
Sequence and Comparative Analysis of the Mouse 1-Megabase Region Orthologous to the Human 11p15 Imprinted Domain
- Patrick Onyango1,2,
- Webb Miller3,
- Jessica Lehoczky4,
- Cheuk T. Leung1,5,
- Bruce Birren4,
- Sarah Wheelan5,7,
- Ken Dewar4, and
- Andrew P. Feinberg1,2,5,6,8
- 1Institute of Genetic Medicine and 2Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA; 3Department of Computer Science and Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, USA; 4Whitehead Institute/MIT Center for Genome Research, Cambridge, Massachusetts 02141, USA; 5Department of Molecular Biology and Genetics and 6Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA; 7Center for Biotechnology Information, National Institutes of Health, Bethesda, Maryland 20894, USA
Abstract
A major barrier to conceptual advances in understanding the mechanisms and regulation of imprinting of a genomic region is our relatively poor understanding of the overall organization of genes and of the potentially important cis-acting regulatory sequences that lie in the nonexonic segments that make up 97% of the genome. Interspecies sequence comparison offers an effective approach to identify sequence from conserved functional elements. In this article we describe the successful use of this approach in comparing a ∼1-Mb imprinted genomic domain on mouse chromosome 7 to its orthologous region on human 11p15.5. Within the region, we identified 112 exons of known genes as well as a novel gene identified uniquely in the mouse region, termed Msuit, that was found to be imprinted. In addition to these coding elements, we identified 33 CpG islands and 49 orthologous nonexonic, nonisland sequences that met our criteria as being conserved, and making up 4.1% of the total sequence. These conserved noncoding sequence elements were generally clustered near imprinted genes and the majority were between Igf2 andH19 or within Kvlqt1. Finally, the location of CpG islands provided evidence that suggested a two-island rule for imprinted genes. This study provides the first global view of the architecture of an entire imprinted domain and provides candidate sequence elements for subsequent functional analyses.
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos. AF313042 to AF313150.]
Genomic imprinting is an epigenetic modification of the gamete or zygote that leads to preferential expression of a specific parental allele in somatic cells of the offspring. The mechanism of imprinting is unknown but it is thought to involve CpG island methylation (Sapienza et al. 1987; Sutcliffe et al. 1994), antisense transcripts (Wutz et al. 1997), short repeat elements (Szebenyi and Rotwein 1994), and/or trans-acting binding proteins that may interact with one or more of these sequences (Bell and Felsenfeld 2000; Hark et al. 2000; Srivastava et al. 2000). One of the most surprising recent discoveries in the study of genomic imprinting is that imprinted genes are grouped in large multigene domains (Lee et al. 1997; Ainscough et al. 1998; Feinberg 1999). In particular, we and others have found that human chromosomal band 11p15 contains at least eight imprinted genes concentrated in an ∼1-Mb domain, of which six are expressed from the maternal allele and two are expressed from the paternal allele (Feinberg 1999). The organization of this domain is somewhat complicated in that we have identified two separate subdomains that are imprinted, separated by a region of three genes that appear to escape imprinting (Lee et al. 1998, 1999). The boundaries of the overall 11p15 imprinted domain are known at both centromeric and telomeric ends because of the presence of at least eight nonimprinted genes that extend beyond the imprinted domain, including NAP2 and NUP98 on the centromeric side, andL23MRP and CTDS on the telomeric side (Rachmilewitz et al. 1993; Tsang et al. 1995; Hu et al. 1996, 1997; Zubair et al. 1997). Thus it is likely that both local and regionalcis-acting elements are involved in the regulation of genomic imprinting. However, almost nothing is known about the identity or location of such regulatory elements, with the notable exception of a region that has been intensively studied upstream of and downstream from the H19 gene (Thorvaldsen et al. 1998; Bell and Felsenfeld 2000; Hark et al. 2000; Srivastava et al. 2000).
Understanding the genomic organization of this domain is also critical to the study of the disorder Beckwith-Wiedemann syndrome (BWS), which causes prenatal overgrowth, birth defects, and predisposition to a wide variety of childhood cancers, most commonly Wilms tumor (Feinberg 1999). We have found that BWS can involve altered imprinting of either of the two subdomains within the 11p15 imprinted domain, one includingH19 and IGF2 and the other including the maternally expressed genes p57KIP2, KVLQT1and paternally expressed LIT1, an antisense orientation transcript within KVLQT1 (Weksberg et al. 1993;Steenman et al. 1994; Lee et al. 1997,1999).
A powerful approach to identifying functionally important sequences is by aligning of orthologous genomic regions. Evolutionarily conserved genes often have similar structure and function and important regulatory elements may be conserved even between distantly related organisms whose genomes may have little or no similarity overall (Elgar 1996; Hardison et al. 1997). When comparing the mouse and human genomes, the average size of syntenic segments is estimated to be 7.1–15 Mb (O'Brien et al. 1999). The mouse ortholog of the entire human 11p15 imprinted domain is contained in a single syntenic block on mouse chromosome 7 (Blake et al. 2000).
We have taken a comparative genomics approach to identify novel genes and potential regulatory elements within the 11p15 imprinted domain. We identified 87 overlapping BACs spanning ∼1 Mb of mouse chromosome 7 that includes the entire imprinted domain and flanking nonimprinted genes. Draft sequence was obtained from a minimal tiling path of five BACs and this sequence could be ordered by comparison with the publicly available human sequence. Deeper coverage mouse sequence was obtained for the region corresponding to an estimated 250-kb gap remaining within the Human Genome Project sequence, so that ∼95% of the sequence across the entire domain could be ordered and analyzed. This work represents the largest ordered and oriented sequence comparison between mouse and human to date and the first comparative sequence analysis of an entire imprinted domain.
RESULTS
Construction of a BAC Contig across the Entire Orthologous Mouse Imprinted Domain
Forty-five overgo probes (Table 1) were pooled and used for hybridization screening (Ross et al. 1999) of high-density BAC clone filters of the 11.2 × genome equivalent RPCI-23 female mouse C57 BL/6J library. Single-colony isolates were recovered from all addresses identified in the primary screen, then rearrayed and replicated onto sets of filters. In a second round of screening, individual copies of the arrayed clones were tested with individual overgo probes to establish the clone–marker relationships (Table 2). The BAC contig was estimated to span 1.2 Mb and includes the entire orthologous mouse imprinted domain, flanked by the NAP2 gene at the centromeric end and theL23MRP gene at the telomeric end (determined by subsequent sequence analysis). Overall, BAC clones contained an average of 7.8 probes per clone, and each probe tested positive against an average of 9.3 redundantly identified clones (data not shown). Marker density across the region, recovered clone depth, and the marker–clone relationships indicated that the entire region had been captured in an overlapping set of clones. A minimal path of clones for genomic sequencing was selected using combined knowledge of marker content and restriction enzyme digestion fingerprint analysis (Marra et al. 1997). A restriction enzyme map was constructed for HindIII (data not shown), which allowed a more refined interpretation of clone order and overlaps. From this a set of five overlapping clones collectively spanning the region were selected for genomic sequencing (Fig.1).
STSs Used to Identify BACs within the Mouse Imprinted Domain Orthologous to Human 11p15
BACs and STSs Marker Content of the Mouse Imprinted Domain Orthologous to Human 11p15
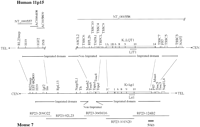
Overview of the imprinted gene domain on human 11p15 and mouse chromosome 7. The organization of the human and mouse domains is depicted, including the locations of the two imprinted subdomains within the region, the locations of the mouse BAC clones that were sequenced and analyzed, and the sources of human sequence for comparison.
For four of the BAC clones (RP23–209o22, RP23–366m16, RP23–101n20, and RP23–124b2) draft quality sequencing and assembly were performed to 5× depth sequence coverage based on NotI/pulsed field gel estimates of clone size (data not shown). Draft assemblies at this level of coverage contain the vast majority of the clone sequence (>90%), with the remaining sequence gaps being small (<1 kb;Bouck et al. 1998). Although the outcome of a draft assembly is a series of sequence contigs of unknown order and orientation, sequence alignments to references (other genomic sequences, genes, etc.) can be used to determine the correct positioning of the draft sequence contigs. Deeper coverage sequencing (10–12×) and assembly, especially using paired forward/reverse reads from sequencing subclones, further reduces the gap number and can generate self-ordered contig sets (Bouck et al. 1998). For the RP23–92l23 clone, deeper coverage sequencing and finishing was performed. This corresponds to the portion of the human genome that has not been sequenced.
Global Comparison of the Mouse and Human Orthologous Imprinted Domain
We used the program PipMaker (Schwartz et al. 2000) to perform a detailed comparison between mouse and human genomic sequences. This analysis is shown graphically in the percent identity plot (PIP) in Figure 2. The reference sequence is mouse and it is oriented from centromere to telomere (the human domain is oriented oppositely). We have used both geometric figures and coloring to annotate the PIP. Structural features in the mouse, including exons, repeats, and CpG islands, are shown above the top line. Evolutionarily conserved elements were identified by PIP analysis. Segments between consecutive gaps in aPipMaker alignment and having ≥50% nucleotide identity are displayed in Figure 2 as short horizontal lines. Exons are considered to be conserved (Fig. 2, green) if they are completely spanned by PipMaker alignments. To determine conserved CpG islands (Fig. 2, orange), we usedBLAST2 to identify segments having ≥50% nucleotide identity. Sequences that do not appear to be an exon, a CpG island, or part of an interspersed repeat identified byRepeatMasker are considered to be conserved (Fig. 2, blue) if they align without a gap for ≥100 bp in thePipMaker alignment with ≥70% nucleotide identity. This criterion, although arbitrary, was used by Loots et al. (2000). Other authors (e.g., Lund et al. 2000; Mallon et al. 2000) have adopted different thresholds. In our analysis, there were eight instances in which a cluster of nearby segments, each meeting this criterion, was merged and considered to be a single conserved region. Novel exons identified by Genscan,GRAIL, or EST identity and confirmed by RT-PCR or Northern blot analysis are also depicted (Fig. 2, red), whether or not they are conserved.

Comparison of mouse and human sequence of the imprinted gene domain. Percent Identity Plot (PIP) showing order and alignment of the entire imprinted domain on mouse chromosome 7 as compared with the orthologous region on human 11p15.5. The mouse sequence is the reference sequence and the short horizontal lines correspond to segments of sequence conservation. Conserved features are color coded as follows: Conserved exons, green; conserved CpG islands, orange; conserved nonexonic sequences not obviously within one of these categories, blue (see text for criteria). Novel genes are shown in red. Where two features apply, two colors are used. The white area is the portion of the human genome sequence that is incomplete but for which mouse sequence was obtained. Vertical black lines show the position of the remaining gaps within the mouse draft assembly sequences. The sequences within these gaps are expected to be <10% (Bouck et al. 1998) of the overall region. Where there is disagreement about nomenclature, exons are numbered arbitrarily (e.g., Igf2).
In all our comparisons, it should be noted that ∼250 kb of the human imprinted domain has not yet been completed (Figs. 1, 2) and that the mouse reference sequence was constructed from draft sequences for four of the five mouse clones spanning this region. As the sequencing efforts of both species give rise to fully accurate and complete data, many of our observations will become more refined, especially with regard to precise physical distances between features. Nonetheless, the accuracy and comprehensiveness of the existing sequences have provided an important resource for the identification of new candidate genes and regulatory sequences.
A global comparison of the human and mouse sequence revealed the presence of 16 known genes: Rl23mrp, H19,Igf2, Ins, Th, Mash2,Tssc6, Tapa1, Tssc4, Trpc5l,Kvlqt1, Lit1, p57KIP2 ,Tssc5, Tssc3, and Nap2 (Fig. 2; Table3). The genomic organization of these genes is, for the most part, comparable between the two species. The total number of exons of known genes is 119 in the human and 112 in the mouse. Of these exons, 110 were conserved. However, some exons were present in the imprinted domain of one species and not the other. For example, mouse Igf2 consists of eight exons whereas the human gene contains one additional exon, and the single-exon encoded ribosomal proteins L26 and L13 were only present in the human and mouse, respectively (Table 4).
Global Sequence Comparison of Human 11p15 and the Orthologous Mouse Domain
Novel Genes in the Imprinted Domain
To assess the level of background sequence similarity between human and mouse, we determined the fraction of noncoding, nonrepetitive mouse sequence that can be aligned to the human sequence using the protocol of Endrizzi et al. (1999) and Zhang et al. (1999). The imprinted domain between Trpc51 and Tssc3 and the nonimprinted domain from Tssc6 to Tssc4 showed a similar fraction of aligned positions (19.6% and 18.8%, respectively). In contrast, the imprinted domain between H19 and Mash2 showed approximately twice the degree of alignment (35.8%), which indicates either that it contains a larger fraction of functional DNA or that neutral mutations are being fixed at a lower rate. Although variable, these numbers are in the range (6.4%–78.1%) observed using the same technique in nine other genomic regions (see Endrizzi et al. 1999, Table 3).
The GC content of the entire domain was less in mouse (47.8%) than that in the human (54.7%). Thirty-three CpG islands were conserved between the two species, and there were approximately twice as many CpG islands in human as there were in the mouse (119 vs. 65). There were an additional 49 conserved nonisland intergenic or intragenic sequences (Tables 3 and 5). Some of these conserved sequences may represent previously unrecognized exons of genes, based on their location, for example, conserved sequences at 67609–67753 (145 nt, 79%) and 82671–82887 (217 nt, 86%) located betweenH19 and Igf2 (Fig. 2; Table 5). However, 39 of the 49 conserved sequences are unlikely to be part of the coding sequence of genes because they did not have high coding potentials following predictions with Genscan orGRAIL. The total sequence represented by all of the nonexonic conserved elements combined was ∼27 kb or 4.1% of the total genomic sequence analyzed.
Conserved Non-exonic Non-CpG Island Sequences
RepeatMasker identified a significantly greater number of repetitive elements in the human sequence than in the mouse (Table 3). Most of this difference was because of the nearly twofold higher fraction of long interspersed nuclear elements in the human sequence (Table 3). In addition, there were threefold more DNA transposon fossils belonging to the medium reiterated repeats (MER) and mariner families. Finally, a VNTR-like repeat, [TGTGAATA(C/T)GCTC(A/G)G]N was located between humanNAP2 and TSSC3 (i.e., at the centromeric end of the imprinted domain) but was not conserved in the mouse. In addition, there were 17.9 tandem copies of a 27-bp motif at mouse positions 126926–127409, upstream of Igf2. A very prominent feature was found at 144–350kb. The region, when masked for interspersed repeats and low-complexity regions using RepeatMasker, shows a striking pattern of alignments between different parts of the region, while having no matches with other genomic sequences in the NCBI databases. Overall, the human imprinted domain has a greater physical size than the orthologous region in mouse (900 kb plus a gap estimated at 250 kb in the human vs. 916 kb in the mouse). This size difference may be partially explained by the increased presence of retroposons. The completion of the human and mouse sequences, in addition to permitting even more refined analyses of the genomic features associated with imprinting, will also be informative in showing how the regions of the two species have been evolving since the time of the mammalian radiation.
Msuit, a Novel Imprinted Transcript Present in Mouse but not Human
Although our primary focus was the identification of conserved sequences, we also observed that several predicted transcripts were present in one species but not the other. For example, by searching dbEST we found that nucleotides 862814 to 864030, approximately 1.9 kb upstream of the mouse p57KIP2 gene, matched EST1179335 (accession no.AA717997; Fig. 2, red). RT-PCR and Northern blot analysis of this EST revealed expression in all fetal and adult tissues, but low stringency Southern blots did not show conservation in human (Fig.4 and data not shown). Given the location of this sequence between p57KIP2 and Tssc5, we thought the transcript might be imprinted despite its lack of conservation. To test this hypothesis, we used a G/C transcribed polymorphism that distinguishes Mus musculus castaneus fromMus musculus musculus, at nucleotide 247 of the EST (Fig. 4). RT-PCR analysis of fetal and adult tissues revealed monoallelic expression, with preferential expression from the maternal allele in all tissues analyzed, indicating that the gene is imprinted (Fig. 4). Based on this result, we designated the gene Msuit1, for mouse-specific ubiquitously imprinted transcript 1.
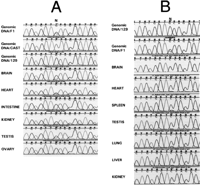
Imprinting analysis of Msuit. F1 cDNA derived from fetal and adult tissues was sequenced from bidirectional crosses of Mus musculus musculus (129/Sv) and Mus musculus castaneus(CAST). A G/C (129/CAST) transcribed polymorphism identified in the genomic DNA at nucleotide 247 was used to distinguish the two alleles. (A) Expression analysis of Msuit in the brain, heart, intestine, kidney, testis, and ovary of F1 obtained from a cross of 129 (mother) and CAST (father). Genomic DNA sequences from each parent and from F1 are included. (B) Expression of Msuit in the brain, heart, spleen, testis, lung, liver, and kidney of F1 from the reciprocal cross. Genomic DNA sequences from paternal parent (129) and F1 are included.
Several Additional Nonconserved Transcripts Unique to the Mouse or Human
Within this region, we identified two transcripts (Fig. 2, red; Table 4) that were unique to the mouse: Ribosomal protein L13(GenBank accession no. NM_016738) located 78 kb telomeric toIns; and EST670599 (GenBank accession no. AA221972), located 14 kb centromeric to Th in the mouse. We also identified five transcripts that were unique to the human (Table 4): Ribosomal protein L26 (accession no. NM_016093) located 15 kb centromeric toTSSC6; EST7905961 (GenBank accession no. AW812967) located upstream of Kvlqt1; EST1100208 (GenBank accession no.AA584837) located 42 kb telomeric to KvLQT1; EST42127 (GenBank accession no. AA337385) located 3 kb telomeric toTAPA1; and EST1422939 (GenBank accession no. AI732937) located 15 kb telomeric of p57KIP2 . Northern blot hybridization and RT-PCR confirmed that all of these were genuine transcripts (Fig. 3; data not shown). Except for the ribosomal proteins and EST670599, which was homologous with the neuronal apoptosis inhibitory protein 3 (Naip3) gene (and thus designated Naip3L1), none of the other five human sequences showed similarity to any sequence in the public databases. Based on the location of these five human transcripts within the minimal region defined by a tumor-suppressing subchromosomal fragment that suppresses the growth of RD cells (Koi et al. 1993), we designated these transcripts tumor-suppressing subchromosomal fragment cDNAs 7, 9, 10, and 11 (Tssc7, Tssc9, Tssc10, andTssc11; TSSC8 is described below) in accordance with our previously established nomenclature (Fig. 1; Table 4)

Expression analysis of novel transcripts in the imprinted gene domain. Human and mouse Northern blots were hybridized with expressed sequence tag (EST) probes. (A) Mouse Northern blot hybridized with EST670599 (accession no. AA221972): 1, heart; 2, brain; 3, spleen; 4, lung. (B) Human Northern blot hybridized with EST1422939 (GenBank accession no.AI732937): 1, spleen; 2, lung; 3, prostate; 4, testes. (C) Human Northern blot hybridized with Ihit, aGenscan-predicted cDNA located betweenH19 and Igf2: 1, heart; 2, brain; 3, spleen; 4, kidney. (D) Human fetal Northern blot hybridized with Ihit, 1, kidney; 2, liver; 3, lung; 4, brain.
Conserved Novel Transcripts
By using PIP matches to search dbEST, we identified a sequence of 332 nt in mouse at nucleotides 660496 to 663300 with 85% identity to human sequence that corresponded to mouse ESTJ1011C10 (accession no.AU041933), as well as to human EST2466762 (accession no. AI933351). This conserved sequence was located 5 kb telomeric to exon 10 ofKvlqt1 (Table 4; Fig. 2, red) and was designatedTssc8. RT-PCR with gene-specific primers showed a transcript in all tissues examined, with transcriptional orientation opposite toLit1, even though Tssc8 lies within Lit1(data not shown). The ESTs do not contain an obvious ORF, nor do they show homology with any known transcripts. Similarly, we identified a mouse EST482800 (accession no. AI594936) located between H19and Rl23mrp that showed 88% sequence identity to human sequence (Table 4, Fig. 2, red). Because this transcript is immediately telomeric to H19, elucidation of its imprinting status may further delimit the telomeric imprinted–nonimprinted subdomain boundary. We designated this transcript Rhit1(R123mrp-H19 interval transcript −1).
Conserved Intergenic Sequences and a Nonconserved Transcript betweenIGF2 and H19
The IGF2 and H19 genes have attracted great interest as a model for imprinting studies (Wolffe 2000), and both genes can undergo loss of imprinting in cancer (Rainier et al. 1993; for review, see Feinberg 1999). Comparison of mouse and human sequence allowed us to order the region from Ins to L23mrp, which existed previously only as draft assembly sequence in the Human Genome Project (Bentley 2000). This analysis revealed the presence and location of several previously unrecognized conserved sequences. These include two CpG islands between Igf2 and Ins and two CpG islands located downstream from H19 (Fig. 2, orange).
In addition, we observed seven conserved nonexonic, nonisland sequences between Igf2 and H19 (Fig. 2, blue; Table 5). RT-PCR did not reveal a product in mouse fetal and adult tissues and there were no matches to EST sequences, which indicates that these may represent conserved regulatory sequences. Consistent with this possibility, the conserved sequences are within the region shown in functional complementation experiments to be necessary to maintain normal imprinting of a transgenic YAC containing both Igf2 andH19 (Ainscough et al. 1997). Finally, Genscan andGRAIL analysis of the mouse sequence between Igf2and H19 revealed several predicted exons that were not previously known. For one of these predicted exons (nucleotides 76583–76864), we detected a strong 1-kb signal on Northern blots derived from both mouse and human RNA from fetal and adult liver and from placenta (Figs. 3 and 5; data not shown). In addition, a similarly sized transcript was apparent in the human brain (Fig. 3). The predicted protein sequence showed no homology with any known sequences and we designated the gene Ihit1 (Igf2-H19 interval transcript-1). Northern blot hybridization indicated that the sequence is conserved in human. However, the precise localization must await the completion of the human sequence between H19 andIGF2.

Genscan-predicted nucleotide and amino acid sequence ofIhit. The transcript is located between H19 andIgf2 in the mouse.
A Two-Island Rule for Imprinted Genes
CpG islands are defined as sequences of ≥200 bp with a GC content (i.e., [G + C]/N > 0.5) and an observed-to-expected CpG dinucleotide content (i.e., [CpG × N]/[C × G] > 0.6; Gardiner-Garden and Frommer 1987). CpG islands are normally unmethylated, but allele-specific methylation of CpG islands appears to mark both the inactive X chromosome (Yen et al. 1984) and many imprinted genes, for example,H19, Snrpn, and Igf2r (Brandeis et al. 1993;Shemer et al. 1997; Wutz et al. 1997). In addition, GC-rich sequences that are not CpG islands (i.e., they meet the first, but not the second criterion above) may also be differentially methylated (termed a differentially methylated region) in the vicinity of imprinted genes, for example Igf2 (Sullivan et al. 1999) and a second site 2–4 kb upstream of the H19 CpG island (Thorvaldsen et al. 1998). Therefore, one of our goals was to identify conserved CpG islands and GC-rich sequences that might serve as a substrate for future experiments to investigate allele-specific methylation.
This analysis revealed 33 conserved CpG islands (Fig. 2, orange), and 28 conserved GC-rich (>50%) sequences (Table 5). Remarkably, eight of nine conserved imprinted genes within the entire domain showed two or more conserved CpG islands upstream of or within the gene (Table6), but all of the six nonimprinted genes were associated with no or one CpG island (Table 6). This difference was statistically significant (p < 0.01, Fisher's exact test). Generally, one conserved CpG island associated with each imprinted gene was located <2 kb upstream of the gene and, in some cases, overlapped the first exon, for example, H19,Igf2, Mash2, Kvlqt1,p57KIP2, Msuit1, Tssc5, andTssc3. Additional conserved CpG islands associated with the imprinted genes were generally located within an intron and often extended into one or both of the adjacent exons.
CpG Island Organization and Allelic Expression
Nonisland Conserved Sequences
We identified 49 nonisland conserved sequences that did not correspond to known exons (Fig. 2, blue; Table 5). These sequences were clustered predominantly around imprinted genes. In particular, within the imprinted gene subdomain that extends from Mash2 toH19 we identified 10 conserved nonisland sequences, seven of which were located between H19 and Igf2 (Fig. 2), and two that were within Igf2. Two additional such sequences were located within 14 kb downstream from H19. Of the remaining 37 nonisland conserved sequences, 36 were located within the imprinted gene subdomain that extends from Tssc3 to Kvlqt1, and 33 of these were within Kvlqt1 itself. Interestingly, 12 of these conserved sequences were located within 44 kb upstream of theLit1 CpG island (Fig. 2), and six of these are GC rich, even though they did not meet the full definition of a CpG island. It will be of interest to determine whether any of these conserved GC-rich sequences are differentially methylated between the two parental chromosomes, given that the CpG island immediately upstream ofLit1 is not conserved between human and mouse.
DISCUSSION
In this report, we have described the first sequencing and comparative analysis of an entire imprinted gene domain between human and mouse. If one excludes a gap that remains within the human genome sequence, which we have sequenced in the mouse, and smaller gaps within the mouse sequence, this analysis includes 915 kb of mouse and 900 kb of human, the largest comparative sequencing analysis of a single ordered and oriented domain to date. The majority of the mouse sequence analyzed in this study reflects draft sequence assemblies (Collins et al. 1998). The value of the draft sequence, which is anticipated to provide >90% coverage (Bouck et al. 1998), has been greatly enhanced through the availability of sequence from an orthologous region of a second species.
The order and orientation of the mouse sequence contigs could be established through alignment with respect to the human sequence, allowing us to clearly establish positional information for the conserved sequence elements. In this case, the available human sequence was finished, but for organisms for which the evolutionary distance is similar to that between human and mouse, comparable utility can be obtained when each of the sequences is draft (K. Dewar and W. Miller, unpubl. ).
We found 16 conserved known human genes that were made up of 119 exons in the human and 112 in the mouse. Of these, 110 (98%) were conserved. There were also several transcripts present in this region in one species but not the other, including ribosomal protein L26 in human, ribosomal protein L13 and a homolog of Naip3(Naip3L1) in mouse, and several ESTs unique to one species or the other. We showed that one of the sequences unique to the mouse was imprinted, and we designated it Msuit1, for Mouse-specific ubiquitously imprinted transcript-1. An intriguing potential mechanistic explanation for the imprinting of Msuit1 is that the location of a gene within this domain may subject it to long-rangecis-acting regulatory sequences that are responsible for allele-specific silencing, such as chromatin alterations acting at a distance, similar to telomere silencing in yeast or to position effect variegation in Drosophila.
One of the most striking conclusions of this analysis is that the number of conserved sequences outside the known coding exons and interspersed repeats is small. There were 82 such sequences, with an average length of 337 bp, thus making up ∼4.1% of the total noncoding sequence throughout the domain. The sequence analysis ofLoots et al. (2000) found 91 conserved sequences (each ≥100 bp of 70% identity) distributed >900 kb of noncontiguous draft assembly sequence, although the fraction of sequence this represents was not reported. Conservation of 1% of noncoding sequence was also reported over a relatively short interval (92 kb; Jang et al. 1999). Thus comparative sequencing may be a powerful strategy for identifying the critical nonexonic regulatory sequences that would be difficult to determine by analysis of a single genome.
Of these 82 sequences, 33 (42%) were CpG islands and 28 were GC-rich sequences in both species. Thus 61 of 82 (74%) of the conserved nonexonic sequences were either GC rich or were true CpG islands. This provides further evidence of an important role for DNA methylation in the regulation of genes throughout this domain. Consistent with this idea, at least some of these sequences appear to show partial methylation in genomic DNA (P. Onyango and A.P. Feinberg, unpubl.), including the CpG islands, which are normally unmethylated except for the inactive X-chromosome and imprinted genes (Yen et al. 1984;Brandeis et al. 1993; Shemer et al. 1997; Wutz et al. 1997). We are currently determining which of these sequences might show allele-specific methylation.
The location of these conserved sequences is also of particular interest in that they are not randomly distributed. We had previously shown that the imprinted domain is itself divided into two imprinted subdomains in human (TSSC3 to KVLQT1, andASCL2 to H19), with a region of little or no imprinting between them (TSSC4 to TSSC6) (Lee et al. 1998; Feinberg 1999). All but one of the conserved sequences fell within one of the two imprinted subdomains. This observation provides further support for a role of these sequences in the regulation of genomic imprinting.
Curiously, we found that the imprinted genes tended to be associated with two or more CpG islands. This also appears to be true for imprinted genes on other chromosomes (Yen et al. 1984; Brandeis et al. 1993; Shemer et al. 1997; Wutz et al. 1997), although, to our knowledge, this has not been commented on in the literature, likely because interspecies global sequence comparisons have not been possible. We suggest that there may be a two-island rule for imprinting, that is, in most cases more than one CpG island is required to maintain normal imprinting. Perhaps the additional CpG island is related to a second methylation mark or, alternatively, to the presence of antisense transcripts associated with these genes. The latter appears to be the case for Kvlqt1, Igf2r, andIgf2.
This analysis also revealed that a CpG island upstream of the humanLit1 antisense RNA is in fact not conserved in the mouse, even though it shows differences in allele-specific methylation and alterations in BWS. However, we identified several GC-rich sequences, 5–44 kb upstream of this CpG island that are >70% conserved between human and mouse. Preliminary analysis suggests that at least one of these sequences also shows allele-specific methylation (P. Onyango and A.P. Feinberg, unpubl.) and thus it might be important in normal imprint regulation or disease. Another potentially important sequence is a 75% conserved CpG island 4 kb upstream ofp57KIP2 . In contrast to the CpG island withinp57KIP2 , which is unmethylated in humans, this newly identified sequence is partially methylated in humans (P. Onyango and A.P. Feinberg, unpubl.).
The mouse Igf2 and H19 genes have attracted a great deal of interest, but the sequence between them has been previously unknown. The human sequence between these genes has been reported by the Human Genome Project in six unordered fragments. We were able to order the human interval between IGF2 and H19 by comparison to mouse sequence. This analysis revealed 10 conserved sequences in this interval, including three CpG islands. A novel gene termed Ihit also lies within this interval, at least in the mouse.
Finally, an intriguing concept in the study of genomic imprinting is the idea of a large genomic domain that might be regulated hierarchically, with some local elements regulating individual genes and other elements having more global effects. Such an idea is consistent with the imprinting center deletions observed in Prader-Willi and Angelman syndromes, which disrupt imprinting over several megabases. Similarly, we have observed patients with BWS and loss of imprinting affecting either LIT1 or IGF2 but not both, and others with loss of imprinting in both gene regions (Lee et al. 1999; DeBaun et al., in prep.). It will thus be of interest to examine the conserved sequences identified here not only in normal tissues, but also in disease tissues, to gain insight into their potential role as more global cis-acting regulators of gene expression.
METHODS
Isolation of a 10×-Depth BAC Contig from Mouse Chromosome 7, Identification of a Minimal Tiling Path, and Sequencing of the Mouse Contig
An overgo hybridization protocol (Ross et al. 1999) was used for probes generated from gene sequences of the imprinted region. Forty-five overgos were pooled and screened against high-density BAC clone filters of a 11.2× genomic equivalent female mouse C57 BL/6J genomic library (RPCI-23; BAC/PAC Resources, Oakland CA;www.chori.org/bacpac/). Single-colony isolates were recovered from all positive well addresses, rearrayed into a 384-well microtitre plate, and then duplicated onto a series of filters (HybondN+, Amersham). Each overgo probe was tested against a rearrayed copy to establish the marker and clone relationships. Using marker-clone content andHindIII fingerprint information (Marra et al. 1997) a set of five minimally overlapping clones were selected for sequencing (GenBank accessions nos. AC013548, AC012382, AC015800, AC012540, and AC023248). Draft sequence assembly of all the clones was performed by ligating mechanically sheared 2-kb fragments of BAC DNA into an m13 sequencing vector, followed by random shotgun sequencing at 5× coverage of the estimated clone size, and then assembly. To increase sequence contiguity and establish the order and orientation of the sequence within AC012382, an additional subclone library of 4-kb fragment size was prepared and sequenced in a plasmid sequencing vector. Plasmid subclones were sequenced from both ends to an additional 5× coverage and integrated into the assembly. Sequence gaps and ambiguities were subsequently resolved using standard finishing techniques (Wilson and Mardis 1997). We were able to order and align the mouse draft sequences with the human by performing both a PIP comparison and an analysis using a novel NCBI toolkit termed Alignment Construction Utility and Tools Environment (ACUTE). ACUTE is capable of generating, viewing, and analyzing discontinuous or overlapping sequence alignments. The mouse draft assembled sequence, although multipass and >99.9% accurate, was in unordered fragments, and the human sequence was in three large pieces, with gaps of unreported size. The initial set of mouse–human alignments was used to order and orient the mouse draft sequence. Approximately 95% of the sequence could be unambiguously ordered this way to generate an ordered and oriented sequence spanning the entire imprinted region. Similarly, the human sequences could be ordered, oriented, and concatenated. The sequences used in our analysis can be obtained at http://www.jhmi.edu/feinberg_lab orhttp://bio.cse.psu.edu/. A gap remains in the human sequence spanning the TH gene. Therefore, in this area, deeper coverage mouse sequence was obtained. Thus comprehensive sequence was generated over the entire imprinted domain and comparison between mouse and human could be performed over all but the portion not yet completed by the Human Genome Project.
Global Comparison of the Mouse and Human Sequences
To compare the mouse and human sequences over the entire imprinted domain we used PipMaker(http://bio.cse.psu.edu/PipMaker/). The program was run in a manner constraining matches to be both conserved and colinear between the two species. Matches of a desired minimum length and percent identity lying between consecutive gaps in a PipMakeralignment were found with a program called strong_hits, which can be downloaded from the PipMaker site. The human sequences were retrieved from GenBank (accession nos. NT_000558, NT_000557, and AC006408). We used the concatenated mouse sequence as the reference sequence in PipMaker analysis. To eliminate spurious matches resulting solely from low and high complexity repeats, we masked the mouse sequence usingRepeatMasker(http://ftp.genome.washington.edu/cgi-bin/RepeatMasker) before performing the PipMaker analysis.RepeatMasker was also used to deduce the repeat content for the sequences from each species. Tandem repeats were identified with the program Tandem Repeats Finder(http://c3.biomath.mssm.edu/trf.html; Benson 1999).
Gene Prediction
To identify potential genes in both the mouse and the human sequences we used a four-step approach. First, we masked the sequences for high complexity repeats usingRepeatMasker. Second, repeat-masked sequences were analyzed for exon content using Genscan(http://ccr-081.mit.edu/Genscan.html),GRAIL (http://grail.lsd.ornl.gov/Grail-1.3) and PipMaker. Third, we used all the predicted coding sequences or highly conserved sequences from step one to search GenBank databases. The fourth step involved directBLAST database searches using fragments of either the mouse or human sequences
Identification of Conserved Sequences
CpG islands were found by a simple program, written inC, that looks in 200-residue windows for regions that meet the definition of Gardiner-Garden and Frommer (1987). Conserved sequences were identified as described in the text.
Imprinting Analysis
Mice were purchased from Jackson Laboratory. We crossed inbredMus musculus (129/Sv) to inbred Mus musculus castaneus (CAST/Ei) to obtain F1 mice with polymorphic genotype. To identify polymorphisms we amplified by PCR and sequenced genomic DNA from F1, 129Sv, and CAST/Ei. PCR conditions were as follows: 2 min at 95°C; then 40 cycles each of 1 min at 95°C, 30 sec at 60°C, 1 min at 72°C; then 9 min at 72°C. RNA was extracted from tissues of F1 animals derived from crosses from both directions using the protocols outlined below. Total RNA was isolated using RNeasy minikit from Qiagen. To eliminate DNA contamination from RNA preparations, samples were treated with preamplification-grade DNase I (GIBCO) according to supplied protocols. RT-PCR was performed using the Superscript II preamplification system (GIBCO) and was performed for each sample in the presence and absence (negative controls) of RT. Samples were sequenced only when no bands were obtained with the negative controls. The primers used for the imprinting analysis were ESTAA7179-F: 5′-AAGCAAGTGATGCAAGCATCC-3′ and ESTAA7179-R: 5′-ACTCCACACTTATTTGTGACC-3′. DNA and cDNA sequencing was run on an ABI-377 automated sequencer following protocols recommended by the manufacturer (Perkin-Elmer).
Northern Blots
Multiple-tissue Northern blots were purchased from Clontech. Hybridization and washes were performed according to manufacturer's recommendations. Blots were exposed to X-Ray films for 1–14 days.
Acknowledgments
We thank Eric S. Lander for encouragement and support, members of the WI/MIT Center for Genome Research and UTSW Genome Science and Technology Center for genomic sequencing of the mouse and human regions, respectively, and the members of the Feinberg laboratory for helpful discussions and technical assistance. This work was supported by grants from the National Institutes of Health to A.P.F., W.M., and E.S.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵8 Corresponding author.
-
E-MAIL afeinberg{at}jhu.edu; FAX (410) 614-9819.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.161800.
-
- Received August 24, 2000.
- Accepted September 19, 2000.
- Cold Spring Harbor Laboratory Press








