangements such as translocation or large inversion are identifiable, but small interstitial rearrangements remain undetected. The fine comparison of chromosome maps will be improved greatly by sequencing entire genomes from closely related species. In eukaryotes, large sequence datasets were only available for organisms too distantly related to yield valuable insights into the evolution of chromosome maps. A recent sequencing project, designated Génolevures, has laid the foundations of a true comparative genomics within a define eukaryotic phylum, the Hemiascomycetes (http://cbi.labri.u-bordeaux.fr/Genolevures; Souciet et al. 2000). In this study, a low coverage random sequencing has been performed on 13 different yeast species. Among them, Saccharomyces bayanusvar. uvarum (abbreviated here as S. uvarum) is the closest to S. cerevisiae. It belongs to theSaccharomyces sensu stricto complex (Vaughan-Martini and Martini 1998). A previous study on the chromosomal evolution between the six species belonging to this complex has shown that their genomes, each composed of 16 chromosomes, differed by a limited number of chromosomal translocations (Fischer et al. 2000). For instance, three reciprocal and one nonreciprocal translocations have been characterized between the genomes of S. cerevisiae and S. uvarum. The average amino acid identity between their ORF products is approximately 80% (Malpertuy et al. 2000b), which compares to the level of divergence between mouse and human (86% amino acid identity;Makalowski and Boguski 1998). However, the synteny is very well conserved between these two genomes as 98% of the genes in S. uvarum have retained the same neighboring relationships as inS. cerevisiae (Bon et al. 2000; this work). We chose to study the evolution of gene order between these two genomes presenting a highly similar genetic organization to identify the primary events leading to changes in gene order before the accumulation of numerous rearrangements erases the traces of the initial events. The sequence data generated by the 0.4× coverage of the S. uvarum genome allowed us to follow gene by gene the mechanisms leading to gene order reshuffling between the two genomes by combining a computational analysis of the synteny among the 1810 gene couples identified in Bon et al. (2000) and an experimental approach to validate and map the synteny breakpoints. The results presented here uncover the central role played by the duplications in the evolution of gene order. Most of the synteny breakpoints corresponded to localized changes resulting from the ancient duplication of few genes followed by differential loss of the duplicated copies in the two species. Although it is commonly believed that gene order along chromosomes is reshuffled by large chromosomal rearrangements such as translocations and inversions, we show that these rearrangements have only a minor effect on gene order evolution between the genomes of S. cerevisiae and S. uvarum.
RESULTS
We describe the identification of a set of 35 nonsyntenic gene couples between S. uvarum and S. cerevisiae. These gene couples were identified from paired sequences obtained from both ends of the plasmid inserts of the S. uvarum library and not from sequences assembled from individual shotgun reads, thus avoiding the problem of contig misassemblies. We then describe in detail the events that lead to gene order changes. We show that chromosomal translocations account only for three of the nonsyntenic gene couples. In contrast, 10 couples clearly result from gene loss within ancestral duplications. These synteny breakpoints correspond either to S. cerevisiae singletons localized within and outside of the previously recognized ancestral block of duplication, which still exist as duplicates in the S. uvarum genome, or to alternate loss between S. cerevisiae and S. uvarum of the two copies of anciently duplicated genes. The duplication-loss mechanism also is invoked to explain the two cases of local gene inversions without loss of synteny. Finally, the remaining nonsyntenic gene couples correspond to rearrangements that are also compatible with a duplication-loss mechanism.
Identification of a Set of 35 Nonsyntenic Gene Couples
In this work, we have characterized 35 different nonsyntenic gene couples between the genomes of S. cerevisiae and S. uvarum. The “nonsyntenic gene couples” were defined here as two neighboring ORFs in the S. uvarum genomic library inserts whose homologs lie on different S. cerevisiae chromosomes or, by extension, on the same chromosome but very distant from each other.
In the previous work by Bon et al. (2000), BLASTXcomparisons of 5140 paired-end random sequence tags (RSTs) of S. uvarum against the S. cerevisiae proteome defined 1810 nonredundant gene couples, 1776 couples with two nonambiguous matches and 34 couples with a least one ambiguous match. Ambiguity results from the fact that S. cerevisiae genes may form family of paralogs. Among the 34 pairs corresponding to ambiguous matches, we considered here 16 gene couples as nonsyntenic because none of the matches of the first ORF is located next to any of the matches of the second ORF in the genome of S. cerevisiae (Table1). The colinearity of each of these 16 plasmid inserts with the S. uvarum genome was shown by PCR amplification of the corresponding regions in the S. uvarumgenomic DNA (data not shown).
Nonsyntenic Gene Couples Having Ambiguous Matches
Thirty-eight nonsyntenic gene couples originally were identified among the 1776 couples with nonambiguous matches (Bon et al. 2000). A closer look at these sequences allowed us to correct previous annotation mistakes (see Methods) leaving 30 couples to study. Hybridization experiments on pulse-field gel electrophoresis (PFGE) separated S. uvarum chromosomes revealed that another 12 gene couples corresponded to cloning chimeras (see Methods). Finally, an additional couple was identified during the course of this work,SuYML051w-SuYJR057w (Su for S. uvarumhomolog to YML051w and YJR057w, respectively; Table 2), bringing the number of true nonsyntenic gene couples to 19. The nonchimerical nature of these 19 clones was shown by PCR amplification of the corresponding regions in the S. uvarum genomic DNA and/or by hybridization of the two ORFs onto the same S. uvarum chromosome. These nonsyntenic gene couples were examined in detail to understand the type of mechanism responsible for the loss of synteny between S. cerevisiae and S. uvarum.
Nonsyntenic Gene Couples Obtained from Non-Ambiguous Matches, Inversions, and Deletion
Loss of Synteny by Chromosomal Translocation
One nonreciprocal and three reciprocal translocations have been characterized previously between the S. cerevisiae and theS. uvarum genomes, leading to seven synteny breakpoints (Fischer et al. 2000). Three of these translocation breakpoints also were identified during the course of the Génolevures project (Bon et al. 2000). This number is in good agreement with the expected value of 2.8 given by the genome coverage of 0.4× (0.4 × 7 = 2.8). However, among the 35 synteny breakpoints characterized in this work, only three nonsyntenic couples are explained by translocations, which represents <10% of the total number of changes in gene order.
The breakpoint of the translocation SuIItIVR(translocation of a piece of the right arm of chromosome IV onto chromosome II, according to the nomenclature of chromosomes defined inFischer et al. (2000)), comprising the pairSuYBR030w-SuYDR012w was characterized previously and sequenced (Ryu et al. 1998). The authors showed that this translocation results from an ectopic recombination event between two copies of a duplicated gene, RPL2A and RPL2B.
Two other nonsyntenic couples correspond to the two breakpoints of the same reciprocal translocation, involving chromosomes VIII and XV (Table2). Both regions were amplified from S. uvarum total DNA, sequenced (accession numbers AJ316068 and AJ316069) and compared with the corresponding S. cerevisiae sequence (Fig.1). On chromosome VIII in S. cerevisiae, the translocation breakpoint is localized in a region between YHR014w (SPO13) and YHR015w(MIP6) comprising two tRNA genes (tS[AGA]H and tQ[UUG]H) and three solo long terminal repeats (LTRs) from the retrotransposon Ty1 (truncated copies of 119, 241, and 324 bp, respectively). On chromosome XV, there is no tRNA, no LTR, or any repeated sequences in the intergenic region between YOR018w (ROD1) andYOR019w. Comparison of the junction sequences in S. uvarum with the corresponding regions in S. cerevisiaerevealed that the translocation has occurred between the tS(AGA)H tRNA gene and a full-length LTR related to the delta sequence of Ty1 (331 bp flanked by the characteristic 5-bp inverted repeat TGTTG) on chromosome VIII and within the intergenic regionROD1-YOR019w on chromosome XV (Fig. 1). This translocation breakpoint lies in a region of 377 and 524 bp on chromosome SuVIIItXV and SuXVtXIII, respectively, where no homology with any LTR or repeated sequences were recognizable. It is noteworthy that no sequence identity is found between the two recombining regions, either in S. uvarum or in S. cerevisiae. Thus, this translocation results either from an illegitimate recombination mechanism between nonhomologous sequences or from homologous recombination between sequences that have subsequently diverged. During the Génolevures program, homology withYHR016c (S. cerevisiae chromosome VIII) andROD1 (S. cerevisiae chromosome XV) also was found at both ends of the same plasmid insert (RSTs AT0AA013C08D1 and AT0AA013C08T1) in the library of S. servazzii, a species that does not belong to the sensu stricto complex (Casaregola et al. 2000). This finding supports the idea that the S. uvarum chromosomes represent the ancestral form and that the translocation has occurred in the S. cerevisiae lineage.
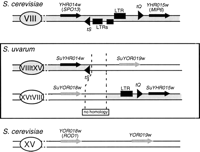
Junctions of the reciprocal translocation between chromosomes VIII and XV. Chromosome numbers are indicated in the ovals that represent the centromeres. Both Watson and Crick strands are represented, and the genes are symbolized by arrows. Sequences from chromosome VIII and XV of S. cerevisiae are drawn in gray and white, respectively. The tRNA genes are abbreviated as tS and tQ (see text) and depicted as triangles. (black boxes) LTRs from Ty1. These two corresponding regions of S. uvarum chromosomes were sequenced (accession numbers AJ316068 and AJ316069).
Loss of Synteny by Gene Loss within Ancestral Duplications
The loss of synteny between two genes also may result from the ancient duplication onto two different chromosomes of one or both genes of the couple followed by a differential loss of the duplicated copies in the two species studied. Now, the analysis of the nonsyntenic couples between S. cerevisiae and S. uvarum offers us a possibility to examine the importance of this mechanism in genome evolution. Three distinct situations were encountered.
First, we found unique genes within the S. cerevisiaeancestral blocks of duplication that correspond to duplicated genes in the S. uvarum genome. Blocks of ancestral duplication were identified in the S. cerevisiae genome as arrays of duplicated genes, called paralogs, interspersed with genes that are not duplicated (Lalo et al. 1993; Goffeau et al. 1996; Coissac et al. 1997; Mewes et al. 1997; Wolfe and Schields 1997). As it is hypothesized that random deletion of one of the two copies occurred after the initial duplication event (Achaz et al. 2000; Keogh et al. 1998), block duplication followed by individual gene loss can result in loss of synteny. This was observed in two distinct situations, the first within ancestral block of duplication 3 (according to the nomenclature of the blocks defined in Wolfe and Schields [1997]) and second on the border of duplication block 8 (Fig. 2, A and B, respectively). In S. cerevisiae, YDR037w (KRS1) and YBR164c (ARL1) exist as singletons. In S. uvarum, both genes were found as duplicates that originated from the ancestral duplication of blocks 3 and 8, respectively. This was shown by identification by dot-matrix analysis (see Methods) of traces of the second copy of these genes within the S. cerevisiaegenome. These traces were named relics to account for the ancestral presence of a duplicated gene in the present-day corresponding intergenic region. A relic of KRS1 was identified onto chromosome II within the duplication block 3, and a relic ofARL1 was found onto chromosome XVI on the border of duplication block 8 (Fig. 2). The localization of the ARL1relic shows that the ancestral duplication between chromosomes II and XVI probably was more extended than the block 8 characterized inS. cerevisiae. The presence of these relics in the S. cerevisiae genome proves that some of the singletons within the present-day duplicated blocks existed originally as duplicates. Moreover, it shows that gene loss within the duplicated blocks occurred, at least in these cases, by accumulation of mutations rather than deletion of one copy. For instance, the relic ofKRS1, which shows the highest sequence conservation of all relics identified in this work (see relics on Figs. 2, 5, and 6 for comparison), differs from the active copy of KRS1, which is 1776 nucleotides long, by 293 point mutations introducing 25 stop codons and 59 insertions/deletions from 1 to 113 nucleotides.
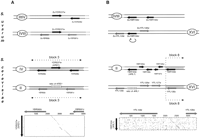
Loss of duplicated genes within S. cerevisiae ancestral duplication blocks. Symbols are the same as in Fig. 1. Shaded parts of the chromosomes correspond to duplication blocks in S. cerevisiae (Wolfe and Schields 1997). The relics ofYDR037w (KRS1) and YBR164c (ARL1) identified in theS. cerevisiae genome are symbolized by the small vertical lines. (A) Loss of the second copy of YDR037w within the S. cerevisiae duplication block 3 on chromosome II:YDR037w is a singleton in the S. cerevisiae genome whereas it is duplicated in S. uvarum. One of the two copies was isolated next to YDR038c, as expected, whereas the second copy was identified between SuYBR060c and SuYBR061c. A relic of the second copy of YDR037w was detected by DNA dot matrix by aligning the sequence of the intergenic region betweenYBR060c and YBR061c with the sequence ofYDR037w. The stringency/window parameters of the dot matrix were set at 15/23. (B) Loss of the second copy ofYBR164c on the border of the S. cerevisiaeduplication block 8 on chromosome XVI. YBR164c is a singleton in S. cerevisiae whereas it is duplicated in S. uvarum. One of the two copies was isolated associated toSuYBR166c whereas the other copy is located betweenSuYPL108w and SuYPL109w. In S. cerevisiae SuYPL108w and SuYPL109w lie outside of the duplicated block 8. In S. uvarum, the second copy ofSuYBR164c is inverted with respect to the centromere (as symbolized by the curved arrow), as is the case for the whole of block 8 in S. cerevisiae. A relic of the second copy ofYBR164c was identified in the S. cerevisiae genome in the intergenic region between SuYPL108w andSuYPL109w. The stringency/window parameters of the dot matrix were set at 11/19.
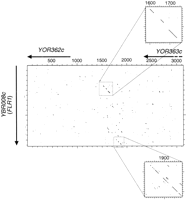
DNA dot matrix between YBR008c (FLR1) and theYOR362c-YOR363c region. The stringency/window parameters of the dot matrix were set at 15/23 for the main matrix and for thetop magnification and at 13/23 for the bottommagnification.
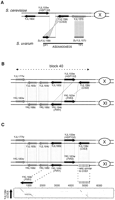
Local gene inversions. Symbols are the same as in Figure 1. (black arrows) Genes that are members of the same gene family. (A) Local gene inversion of SuYJL158c. The chromosomal neighborhood of YJL158c (CIS3) in S. cerevisiaechromosome X is presented at the top of the figure. At the bottom, hits are depicted that were obtained from bothBLASTX (dark shading) and BLASTN (light shading) comparisons of the two S. uvarum paired RSTs (AS0AA004E05DP1 and AS0AA004E05TP1) with the S. cerevisiaegenome. (B) Structure of the duplicated block 40 in S. cerevisiae. Only those genes being duplicated between and/or within the two blocks are represented. The shaded areas between genes represent the BLASTP-based identification of the paralogous partners (Wolfe and Schields 1997). (C) Identification of a relic of YJL158c. The relic on S. cerevisiae chromosome XI is symbolized by the small vertical lines and the corresponding DNA dot matrix is presented below (stringency/window set a 15/23). The shaded area corresponds to the new paring of duplicates in block 40.
The second situation encountered was duplicated genes in S. uvarum that correspond to singletons localized outside of the recognized block of duplication in S. cerevisiae. Some ancestral blocks of duplication have escaped from comparison of theS. cerevisiae genome with itself. They were recognized only from comparisons with other species (Llorente et al. 2000). This situation is illustrated by the ORF YKR097w (PCK1), which exists as a singleton in S. cerevisiae , but was found duplicated in the S. uvarum genome (Fig.3A). The partial sequences of the twoS. uvarum homologs to PCK1 (SuYKR097w) overlap over 213 nucleotides and share only moderate sequence identity (44% in nucleotides with many small insertions/deletions), which is fully compatible with the presence of two distinct loci. In addition, hybridization of the SuYKR097w probe (see Methods) revealed both chromosomes SuXI and SuVIIΔL (not shown), confirming that this gene is duplicated in the S. uvarum genome. The localization of the two copies of the duplicatedSuYKR097w gene in S. uvarum does not correspond to any identified duplicated block in S. cerevisiae.BLASTX comparison against the S. cerevisiaeproteome revealed that the second copy of SuYKR097w is truncated in both the 5′ and in the 3′ regions of the gene, before residue 357 and after residue 506 (compared with the 544 residues ofPCK1 in S. cerevisiae, Fig. 3B). In addition,BLASTN comparison against the S. cerevisiaegenome revealed that the region downstream of the homology withPCK1 shares homology alternatively with chromosome VII and XI of S. cerevisiae. This mosaic region of two S. cerevisiae chromosomes probably results from an ancient duplication block whose trace is not detectable anymore in the S. cerevisiae genome. Despite the big size of the intergenic region between YGL006w (PMC1) and YGL007w inS. cerevisiae (2283 bp vs. an average of 500 bp [Dujon 1996]), no relic of PCK1 could be found at this position.
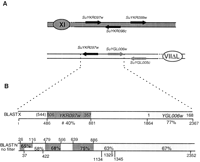
Loss of a duplicated gene in S. cerevisiae outside previously recognized duplication blocks. Symbols are the same as in Fig. 1, and the S. uvarum sequences homologous with S. cerevisiaechromosomes XI and VII are drawn in dark and light gray, respectively. (A) Chromosomal mapping of the two copies ofSuYKR097c in S. uvarum genome. SuYKR097c was isolated twice paired with SuYGL006w and once associated withSuYKR098w and SuYKR099w. (B)BLASTX and BLASTN comparisons (BLOSUM62) between the S. uvarum SuYKR097w-SuYGL006w region and the S. cerevisiaegenome. The sequence of the synteny breakpoint on chromosome VIIΔL was assembled from sequences of overlapping RSTs from the Génolevures projects (XAS0AA01F03TP1, AS0AA19G08DP1, and XAS0AA01F03DP1). Percentage of identity as well as coordinates of the hits are indicated.
Finally, the third category of rearrangement belonging to the duplication-loss mechanism corresponds to genes that seem to have transposed from one chromosome in S. cerevisiae to another chromosome in S. uvarum. In fact, a closer look at these synteny breakpoints revealed that they correspond to an unnoticed ancestral coduplication of two genes (or more) on two different chromosomes, followed by alternate gene loss in the two species. This leaves only one copy of each gene in each of the two genomes but localized on two different chromosomes. This mechanism is documented here by the following three cases.
The first example is the alternate loss of YML051w(GAL80) between S. cerevisiae and S. uvarum. Although GAL80 belongs to chromosome XIII ofS. cerevisiae, its S. uvarum homolog (SuYML051w) map onto chromosome SuX (Fig.4A). This situation would result from the duplication of GAL80 in the ancestry of S. cerevisiaeand S. uvarum followed by the alternate loss of the two copies in the two species. This mechanism is strongly supported here by the identification in Kluyveromyces thermotolerans and inK. lactis of series of nonsyntenic couples whose homologs lie alternatively on chromosome X and XIII in S. cerevisiae(Bolotin-Fukuhara et al. 2000; Malpertuy et al. 2000a; Fig. 4B). These gene couples can be assembled in a single map of 12 genes corresponding to a mosaic of 2 S. cerevisiae chromosomes called atrans-chromosomal series (Llorente et al. 2000; Fig. 4C). The alternate localization of these genes along two different chromosomes in S. cerevisiae would be the result of an ancient duplication of the whole region onto chromosomes X and XIII, followed by a massive loss of the duplicates alternatively on each chromosome. This region was not known as an ancestral duplication block before. Indeed, it is located exactly between block 28 and block 42 on chromosome X and between block 44 and block 19 on chromosome XIII. The only remaining trace of this ancestral duplication in the present-day S. cerevisiae genome is the presence of the two member gene family YML047c/YJR054w (Fig. 4C).
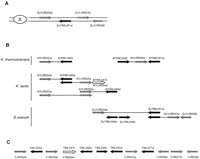
Alternate loss of YML051w between S. cerevisiae andS. uvarum. (Gray and black arrows) ORFs homologous with genes located on S. cerevisiae chromosome X and XIII, respectively. (White arrow) An ambiguous match with both YML047c andYJR054w. (A) Chromosomal mapping ofSuYML051w: the ORF SuYML051w is associated toSuYJR054w and to SuYJR057w/SuYJR058c in two different plasmid inserts of the S. uvarum library (Table 2). TheSuYML051w probe hybridized onto chromosome SuX (hybridization not shown). (B) Trans-chromosomal series: gene couples corresponding to a mosaic of S. cerevisiaechromosomes X and XIII identified in three different Génolevures species. (C) Hypothetical ancestral gene order before duplication: compilation of the gene couples identified in the three species.
The second case corresponds to the alternate loss of YBR008c(FLR1) between S. cerevisiae and S. uvarum. The S. uvarum homolog to FLR1 (SuYBR008c) was isolated in the S. uvarum library twice associated withSuYOR363c (Table 2) but in an inverted orientation. Both ORFs were localized on S. uvarum chromosome VIIItXV by hybridization onto PFGE karyotype (the terminal 700 kb from the right arm of chromosome XV encompassing SuYOR363c is translocated onto chromosome VIII [Fischer et al. 2000], data not shown). According to this localization, one can make the assumption thatSuYOR363c has kept the same orientation relative to the centromere than YOR363c in S. cerevisiae. This would imply that SuYBR008c would be in an inverted orientation relative to the centromere of the S. uvarum chromosome VIIItXV. This is supported by the identification by DNA dot matrix of a weak sequence similarity between FLR1 and the intergenic region between YOR362c and YOR363c but in an inverted orientation (Fig. 5). This trace is a relic of an ancestral copy of FLR1. Thus, FLR1 must have existed as duplicates and the copy on chromosome XV was lost in S. cerevisiae but kept in S. uvarum whereas the copy on chromosome II was lost in S. uvarum but kept in S. cerevisiae. Moreover, FLR1 belongs to the block 3, which is duplicated on chromosome IV (Wolfe and Schields 1997). This means that this gene has been involved in a least two successive duplication events during the yeast genome evolution.
The last case of alternate gene loss is illustrated by the ORFYIL089w, belonging to S. cerevisiae chromosome IX and whose S. uvarum homolog was isolated in the same plasmid insert that SuYAR019c (Table 2). Both corresponding probes hybridized onto chromosome SuI. No relic of YIL089wwas found next to YAR019c on S. cerevisiaechromosome I. However, the corresponding intergenic region of 1721 bp is big enough to have accommodated a copy of this gene at one point of its evolution.
In conclusion, from the set of 19 nonambiguous synteny breakpoints analyzed, 10 gene couples correspond to six distinct situations of differential loss of duplicates. In addition, a similar mechanism can be invoked to explain local gene inversions as well as most, if not all, of the composite rearrangements described below.
Duplication-Loss and Local Inversion
Two cases of local gene inversion, without loss of synteny, that could result from a duplication-loss mechanism were identified (see Methods). SuYJL158c (S. uvarum homolog toCIS3) and SuYOR182c (S. uvarum homolog toRPS30B) were inverted relative to their respective centromeres (Table 2). It is noteworthy that both CIS3 and RPS30Bbelong to ancestral blocks of duplication 45 and 40, respectively, and have a paralog in the corresponding duplicated region. As detailed below, this is indicative of a relationship between duplication and inversion.
The detailed analysis of the SuYJL158c inversion was very informative about the molecular mechanism responsible for local gene inversion. In S. cerevisiae, CIS3 and its two neighboring genes, YJL159w (HSP150) and YJL160c, are members of the same gene family (also comprising YKL163w[PIR3] and YKL164c [PIR1], see below). Sequence similarity between the three corresponding proteins is restricted to their N-terminal region, from residues 20 to 75, of 227 in total. However, the match detected between the S. uvarum RST (AS0AA004E05DP1) and CIS3 corresponds to the C-terminal region of the protein, between residues 129 to 227 (Fig.6A). As there is no sequence similarity in this region between Cis3p, Hsp150p, and Yjl160p, the corresponding ORF in S. uvarum was annotated as a nonambiguous ortholog toCIS3 and designated SuYJL158c, although its orientation was inverted. However, on the S. uvarum RST, downstream of the homology with CIS3, sequence similarity was detected at the nucleotide level with the intergenic regionCIS3-HSP150 but in proper orientation relative to the rest of the insert. This pattern revealed that the match with CIS3in the S. uvarum RST corresponds to the chromosomal region of HSP150 in S. cerevisiae (Fig. 6A). In S. cerevisiae, the gene family comprising CIS3, HSP150, YJL160c, PIR3, and PIR1 is fully included in the duplicated block 40. The pairs CIS3-PIR3and HSP150-PIR1 defined in Wolfe and Schields (1997) show an inverted orientation relative to the rest of the block. andYJL160c has no paralog in block 40 (Fig. 6B). A closer look at these chromosomal regions allowed us to identify a relic in the intergenic region between YKL162c and PIR3, which corresponds to the true structural paralog to CIS3. It follows that the true structural paralogs to HSP150 andYJL160c are in fact PIR3 and PIR1, respectively, and that there was no physical DNA inversion between the two chromosomal regions (Fig. 6C).
Altogether, these findings strongly support a model of local gene inversion based on an initial event of duplication in the common ancestor to S. cerevisiae and S. uvarum, producing a tandem inverted repeat of one gene (Fig.7). After the speciation process, the two copies of this gene could either be differentially lost between the two species or diverge independently from their respective paralogs in the other species. This would lead to an apparent gene inversion relative to the neighboring genes but without any physical DNA inversion between the two species.
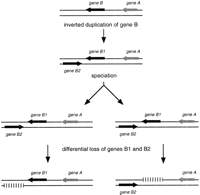
Model for local gene inversion. Inverted duplication of geneB in the common ancestor of two different present-day species, followed by speciation and differential loss (or divergence) of the two copies between the two species produces an apparent inversion of gene B relative to gene A.
Composite Rearrangements
Six different nonsyntenic gene couples correspond to three composite rearrangements between S. uvarum and S. cerevisiaegenomes. In each case, multiple events have accumulated in a given chromosomal region. Two of these complex events involved sequences localized in the S. cerevisiae subtelomeric regions (Table 2).
The first composite rearrangement correspond to the triplication ofYMR090w in the S. uvarum genome associated with the transposition of YMR091c (NPL6) from S. cerevisiae chromosome XIII to S. uvarum chromosome XII.SuYMR090w was found in three different plasmid inserts from the library associated once to SuYER157w and twice toSuYLR039c (Fig. 8A). BothSuYER157w and SuYMR090w probes hybridized onto chromosome VtVII of the S. uvarum karyotype, and two additional signals were obtained onto chromosomes XIII and IX with theSuYMR090w probe (not shown). The SuYMR091c hybridizes with the S. uvarum chromosome XII (not shown). In S. cerevisiae, no relic of NPL6 could be identified next toYLR039c, despite a very large intergenic region of 1862 bp between YLR038c and YLR037c (i.e., in the region where SuYMR091c was identified in S. uvarum, Fig.8A). Similarly, no relic of YMR090w was found next toYER157w, the intergenic region being 1664 bp long. The differences in the chromosome map characterized at these loci imply the accumulation of at least three rearrangements, a triplication ofSuYMR090w (most probably by two successive duplications) as well as the transposition of NPL6 from chromosome XIII to chromosome XII, which possibly corresponds to an ancestral duplication of this gene followed by alternate gene loss in the two species.
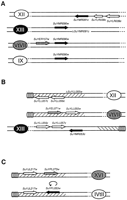
Composite rearrangements. Symbols are the same as in Fig. 1. The shading of the arrows corresponds to the shading of the centromeres. (A) Nonsyntenic gene couples SuYER157w-SuYMR090w andSuYMR091c-SuYLR039c. (ΔSuYMR091c) The absence of hybridization of the corresponding probe onto chromosomeSuXIII. (B) Nonsyntenic gene couples SuYLL055w-SuYEL071w and SuYLL057c-SuYMR053c. TheS. uvarum ORFs homologous with S. cerevisiae subtelomeric genes lie in hatched areas that represent, by analogy with S. cerevisiae, the subtelomeric regions in S. uvarum. Similarly, the contiguous ovals symbolize S. uvarum telomeres.SuYLL056c and SuYLL057c are not represented in a hatched area on chromosome SuXIII because they were associated to SuYMR053c whose ortholog in S. cerevisiae is not localized in the subtelomeric regions. (C) Nonsyntenic gene couples SuYJL217w-SuYPL273w and SuYJL217w-SuYFL053w. Same representation for subtelomeres and telomeres as in (B). (Curved arrow) Inversion of the orientation of SuYFL053w.
The second composite rearrangement involves a transposition ofYLL055w in conjunction with a duplication of the two neighboring genes YLL056c and YLL057c (Fig. 8B). Hybridization experiments onto S. uvarum PFGE karyotypes revealed that SuYLL055w is absent from chromosome XII and transposed onto chromosome VtVII. In addition, both SuYLL056cand SuYLL057c were found to be duplicated onto chromosome XIII. These chromosome map rearrangements involved genes localized in the subtelomeric regions of chromosomes V and XII in S. cerevisiae. At least two different rearrangements could be invoked, an ancestral duplication of YLL055w onto chromosome VtVII followed by the loss of the original copy on chromosome XII, and a coduplication of YLL056c and YLL057c onto chromosome XIII. However, the failure to observe any relic of these events in the corresponding intergenic regions in S. cerevisiae does not allow us to retrace precisely which rearrangements reshaped the ancestral gene order either in the S. cerevisiae or in theS. uvarum lineages.
The last composite rearrangement represents both a transposition and a duplication of YJL217w (Fig. 8C) as well as an inverted transposition of YFL053w (DAK2) in the S. uvarum genome. The SuYJL217w probe hybridizes onto both chromosomes SuXVI and SuIVtII (and not onto chromosome X as in S. cerevisiae). This ORF is associated with SuYPL273w and SuYFL053w on two different plasmid inserts. Although the SuYPL273w probe hybridizes onto S. uvarum chromosome XVI as in S. cerevisiae, the SuYFL053w probe hybridizes onto chromosome IVtII (and not on chromosome VI as in S. cerevisiae, not shown). In addition, SuYFL053w shows an inverted orientation relative to the centromere compared with its S. cerevisiaecounterpart. These three genes, YJL217w, YPL273W, and DAK2, are localized in the S. cerevisiaesubtelomeric regions, and no relic of these genes could be identified. Once again, differences in the genetic organization of these loci are compatible with a duplication-loss mechanism.
These composite rearrangements correspond to a juxtaposition of individual synteny breakpoints, most of which, if not all, are explainable by the above mechanism of duplication loss. However, it is difficult to discriminate between an accumulation of successive rearrangements and a burst of simultaneous rearrangements in a given chromosomal region. Note that even between two genomes with a highly similar genetic organization some cases of accumulation of rearrangements impede to retrace precisely the primary events leading to a loss of synteny.
DISCUSSION
From a 0.4× random sequencing coverage of the S. uvarumgenome, a total of 35 synteny breakpoints were identified among 1810 gene couples examined. This figure when extrapolated to the whole genome predicts a total of approximately 80 synteny breakpoints between the genomes of S. cerevisiae and S. uvarum. The overall conservation of synteny between these two species then could be estimated at 98% (35 of 1810 gene couples in total).
It is commonly accepted that gene order along chromosomes is reshuffled by chromosomal rearrangements such as translocations, duplications, deletions, and inversions. These chromosomal rearrangements physically affect the presence, the orientation, and/or the localization of a DNA segment within the chromosomes. On the contrary, differential gene loss within a previously duplicated segment does not involve any recombination mechanism except for the initial duplication event. As described in this work, gene loss is mainly caused by the accumulation of mutations within the coding sequences. In some cases, traces of an ancient presence of a copy of a gene could still be detected at the nucleotide level by DNA dot-matrix analyses but not at the amino acid level by BLASTX comparisons as it is classically the case for pseudogenes. These traces were named relics rather than pseudogenes because all the characteristics of an ORF have been erased by the high number of mutations accumulated in these sequences. This mechanism of gene erasing was found to affect one copy of an anciently duplicated gene (or possibly one member of a gene family). Sequence conservation between the relic and the present-day active copy of the anciently duplicated gene is variable (see Figs. 2and 5 for comparison). In some cases, we were not able to detect any relic despite large intergenic regions at the synteny breakpoints. This may be because of an accumulation of mutations in these regions to such an extent that there is not enough resemblance left with the ancestral gene to be detected by dot matrix. The inactivation of one copy of a duplicated gene, presumably by a nonsense mutation, would not be counterselected, and the subsequent accumulation of base substitutions would follow the mutation rate. Duplication creates an unstable transient stage; one of the two copies will disappear. This idea is fully compatible with the prediction based on the degree of nucleotide divergence between duplicated genes that most duplicates are silenced by accumulation of mutations very early after the duplication event (Lynch and Conery 2000).
One can ask what the relative contribution of duplications followed by differential gene loss compared with the other chromosomal rearrangements in the evolution of gene order between S. cerevisiae and S. uvarum is. Except for the duplication events that underlie most of the changes in gene order, the contribution of chromosomal rearrangements appeared to be limited to three translocations. It is noteworthy that no case of large inversion has been found among the 1776 nonambiguous gene couples indicating that inversion of large DNA segments is rare within the evolutionary distance between S. cerevisiae and S. uvarum. This might be a general difference between eukaryotes and prokaryotes in which inversions are predominantly large (Huynen et al. 2001). Contrary to what is commonly reported, chromosomal rearrangements played only a minor role in gene order evolution. The 34 remaining events (32 couples of nonsyntenic genes plus two cases of local gene inversion) resulted from or were compatible with a segmental duplication followed by differential gene loss (or divergence) of the duplicated genes.
During the Génolevures program, the proportion of local gene inversions with conservation of synteny was calculated for the 13 species and revealed that this phenomenon remains rare over relatively long evolutionary distances (from S. cerevisiae to mostKluyveromyces species) but becomes prominent over longer evolutionary distances (between S. cerevisiae andPichia, Candida, Debaryomyces, andYarrowia species; Llorente et al. 2000). This is in good agreement with comparative studies on gene order/orientation evolution between S. cerevisiae and S. bayanus, S. servazzii, and S. kluyveri, on one hand (Langkjaer et al. 2000) and between S. cerevisiae and Candida albicans, on the other hand (Seoighe et al. 2000). The former showed that the impact of gene inversion was very limited between closely related species whereas the latter showed that small size inversions were a major cause of genome reorganization between distantly related species. Our results call for an important consideration regarding the mechanism by which small inversions occur. The characterization of tandemly inverted duplicated genes at the place where local gene inversions occurred supports a model based on an initial inverted duplication event followed by differential divergence or loss of the two copies of the duplicated gene in two different species (Fig. 7).
Surprisingly, 16 of 35 (46%) nonsyntenic gene couples comprised ORFs homologous with S. cerevisiae subtelomeric genes whereas the subtelomeric regions represent less that 10% of the nuclear DNA inS. cerevisiae. This overrepresentation of subtelomeres within the nonsyntenic gene couples is largely attributable to the pairs obtained from ambiguous matches (Table 1). The reason for this is that subtelomeric regions in S. cerevisiae are composed of a variety of tandem and dispersed repeated sequences as well as several large gene families (Louis 1995). The average sequence similarity between the different members is much higher in the subtelomeric families than in the internal ones (except for the ribosomal protein genes). Thus, the high proportion of synteny breakpoints in the subtelomeric regions could be because of the large size of the families that would provide numerous possibilities to differential gene loss leading to synteny breakpoints possibly because of a low selection pressure on the copy number within large families and because the subtelomeric genes are not essential. This is supported by the large proportion of nonsyntenic gene couples found among the ambiguous couples (16 of 34 couples with a least one ambiguous match), which reflects a significantly lower conservation of synteny within gene families than average (53% compared with 98%). Concomitantly, the low level of sequence divergence within members of the subtelomeric families would permit a high level of recombination leading to changes in the order of flanking genes without detrimental effects because of the subtelomeric localization of these sequences.
In conclusion, our results showed that at the macroscopic level, the genomes of S. cerevisiae and S. uvarum are mainly colinear, few translocations being the only large chromosomal rearrangements detectable (Fischer et al. 2000). However, if one looks at a higher magnification, numerous synteny breakpoints are present between these genomes, but they correspond to local rearrangements of gene order most of which result from ancient duplication events. These findings lead us to distinguish between microsynteny and macrosynteny breakpoints. The former is restricted to events encompassing only a few genes such as segmental duplication followed by differential gene loss whereas the latter corresponds to chromosomal rearrangements such as translocations encompassing many genes. Evolution of gene order in the yeast genome is mainly caused by the accumulation of numerous microsynteny breakpoints. Macrosynteny breakpoints could be either disadvantaged because of mechanistic constraints or counter-selected because of chromosomal missegregation at meiosis. Differences between macro- and microsynteny could also be important in speciation. It has been shown that macrosyntenic rearrangements are not a prerequisite for speciation in yeast (Fischer et al. 2000). However, the microsyntenic rearrangement induced by recurrent gene losses within ancestral duplications would provide an efficient way to reproductive isolation. If gene duplications are followed by geographic separation and subsequent alternative losses of the duplicated copies in the two populations, mating of the previously isolated populations will produce an double-null homozygote nonviable progeny (Lynch and Conery 2000). In this regard, microsyntenic rearrangements could be more effective than the macrosyntenic changes in establishing a postmating reproductive barrier between isolated populations.
METHODS
Computational Analyses and Reexamination of the Original Sequence Data Set
All the sequence data utilized in this work are directly accessible via the Génolevures web site:http://cbi.labri.u-bordeaux.fr/Genolevures/Genolevures.php3, except for the sequences of the translocation junctions determined in this work (accession nos. AJ316068 and AJ316069). The paired-end sequence data from the Génolevures project consist of very long sequence tags (RSTs) of 910 bp on average from both ends of the 4–5-kb plasmid inserts (Artiguenave et al. 2000). Annotation of the paired-end sequences by comparison to S. cerevisiae allowed the identification of 1810 pairs of neighboring genes in S. uvarum(Bon et al. 2000). The BLASTX annotations of the 38 nonsyntenic genes couple previously identified were rechecked. This control revealed that three of the 38 gene couples corresponded to incorrect validation of nonsignificant hits against the S. cerevisiae proteome (matches with YOR022c,YPR194c, and YBL082c in the gene couplesSuYHR176w-SuYOR022c, SuYPR194c-SuYCR014c, andSuYPR072w-SuYBL082c were not significant). Two other S. uvarum ORFs (SuYJR045c and SuYOL130w), initially annotated as nonambiguous homologs to S. cerevisiae genes, are each related to a paralogous gene in S. cerevisiae, which preserves the synteny with the rest of the insert. Thus,SuYEL031w-SuYJR045c and SuYFL048c-SuYOL130wcorrespond to the syntenic couples, SuYEL031w-SuYEL030wand SuYFL048c-SuYFL050c. In addition, homologies with the two paralogous genes SuYCR106w and SuYLL054c were defined as a nonsyntenic gene couple whereas they represent in fact two overlapping regions of the same ORF (either SuYCR106w orSuYLL054c). Two other ORFs (SuYBL017c andSuYOL130w) were annotated as clear homologs with S. cerevisiae genes whereas each of them significantly matched to several members of a gene family, none of which were syntenic with the rest of the inserts. Therefore, these pairs belong to the ambiguous match category defined in the first section of Results (Table 1).
We identified two cases of local gene inversion within the set of syntenic couples (Table 2). The inverted gene of the couple was identified using the rationale defined in Llorente et al. (2000).
In the S. cerevisiae genome, we sought traces of ancient duplicated copies of genes (called relics) by aligning the S. cerevisiae sequence corresponding to the synteny breakpoint inS. uvarum with the sequence of the S. cerevisiae gene orthologous with the S. uvarum nonsyntenic gene. These alignments were realized by DNA dot matrix (DNA Strider 1.3f11), which allowed us to detect contiguous series of a very short stretch of sequence similarity. Parameters of the matrices are indicated in the legends to the figures.
Experimental Methods
The S. uvarum strain from the Génolevures program 623–6C ura3–1 (or CLIB533) is derived from NCYC 623 (NRRL Y11845, CBS 7001; Bon et al. 2000). S. uvarum total genomic DNA was extracted according to Johnston (1988) and the nonchimeric nature of the plasmid inserts containing nonsyntenic gene couples was shown by long-range PCR from total DNA with the Ex-Taq kit (Takara) using the conditions recommended by the supplier and oligonucleotide primers (sequences available on request).
Chromosome plugs were prepared as described in Louis (1998). PFGE karyotypes were performed in a Rotaphor R23 tank (Biometra) using the following program: run time 65 h, pulse time from 140 to 180 sec (linear ramping), angle 110°, 140 volts, 0.9% agarose gel (Seakem GTG) in Tris-Borate-EDTA buffer 0.25×.
S. uvarum ORFs used as probes were PCR-amplified from plasmid DNA of the S. uvarum library (Bon et al. 2000) clones using the Wizard Plus SV minipreps kit (Promega) with primers internal to the coding regions (sequences available on request). These PCR products were gel-purified with the Nucleospin Extract kit (Macherey-Nagel) and nonradioactively labeled with the Gene Images kit (Amersham). Southern transfers of PFGE karyotypes were performed onto Hybond N+ membrane (Amersham), probed, and detected according to the Gene Images manual. A total of 12 chimeric clones were identified among the gene couples originally annotated as nonsyntenic by hybridization of each of the two probes from the same gene couple onto different S. uvarum chromosomes (SuYOR304w-SuYPR160w,SuYPL002c-SuYML075c, SuYPR162c-SuYML002w,SuYLR392c-SuYER111c SuYJL046w-SuYOR119c,SuYPL216w-SuYJL099w, SuYLL003w-SuYER080w,SuYCL031c-SuYBR177c, SuYBR269c-SuYJR152w,SuYAL028w-SuYPR015c, SuYNL059c-SuYDR177w, andSuYLL029w-SuYOR049c).
Acknowledgments
This work was supported by the CNRS as part of the network GDR 2354 Génolevures II. We thank our colleagues from the Génolevures network and especially J.L. Souciet. We also thank B. Llorente for critical reading of the article as well as our colleagues from the Unité de Génétique Moléculaire des Levures for fruitful discussions. B.D. is a member of the Institut Universitaire de France.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL fischer{at}pasteur.fr; FAX 0-33-1-40-61-34-56.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.212701.
-
- Received August 27, 2001.
- Accepted October 10, 2001.
- Cold Spring Harbor Laboratory Press








