
Comparative Genome Analysis of the Mouse Imprinted Gene Impact and Its Nonimprinted Human Homolog IMPACT: Toward the Structural Basis for Species-Specific Imprinting
- Kohji Okamura1,6,
- Yuriko Hagiwara-Takeuchi1,6,
- Tao Li2,
- Thanh H. Vu2,
- Momoki Hirai3,
- Masahira Hattori4,
- Yoshiyuki Sakaki1,4,
- Andrew R. Hoffman2,7, and
- Takashi Ito1,5,7
- 1Human Genome Center, Institute of Medical Science, University of Tokyo, Tokyo 108-8639, Japan; 2VA Palo Alto Health Care System and Stanford University School of Medicine, Palo Alto, California 94304, USA; 3Department of Integrated Biosciences, Graduate School of Frontier Sciences, University of Tokyo, Tokyo 113-0033, Japan; 4Human Genome Research Group, RIKEN Genomic Sciences Center, Wako, Saitama 351-0198, Japan; 5Division of Genome Biology, Cancer Research Institute, Kanazawa University, Kanazawa 920-0934, Japan
Abstract
Mouse Impact is a paternally expressed gene encoding an evolutionarily conserved protein of unknown function. Here we identified IMPACT, the human homolog of Impact, on chromosome 18q11.2–12.1, a region syntenic to the mouseImpact locus. IMPACT was expressed biallelically in brain and in various tissues from two informative fetuses and in peripheral blood from an informative adult. To reveal the structural basis for the difference in allelic expression between the two species, we elucidated complete genome sequences for both mouse Impact(∼38 kb) and human IMPACT (∼30 kb). Sequence comparison revealed that the two genes share a well-conserved exon–intron organization but bear significantly different CpG islands. The mouse island lies in the first intron and contains characteristic tandem repeats. Furthermore, this island serves as a differentially methylated region (DMR) consisting of a hypermethylated maternal allele and an unmethylated paternal allele. Intriguingly, this intronic island is missing from the nonimprinted human IMPACT, whose sole CpG island spans the first exon, lacks any apparent repeats, and escapes methylation on both chromosomes. These results suggest that the intronic DMR plays a role in the imprinting ofImpact.
[The sequence data described in this paper have been submitted to the DDBJ/EMBL/GenBank data library under accession nos. AB026264, AF232228, and AF232229.]
A small number of mammalian genes are expressed in a parent-of-origin-dependent manner (Morison and Reeves 1998). They are called imprinted genes, or genes subject to genomic imprinting. That both maternal and paternal genomes are required for the accomplishment of normal development in mammals is assumed to be caused by the presence of imprinted genes playing essential roles in development (Solter 1998). Imprinted genes identified so far include those that regulate proliferation and differentiation of the cell and play pivotal roles in early development, postnatal growth, and behavior of the animal (Morison and Reeves 1998). It is thus not so surprising that aberrations in imprinted genes can cause a variety of pathological states. Besides the characteristic congenital defects such as Prader-Willi syndrome, Angelman syndrome, and Beckwith-Wiedemann syndrome, common diseases including atopic hypersensitivity, diabetes mellitus, bipolar affective disorder, and various malignant tumors are assumed to involve genomic imprinting in their pathogenesis (Nakao and Sasaki 1996). Therefore, the imprinted genes have been subjected to intensive studies with both biological and medical interest.
Nevertheless, why and how some mammalian genes are imprinted still remain largely unknown (Barlow 1997; Reik and Surani 1997; Constancia et al. 1998; Solter 1998; Feil and Khosla 1999; Tilghman 1999). A number of hypotheses have been offered to explain biological roles for genomic imprinting. However, the fact that some genes demonstrate species-specific (Kalscheuer et al. 1993; Pearsall et al. 1996;Riesewijk et al. 1996a,b) and polymorphic imprinting (Xu et al. 1993;Jinno et al. 1994; Bunzel et al. 1998) makes it difficult to develop a unified model. The importance of DNA methylation was clearly demonstrated by gene targeting experiments in which the mice lacking DNA methyltransferase activity show aberration in monoallelic expression of imprinted genes (Li et al. 1993). Regions showing parent-of-origin-dependent DNA methylation (i.e., methylation imprints) are often found in imprinted genes and hence are assumed to play a critical role in this epigenetic process (Constancia et al. 1998; Feil and Khosla 1999). These differentially methylated regions (DMR) may or may not be conserved between species. No organizational or sequence similarity was found between the DMRs of the imprinted mouse and humanH19 loci (Jinno et al. 1996). Although the mouseIgf2r and human IGF2R genes share highly conserved intronic CpG islands containing numerous large direct repeats that are methylated following maternal transmission, Igf2r is monoallelically expressed, but IGF2R is not (Smrzka et al. 1995). Organizational similarity is thus not a sine qua non for the conservation of imprinting between species.
To obtain further insights into the mechanisms of genomic imprinting, comparative studies on more imprinted genes should be useful. To facilitate the identification of novel imprinted genes, we developed a unique screening method designated as the Allelic Message Display (AMD) (Hagiwara et al. 1997). Using the AMD, we identified a novel paternally expressed gene Impact on mouse chromosome 18 (Hagiwara et al. 1997). The predicted protein product of Impact belongs to the YCR59c/yigZ hypothetical protein family, or Uncharacterized Protein Family 29 (UPF0029) (Doerks et al. 1998), which is composed of yeast and bacterial hypothetical proteins sharing a remarkably conserved domain. Despite its significant evolutionary conservation, no clues are currently available regarding the function of members of this family.
To characterize this gene further, we isolated cDNA forIMPACT, the human homolog of Impact, and determined its chromosomal localization, the tissue distribution of its mRNA, and its allelic expression status. Intriguingly, the human IMPACTwas shown to be expressed in a biallelic manner. Because the two genes encode highly conserved proteins, they may well share a common genome organization, and hence the comparison between the two may readily pinpoint structural elements crucial for genomic imprinting. We thus determined complete nucleotide sequences for both genes. This comparative genome analysis revealed a characteristic element, which is found in the imprinted mouse gene but is missing from its nonimprinted human counterpart. This element is methylated in a parent-of-origin-dependent manner and hence may play a role in the imprinting.
RESULTS AND DISCUSSION
Identification of cDNA for Human IMPACT
Combinatorial use of human EST clones showing homology to mouseImpact, cDNA library screening, and rapid amplification of cDNA ends (RACE) cloning allowed us to deduce a contiguous sequence 3683 bp long for the putative human homolog of Impact. To verify this reconstructed structure, we designed oligonucleotide primers from this sequence and used them for RT-PCR. We readily obtained PCR products of expected sizes, which were subjected to direct cycle sequencing to eliminate the effect of potential misincorporation during PCR. The confirmed sequence was deposited in the database under the accession number AB026264. We assume that it is a full- or nearly full-length structure, because it is fairly coincident with the length of the cognate transcript estimated by Northern blot hybridization (see below) and because clones extended further in the 5′ direction were obtained by neither library screening nor RACE.
The cDNA bears an open reading frame of 960 bp, and can encode a protein composed of 320 aa (amino acids) (Fig.1A). The predicted product shows significant homology with that of Impact (82.2% identity, 96.6% similarity) as well as its Xenopus homologXimpact (Yamada et al. 1999) (60.0% identity, 87.5% similarity). The predicted product also has high homology with members of the YCR059c/yigZ hypothetical protein family, or the UPF0029 family, composed of proteins of unknown functions (Fig. 1B). As shown in Figure2B, the region termed B is found in hypothetical proteins from various bacteria and Arabidopsis, and is strikingly conserved among the members. In contrast, the eukaryote-specific regions A and D are less conserved, although the region A contains a GI domain, which has recently been found to mediate a specific protein–protein interaction (Kubota et al. 2000). The cDNA is homologous to that of Impact not only in the ORF but also in the 3′ UTR (total: 60.4 %). In particular, the 180-bp region derived from the 3′ extremity is 94.4% homologous to that of mouseImpact. Such a high homology suggests a role for this segment in, for instance, post-transcriptional processing or regulation. Judging from these features, we designated the gene from which this cDNA is derived as IMPACT, for the human homolog ofImpact.

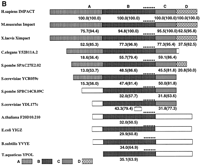
Predicted structures of IMPACT homologs. (A) Multiple alignment of IMPACT homologs. Identical residues are expressed as white letters in black, and similar residues are shaded. Aligned proteins are Homo sapiens IMPACT (accession no.AB026264), Mus musculus Impact (accession no. D87973),Xenopus laevis Ximpact (accession no. AB020319), and hypotheical proteins including Caenorhabditis elegans Y52B11A.2 (accession no. CAA21718), Schizosaccharomyces pombe SPAC27E2.02 (accession no. CAB11676), S. pombe SPBC14C8.09C (accession no.CAA18427), Saccharomyces cerevisiae YCR059c (accession no.CAA42287.1), S. cerevisiae YDL177c (accession no. Z74225),Arabidopsis thaliana F20D10.210 (accession no. CAB37549.1),Escherichia coli YIGZ (accession no. AAC76851), Bacillus subtilis YVYE/YVHK (accession no. CAB15568/AAC44936), andThermus aquaticus YPOL (accession no. P32438). (B) Schematic presentation of members of UPF0029 hypothetical protein family (PROSITE PS00910). The region B is the core region that is highly conserved among human, mouse, yeast, and various bacteria. They all contain a characteristic signature G-x(2)-[LIMV](2)-x(2)-[LIMV]-x(4)-[LIMV]-x(5)-[LIMV](2)-x-R-[FYW](2)-G-G-x(2)-[LIMV]-G (PROSITE PDOC00707) indicated by asterisks (*). The regions A and C are found only in eukaryotes except for one from A. thaliana, which is rather classified as a prokaryotic type. The number in each box is the identity expressed as a percentage of the corresponding region of the putative IMPACT product. The number in each parentheses indicates the similarity.

Chromosomal localization of human IMPACT. (A) FISH analysis of human chromosomes with a biotinylated IMPACT cDNA probe. The doublet signals on both of the chromosome 18s were seen in 4 cells, and the singlet signals on one of the chromosome 18s were found in 19 metaphase cells of the 54 cells inspected. No other hybridization signals were detected. The arrows indicate the doublet signals. (B) G-Banded pattern of the same cell. (C) Idiogram of human chromosome 18 with the location of the IMPACTlocus.
Chromosomal Localization of Human IMPACT
To determine the chromosomal locus of IMPACT, the cDNA clone was used as a probe for fluorescence in situ hybridization (FISH). As shown in Figure 2, clear doublet signals were unequivocally detected on the proximal region of chromosome 18 (Fig. 2A), but not on any other chromosome. Judging from the banding pattern of the same metaphase spread (Fig. 2B), the locus of IMPACT was determined to be chromosome 18q11.2–12.1 (Fig. 2C). This region is syntenic to mouse chromosome 18 A2–B1, the locus to which mouse Impactwas mapped (Hagiwara et al. 1997). These results also support the hypothesis that IMPACT is the ortholog of Impact.
Tissue Distribution of Human IMPACT mRNA
To examine the tissue distribution of IMPACT mRNA, we performed Northern blot hybridization using a probe derived from its ORF. The probe detected two messages: One is ∼3.9 kb long, showing good coincidence with the cDNA, and the other is ∼2.1 kb (Fig.3A). Both are detected in all the tissues examined and display an identical tissue preference pattern (Fig. 3A). We found that a 3′-UTR probe detected only the longer RNA (Fig. 3B) and thus assume that the shorter RNA is generated through differential polyadenylation.
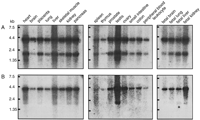
Tissue distribution of human IMPACT mRNA. Northern blot hybridization using RNAs from the indicated tissues was performed with hybridization probes derived from the ORF (A) or 3′ UTR (B) of IMPACT.
Although a modest tissue preference was observed in the distribution ofIMPACT mRNA (Fig. 3), its expression is basically ubiquitous. This is in marked contrast with mouse Impact, which is preferentially expressed in adult brain (Hagiwara et al. 1997). These results may raise the possibility that the clone we obtained is derived from a paralog rather than the ortholog of Impact. However, the striking structural conservation observed not only in the ORF but also in the 3′ UTR, the syntenic localization, and the well-conserved exon–intron organization (see below) support our idea that IMPACT is the ortholog of Impact. Also, every effort to find other homologs of Impact has been, so far, unsuccessful. We thus assume that the IMPACT reported here is the human ortholog of mouse Impact.
Allelic Expression of Human IMPACT
To determine the allelic expression status of IMPACT, nucleotide polymorphisms must be found in the transcribed region. We thus sequenced the 3′ UTR of IMPACT amplified from genomic DNAs from seven Caucasian, seven African-American, and six Asian subjects. Consequently, we found four single nucleotide polymorphisms (SNPs) in this region: 1155 C/T, 2070 T/G, 2180 A/G, and 3104 G/T nucleotide variants.
The C/T heterozygotes at position 1155 (1155 C/T) were not found in the 48 Japanese examined by direct sequencing (data not shown). Similarly, screening by single-nucleotide allele-specific primer extension (SNAS) assay (Vu and Hoffman 1997) revealed only one C/T heterozygote (fetus no. 13527) from a total of 44 fetal subjects (not shown). Because this SNP was originally identified in three out of the seven African-Americans used in the initial screening, it may be enriched in this population. We thus screened peripheral blood DNAs from 56 adult African-Americans and identified five heterozygotes, but, owing to the low expression in peripheral blood leukocytes, our RT-PCR detected theIMPACT transcript in only two of the five informative cases. To examine the allelic expression of IMPACT in these three cases (i.e., one fetus and two adult blood samples), the SNAS assay was applied to the cDNA synthesized from each specimen. As shown in Figure4A, both C and T alleles were detected in fetal brain and adrenal gland (lanes 3, 5: brain; lanes 8, 10: liver; and lane 13: adrenal tissues). No detectable PCR products were observed in the negative controls (Fig. 4A, lanes 4, 6, 8, 11, 14). Thus, humanIMPACT was expressed equally from both parental alleles in these fetal tissues. Similarly, both parental alleles were expressed in one adult blood sample (Fig. 4A, lanes 16, 19) with no detectable bands in negative controls (Fig. 4A, lanes 17, 20). However, in the other blood specimen derived from another adult, the C alleles may appear to be preferentially expressed (Fig. 4A, lane 19), taking the faint T band that appeared even in the control reaction into account (Fig. 4A, lane 21). Although the allelic expression at lower expression level has to be carefully evaluated, this might indicate leaky imprinting ofIMPACT in this particular individual. Another possibility causing apparent preferential allele expression would be cell type-specific imprinting.
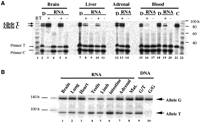
Allelic expression of IMPACT. (A) SNAS assay for allelic expression of IMPACT. The cDNAs from brain, liver, and adrenal gland from the informative 1155 C/T heterozygote fetus and two adult blood samples were subjected to the SNAS assay (labeled as RT+ in lanes 3, 5, 8, 10, 13,16, 19). The SNAS assay products from the genomic DNA are labeled as D (lanes 2, 7, 12,15, 18), whereas those from mock reverse-transcribed samples are shown as the negative control (labeled as RT− in lanes4, 6, 9, 11, 14,17, 20). A control of brain cDNA from a C/C subject is shown in lane 21 (designated as C). Lanes 1 and22 contain a 10-base ladder marker. The positions of the extended product and primer for each allele are indicated. (B) Allelic expression of IMPACT by PCR-RFLP. The cDNAs from the 3104 G/T heterozygous fetus (fetus no. 13466, lane 9) were amplified by primers 119 and 220, and then labeled by primer 219 (see Methods). Digestion with ApoI revealed the two parental alleles (G and T, 140 bp and 92 bp, respectively). Lanes 1–7are cDNAs from the indicated fetal tissues. Lane 8 is cDNA from the maternal endometrium. Genomic DNAs from the informative fetus (lane 9) and a G/G homozygote (lane 10) are shown as controls.
The 2070 T/G polymorphism was identified in one of the seven Caucasian samples by DNA sequencing. We performed the SNAS assay to genotype 48 DNA samples (including 44 fetuses) and found that all 48 were T/T homozygotes (data not shown). This SNP, therefore, appears to be quite rare. The 2180 A/G polymorphism that creates a polymorphicMfeI restriction site (CAATTG) was identified in two of the seven African-Americans by DNA sequencing. We used PCR-RFLP to genotype 44 fetuses and found one heterozygote (fetus no. 13527) and 43 A/A homozygotes (data not shown). This fetus also had the 1155 C/T polymorphism that had already been analyzed by the SNAS assay described above.
The 3104 G/T polymorphism that creates a polymorphic ApoI restriction site (AAATTT) was also identified in two of the seven African-American DNA samples. By PCR-RFLP analysis, we identified two heterozygotes (fetuses nos. 13466 and 13713) and 42 G/G homozygotes. The fetus no. 13466, whose tissues were available, was analyzed for the allelic expression. As shown in Figure 4B (lanes 1–7), all the tissues examined, including brain, lung, heart, testis, limb, intestine, and adrenal gland, demonstrated biallelic expression of humanIMPACT. In addition, the maternal tissue (endometrium, lane 8) also showed biallelic expression. No detectable PCR products were observed in the mock negative controls (−RT, data not shown).
It should be noted that variations in the relative levels of the two alleles were observed in these tissues (Fig. 4B, lanes 1–7). The control heterozygous genomic DNA also showed a predominance of undigested G allele (Fig. 4B, lane 9). Although the restriction digestion was complete (data not shown), heteroduplexes refractory to the digestion were often observed in PCR-RFLP, which can explain the predominant G allele in the G/T DNA control (Fig. 4B, lane 9) and in various tissues (Fig. 4B, lanes 1–3, 5, 7–8). However, two tissues (testis and intestine: Fig. 4B, lanes 4, 6) showed a predominant expression of the digested T allele. Unfortunately, the maternal subject was also a 3104 G/T heterozygote (Fig. 4B, lane 8), and hence we could not determine the parental origin of the digested T allele.
It is conceivable that the imprinting of IMPACT is leaky and polymorphic, at least, in some tissues or cell types. In this context, it is interesting to note that we identified two rare variant forms ofIMPACT cDNA, each of which bears a unique 5′ end but is expressed much less abundantly. Notably, the polymorphic sites used in the allelic expression studies are shared by both variants, and hence all the isoforms are assayed collectively. Accordingly, the allelic expression status of these minor variants are masked by that of the major transcript. It is thus interesting to examine the allelic expression status of each isoform. Unfortunately, despite our exhaustive screening, we have failed to find any isoform-specific SNP, and hence such studies are currently hampered.
Genome Organization of Mouse Impact and Human IMPACT
The data described above show that the human IMPACT gene is basically expressed in a biallelic manner, although an allelic bias may be observed occasionally in some tissues. This is in good contrast with the mouse Impact gene, which is expressed exclusively from the paternal allele in all the tissues examined (Hagiwara et al. 1997). Because the two genes encode highly conserved proteins, they may well share a common genome organization, and hence the comparison between the two may readily pinpoint structural elements crucial for genomic imprinting. We thus determined complete nucleotide sequences for these genes by means of bacterial artificial chromosome (BAC) cloning (Shizuya et al. 1992) and our unique nested deletion strategy (Hattori et al. 1997). Consequently, we elucidated 37,954 bp of contiguous sequence for mouse Impact (accession no. AF232228) and 29,644 bp for human IMPACT (accession no. AF232229).
The genome structures of mouse Impact and humanIMPACT are depicted in Figure 5with the minimum contigs of the subclones. Alignments of the genome sequences with those of cDNAs revealed that both genes have 11 exons. The average size of the exons is about 100 bp except for the last one, which contains the termination codon and is longer than 2 kb. All of the splice junctions follow the GT–AG rule (Table1) and split the open reading frames at the identical positions between the two species. Thus, the overall genome organizations of these genes are well conserved, thereby providing further evidence for their orthology.

Genomic organizations of mouse Impact and humanIMPACT. Physical maps of mouse and human genes are shown in (A) and (B), respectively. Minimum contig of the subclones to cover each gene is also shown. PCR and direct sequencing closed a gap between human subclones 4 × 32 and 4 × 03. Exons are shown as solid boxes and numbered 1 to 11. Arrows indicate the initiation and termination codons. The positions of SINES, LINE-1, CpG islands, and STSs used for the library screening are also illustrated.
Aligned Exon–Intron Organizations of Mouse Impact and Human IMPACT
A remarkable difference between the two genes was found in their upstream promoter regions. The promoter region and the first exon of human IMPACT constitute the sole CpG island of this gene. In contrast, the corresponding region of the mouse gene is rather AT rich (43% GC). Although the ratio of observed versus expected CpG dinucleotides of this region is 0.35, which is significantly higher than the average for the ∼38-kb region (0.25), it does not meet the criteria for a CpG island.
Instead, mouse Impact has a CpG island in its first intron. The intronic island has many TCGGC sequences and a characteristic tandemly reiterated structure (Fig. 6). It is known that such tandem repeats often associate with imprinted genes (Constancia et al. 1998; Feil and Khosla 1999). Notably, we failed to find any such tandemly repeated structures either in the CpG island or elsewhere in the nonimprinted human IMPACT.
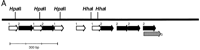

Structure of the CpG island in the first intron of Impact. Tandem repeated structures are schematized by broad arrows (A). The island contains two units designated as 1 and 2, the nucleotide sequences of which are shown in B and C, respectively. Although many polymorphic sites are found between the two mouse strains, two HhaI sites and three HpaII sites are conserved and indicated in the figure. These sites were thus used for the methylation-specific PCR assay in Fig. 7.
Parent-of-Origin-Specific Methylation of the Mouse CpG Island
Because tandem repeats often occur in imprinted genes and are implicated in the establishment of genomic imprinting (Constancia et al. 1998; Feil and Khosla 1999), we examined the methylation status ofIMPACT. For this, we prepared two parental mouse strains,Mus musculus domesticus C57BL/6 (B6) and M. musculus molossinus JF1 (JF), and reciprocal F1 hybrids between the two as described previously (Hagiwara et al. 1997). Then we amplified and sequenced the CpG islands from B6 and JF to search for polymorphisms between the two mouse strains. Fortunately, the island of B6 is 181 bp longer than that of JF, owing to the difference in repeat organization. This allows us to discriminate between the B6 and JF alleles simply by their lengths.
To examine the methylation status, we developed a methylation-specific PCR assay. In this assay, genomic DNAs are first digested with methylation-sensitive restriction endonucleases such as HhaI or HpaII, and then used for PCR to amplify the locus of interest. Although unmethylated targets are cut by the enzymes and will not be amplified, a methylated target survives the digestion to serve as the template for subsequent PCR. In other words, the methylated allele is amplified. Because both B6 and JF alleles for the CpG island of Impact share the same five methylation-sensitive restriction sites, namely, two HhaI sites and threeHpaII sites, we can apply the methylation-specific PCR assay to this island.
We digested the genomic DNAs from B6, JF, (B6 × JF) F1, and (JF × B6) F1 with HhaI, HpaII, orMspI and used them as the templates for PCR spanning the intronic island (Fig. 7A). When native undigested genomic DNAs of the F1 hybrid mice were used as the templates, we readily obtained two bands derived from B6 and JF alleles that can be clearly separated by gel electrophoresis. When the DNAs treated with HhaI or HpaII were used for the PCR, only one of the two bands was obtained. The B6 allele was amplified from (B6 × JF) F1, whereas only the JF allele was detected from (JF × B6) F1 (Fig. 7A). When we usedMspI, a methylation-insensitive isoschizomer ofHpaII, as a control, we could not amplify any bands at all. These results clearly demonstrated that the island is methylated in a parent-of-origin-dependent manner—the silenced maternal allele is hypermethylated, and the active paternal one is undermethylated. Thus, the island serves as a differentially methylated region (DMR) for this gene (Fig. 7C).
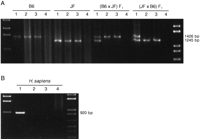
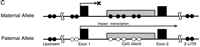
Parent-of-origin-specific methylation of Impact. (A) Methylation-specific PCR assays for the CpG island of mouseImpact. PCR products from native genomic DNA and those digested with HhaI, HpaII, or MspI (lanes1–4, respectively) were subjected to 1.5% agarose gel electrophoresis and stained with ethidium bromide. StyI-digest λ DNA and 1 kb PLUS DNA LADDER (GIBCO BRL) were used as size standards in the left- and rightmost lanes, respectively. The mouse intronic CpG island was analyzed in JF, B6 (B6 × JF) F1, and (JF × B6) F1. (B) Methylation-specific PCR assays for the CpG island of human IMPACT. The human CpG island, which overlaps the promoter region and lacks length polymorphisms, was also analyzed as described in A. (C) Model for the imprinted expression of mouseImpact. Exons are depicted as solid boxes, and CpG islands are shaded. The island is a region of differential methylation, where only the maternal allele is hypermethylated. Closed and open circles stand for hypermethylation and undermethylation, respectively.
We next examined the methylation status of the promoter region, which bears two HpaII sites at positions −55 and −14. We readily amplified the expected DNA fragments from theHpaII-digested genomic DNAs derived from the parental strains and reciprocal F1 hybrids (data not shown). This indicated that the region is at least partially methylated. We thus sequenced the amplified fragments or the methylated allele to know their parental origins by SNP at the position −355 (not shown). When the amplified fragments from undigested genomic DNAs of F1 hybrid mice were used as the templates, we detected both T and C at this position, representing B6 and JF alleles, respectively (not shown). In contrast, we can detect only T from the HpaII-digested DNA from (B6 × JF) F1 and only C from (JF × B6) F1(not shown). These results indicate that the imprinted maternal allele is methylated, and the expressed paternal one is not methylated, like the CpG island (Fig. 7C). We also applied a similar assay to the regions flanking this gene and found that these sites are methylated on both alleles (not shown; see Fig. 7C). Whether the promoter region and the intronic island comprise a single DMR remains to be elucidated through methylation study of this region in early embryonic stages, which is currently underway.
Finally, we applied the methylation-specific PCR assay to the human CpG island, which spans the promoter and the first exon. In contrast to the mouse intronic island and promoter, not only MspI digestion but also HhaI or HpaII treatment completely abolished amplification of the human island, thereby suggesting its undermethylation (Fig. 7B). Furthermore, Southern blot hybridization analyses with the CpG island probe revealed that HpaII digests the island to tiny fragments as efficiently as MspI does (data not shown). These results indicate that the whole island is unmethylated on both chromosomes as are conventional CpG islands (Gardiner-Garden and Frommer 1987).
Taking all these data together, it is clear that the CpG island and the promoter region are subjected to parent-of-origin-dependent methylation and constitute DMRs in mouse, whereas those of the human counterpart are not. The intronic DMR is of particular interest, because it has a characteristic tandemly repeated structure, which is found in many other imprinted genes and is missing in the nonimprinted human counterpart. Although DMR has been implicated in genomic imprinting, its precise role is still controversial; some DMRs have been shown to be necessary for imprinted gene expression, but others seem not to be (Constancia et al. 1998; Feil and Khosla 1999). To elucidate the role of this DMR in genomic imprinting of Impact, we have to examine the temporal coincidence in the establishment of its differential methylation and the monoallelic expression ofImpact. We would be able to undertake much more straightforward tests using transgenic and gene targeting techniques, because the basis for such experiments has already been laid by this study through the elucidation of the genome structure.
METHODS
Isolation of cDNA for Human IMPACT
Based on the sequence of EST ze64c01.r1 showing significant homology to mouse Impact cDNA, we designed oligonucleotide primers and used them for 5′- and 3′-RACE cloning from human brain poly(A)+ RNA and the PCR screening of a pooled fetal brain cDNA library. The IMAGE 363744 cDNA clone bearing the 3′-UTR region of human IMPACT was obtained from Research Genetics and subjected to DNA sequencing. For the final confirmation of the nucleotide sequence, the RT-PCR product for IMPACT was subjected to direct sequencing on both strands using Thermo Sequenase core sequencing kit with 7-deaza-dGTP RPN2440 (Amersham-Pharmacia, UK). The nucleotide sequence of human IMPACT cDNA is deposited in the DDBJ/EMBL/GenBank nucleotide sequence databases under the accession number AB026264.
FISH Mapping of IMPACT
The full-length cDNA for IMPACT was biotinylated by nick translation and hybridized to R-banded chromosomes from cultured lymphocytes of a male donor. Following overnight hybridization at 37°C, the preparations were washed in 50% formamide/2× SSC at 42°C for 15 min, and blocked with 4% bovine serum albumin/4× SSC at 37°C for 30 min as described (Hirai et al. 1996). Signal amplification was achieved using rabbit antibiotin (Enzo Diagnostics), FITC-labeled goat antirabbit IgG (Enzo), and Cy2-labeled donkey antigoat IgG (Amersham-Pharmacia). The chromosomes were counterstained with propidium-iodide. Hybridization signals and banded chromosomes were observed using a fluorescence microscope (Olympus BX, Tokyo) equipped with appropriate filter sets.
Tissue Distribution of IMPACT mRNA
Tissue distribution of the IMPACT transcript was examined by Northern blot hybridization using filters containing poly(A)+ RNAs isolated from multiple adult tissues and fetal tissues (Clontech). The probe was labeled with [32P]dCTP (NEN) using a Prime-It II Random Primer Labeling Kit (Stratagene) and hybridized to the filters overnight at 60°C in 6× SSC, 10× Denhardt's solution, and 1% SDS containing 200 μg/mL salmon sperm DNA. The filters were subsequently washed with 0.2× SSC/0.2% SDS at 60°C for 30 min, and exposed to Imaging-Plates (Fuji Film) to be analyzed on a Fuji BioImaging Analyzer BAS2000 system (Fuji Film). The expression of IMPACT was also analyzed by RT-PCR using multiple tissue cDNA panels (Clontech).
Allelic Expression Analysis of IMPACT by SNAS Assay
We amplified and sequenced the 3′ UTR of IMPACT from genomic DNAs obtained from BIOS Corp. to find polymorphic sites. Three African-Americans were revealed to be C/T heterozygotes at the position 1155. We examined 48 Japanese by direct sequencing. We also examined this site in 44 fetal and 56 African-American adult subjects by SNAS primer extension assay according to the method described previously (Vu and Hoffman 1997). Briefly, purified genomic DNA (or total nucleic acid, TNA) was amplified using primer 179 (5′-TAAGTCAGCCAGTTCAGCATGGAT-3′) and primer 180 (5′-TTAGTTCTCCCAAATAAGCCTGAAAC-3′) to amplify a 120-bp fragment encompassing the polymorphic site. The thermal cycling parameters were as follows: an initial denaturation at 95°C for 1 min, 30 cycles of amplification at 95°C for 10 sec, 55°C for 15 sec, and 72°C for 1 min, and a final extension at 72°C for 5 min. The PCR products were diluted six-fold with water, and aliquots (1.0 μL) were labeled by primer extension using 32P-end-labeled primers 181C (5′-CATAAGTTCTCTA TTTTTGGCAGATG-3′) and 182T (5′-ATAGATCATAAGTTCTC TATTTTTGGCAGATA-3′). The 181C and 182T primers were complementary to the C and T nucleotide at its 3′ end and therefore only extended on the specified C and T alleles under three to five cycles of PCR. Because the 181C and 182T primers were different in size (26 and 32 bases, respectively), their allele-specific extended products (80 and 86 bases) were easily separated on a 5% denaturing polyacrylamide gel. To eliminate the possibility of interference from minute amounts of contaminated genomic DNA, we extensively digested our RNA samples with DNase I (10–20 units of DNase I for each μg RNA, at 37°C for 1 h), and also carried out negative control experiments using mock-reverse-transcribed samples. The SNAS-PCR assays were repeated six times.
Allelic Expression Analysis of IMPACT by PCR-RFLP
To screen for the 3104 G/T heterozygous fetus and to assay for allelic expression, we amplified DNAs and cDNAs using primer 119 (5′-CCTAAAGTCAATTGGCTGG-3′) and primer 220 (5′-ACACGAGCCTGGGCAACATAGA-3′). This amplification yielded a 417-bp fragment encompassing the polymorphic ApoI site. The PCR parameters were as follows: an initial denaturation at 95°C for 1 min, 35 cycles of amplification at 95°C for 20 sec and 65°C for 70 sec, and a final extension at 72°C for 5 min. The PCR products were diluted six-fold with water. Aliquots (1.0 μL) were labeled using 32P-end-labeled primer 219 (5′-TGATCATGGCTCACTGCAGCCTT-3′) and the primer 220. Labeling was performed by eight cycles of PCR (95°C for 1 min, eight cycles of amplification at 95°C for 20 sec, 65°C for 70 sec, and a final extension at 72°C for 5 min). The 32P-end-labeled PCR products (140 bp) were digested with ApoI (New England Biolabs). The 3104G allele was not digested (140 bp), whereas the 4104T allele was digested to yield a 94-bp band.
PCR-Based Genome Walking into the Promoter Regions ofImpact and IMPACT
To walk in genomic DNAs from the 5′ ends of the mouse and human cDNAs, we utilized the GenomeWalker Kit (Clontech). The gene-specific primers used were: mouse first primer, 5′-TTCCTCTTCAGCCATGGTGCTCAGGATC-3′; mouse nested primer, 5′-TGGCAAGCAGCAAATGAATGCAA CTGCG-3′; human first primer, 5′-GGACGGTGTCCTCGTCA ACCATTAACA-3′; human nested primer, 5′-TGGGCCGAC GAAAAACCGGGGTTTCGA-3′. Amplified PCR products were cloned into pT7Blue T-Vector (Novagen) and sequenced using a primer-walk method.
Screening of Mouse and Human BAC Libraries
The PCR screening of pooled mouse and human BAC libraries was performed by Research Genetics . The primers for mouse Impact5′ STS were 5′-GTGGGGTACAGTAAGAGT-3′ (forward) and 5′-TAGTGTAGACTGGGCTCA-3′ (reverse), and those forImpact 3′ STS were 5′-ACGTTTCCCCATTTTACAAG3′ (forward) and 5′-AGTATCACTCACCTGCCCTG-3′ (reverse). The primers for human IMPACT 5′ and 3′ STS were 5′-GGACGGTGTCCTCGTCAACCATTAACA-3′ (forward) and 5′-ACCTGCAGGGTCTGGGCTATTGCCATT-3′ (reverse), and 5′-CGTAGAGTGGGATAGAGGTGGCAGAATG-3′ (forward) and 5′-CTGGAAGATGAAAGATACAT-3′ (reverse), respectively.
Subcloning of Restriction Fragments from BAC Clones
The mouse and human BAC clones were cultured in L-broth in the presence of chloramphenicol (50 μg/mL), and the DNAs were isolated by an automated plasmid isolator PI100 (Kurabo). The crude DNA was treated with RNase followed by purification with salt and polyethylene glycol precipitation. The precipitates were rinsed with 75% ethanol, dried, and then dissolved in TE (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). For the direct sequencing of BAC ends, plasmid DNAs were prepared using Qiagen-tips according to manufacturer instructions.
The BAC DNAs were digested with AvrII, BamHI,NcoI, or XbaI overnight, followed by treatment with Klenow fragments of DNA polymerase I to partially fill the cohesive ends. For instance, AvrII digests were treated with Klenow fragment and 0.2 mM dCTP/dTTP and ligated to the partially filledHindIII site of pSFI-CV2, a cloning vector developed in our previous work (Hattori et al. 1997). Similarly, BamHI,NcoI, and XbaI digests were appropriately filled in and ligated to the partially filled SalI,BamHI, and HindIII sites of pSFI-CV2, respectively. Each ligation mixture was transformed into Escherichia coliDH5α (TaKaRa, Japan). For each digest, we randomly selected 100 colonies, and the plasmid DNAs were prepared and digested withHaeIII for fingerprinting. For the sizing of the inserts of these subclones, we cut them with SfiI and electrophoresed the digests, because each restriction fragment was cloned between the twoSfiI sites of pSFI-CV2.
Construction of the Nested Deletion Libraries
The subclone plasmids prepared as above were digested withSfiI and ligated at a high concentration to generate concatenated DNAs, which were subsequently sonicated to pieces. The variously sized fragments were directionally cloned into pSFI-SV1 and pSFI-SV10 vectors, which we had constructed for the construction of nested deletion libraries (Hattori et al. 1997). The inserts of these clones were amplified by colony PCR and sized by gel electrophoresis. We then chose minimum clones to fully cover the inserts and subjected them to sequencing (Hattori et al. 1997).
DNA Sequencing and Data Assembly
Plasmids and PCR products were sequenced using ABI PRISM BigDye Terminator Ready Reaction Kit (PE Applied Biosystems) by ABI PRISM 377 or 3700 DNA sequencers. For the sequencing of purine-rich regions, we used the ABI PRISM dGTP BigDye Terminator Kit instead of the standard one. Sequence data were analyzed and assembled using the sequence analysis software package SEQUENCHER (Gene Codes). The sequence data for mouse Impact and human IMPACT have been submitted to DDBJ/EMBL/GenBank under the accession numbers AF232228 and AF232229, respectively.
Methylation-Specific PCR Assay
The four kinds of mouse genomic DNAs, each from B6, JF, and their reciprocal F1 hybrids, were prepared from tails (Hagiwara et al. 1997). Human genomic DNA was extracted from peripheral blood. The mouse intronic CpG island was amplified from B6 and JF genomic DNAs using the following primers: 5′-CGGAAGCAATTCAGGAAGTGGGTGGTGT-3′ (forward) and 5′-CCATTTGGGGTCATCCATGAAGTCAGTG-3′ (reverse). Amplified products were directly sequenced to find sequence polymorphisms.
For examination of the methylation status of the island, genomic DNAs were digested with HhaI, HpaII, or MspI overnight in the recommended buffer for each enzyme. Following the heat inactivation of the enzymes at 80°C for 20 min, DNAs were precipitated by ethanol and used as templates for PCR. The assays for the mouse island were performed using primers 5′-CCGTAGCATCACACTACGTA-3′ (forward) and 5′-TCGAACACACACTCGAGGTA-3′ (reverse) with the following thermal cycling parameter: 96°C for 180 sec plus (96°C for 30 sec, 61°C for 40 sec, 72°C for 80 sec) for 5 cycles, (96°C for 30 sec, 58°C for 40 sec, 72°C for 80 sec) for 30 cycles, and 72°C for 180 sec. The PCR for the human island was performed using primers 5′-CCCTAGGAATGTAAAGACGAG-3′ (forward) and 5′-CCAGAAGGAGTGAGATTCGG-3′ (reverse) with the following thermal cycling: 96°C for 180 sec plus (96°C for 30 sec, 63°C for 40 sec, 72°C for 60 sec) for 5 cycles, (96°C for 30 sec, 60°C for 40 sec, 72°C for 60 sec) for 30 cycles, and 72°C for 180 sec. Amplified products were resolved on 1.5% agarose gel electrophoresis followed by ethidium bromide staining. For the mouse promoter region we used primers 5′-GTGGGG TACAGTAAGAGT-3′ (forward) and 5′-TGGCAAGCAGC AAATGAATGCAACTGCG-3′ (reverse) with the following thermal cycling: 96°C for 180 sec plus (96°C for 30 sec, 50°C for 40 sec, 72°C for 60 sec) for 30 cycles, then 72°C for 180 sec. The products were directly sequenced with primer 5′-TCTCCAGCTCTCGTTCAT-3′.
Acknowledgments
We thank Dr. S. Sato (Brain Science Institute, RIKEN) for the generous gift of human genomic DNAs, and the Central Laboratory for Human Embryology Tissue, University of Washington, Seattle, for fetal tissues. We are grateful to K. Oshima, Y. Yamashita, S. Tsuto, and R. Fukawa (RIKEN Genomic Sciences Center) for help in DNA sequencing, and to R. Arai, M. Kondo, M. Tanaka, M. Horishima, T. Aizu, and Y. Matsumura (RIKEN Genomic Sciences Center) for help in construction of the nested deletion libraries. This work was partly supported by Grants-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports and Culture, Japan (MESSC), a Grant-in-Aid for Scientific Research from MESSC, research grants from Science and Technology Agency, Japan, the Japan Society for the Promotion of Science (JSPS), the Research Service of the Department of Veterans Affairs, and NIH Grant DK36054. Both K.O. and Y.H.T. are supported by the Research Fellowship grant from JSPS for Young Scientists.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵6 These authors contributed equally to this work.
-
↵7 Corresponding authors.
-
E-MAIL arhoffman{at}leland.Stanford.EDU; FAX (650) 856-8024.
-
E-MAIL titolab{at}kenroku.kanazawa-u.ac.jp; FAX 81 76 234 4508.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.139200.
-
- Received March 6, 2000.
- Accepted September 28, 2000.
- Cold Spring Harbor Laboratory Press








