
The Sox10Dom Mouse: Modeling the Genetic Variation of Waardenburg-Shah (WS4) Syndrome
- E. Michelle Southard-Smith1,
- Misha Angrist2,
- Jane S. Ellison1,
- Richa Agarwala1,
- Andreas D. Baxevanis3,
- Aravinda Chakravarti2, and
- William J. Pavan1,4
- 1Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health (NIH), Bethesda, Maryland 20892-4472 USA; 2Department of Genetics and Center for Human Genetics, Case Western Reserve University and University Hospitals of Cleveland, Cleveland, Ohio 44106-4955 USA; 3Genome Technology Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892-4431 USA
Abstract
Hirschsprung disease (HSCR) is a multigenic neurocristopathy clinically recognized by aganglionosis of the distal gastrointestinal tract. Patients presenting with aganglionosis in association with hypopigmentation are classified as Waardenburg syndrome type 4 (Waardenburg-Shah, WS4). Variability in the disease phenotype of WS4 patients with equivalent mutations suggests the influence of genetic modifier loci in this disorder. Sox10Dom /+ mice exhibit variability of aganglionosis and hypopigmentation influenced by genetic background similar to that observed in WS4 patients. We have constructed Sox10Dom /+ congenic lines to segregate loci that modify the neural crest defects in these mice. Consistent with previous studies, increased lethality ofSox10Dom /+ animals resulted from a C57BL/6J locus(i). However, we also observed an increase in hypopigmentation in conjunction with a C3HeB/FeJLe-a/a locus(i). Linkage analysis localized a hypopigmentation modifier of the Dom phenotype to mouse chromosome 10 in close proximity to a previously reported modifier of hypopigmentation for the endothelin receptor B mouse model of WS4. To evaluate further the role of SOX10 in development and disease, we have performed comparative genomic analyses. An essential role for this gene in neural crest development is supported by zoo blot hybridizations that reveal extensive conservation throughout vertebrate evolution and by similar Northern blot expression profiles between mouse and man. Comparative sequence analysis of the mouse and humanSOX10 gene have defined the exon–intron boundaries ofSOX10 and facilitated mutation analysis leading to the identification of two new SOX10 mutations in individuals with WS4. Structural analysis of the HMG DNA-binding domain was performed to evaluate the effect of human mutations in this region.
Comparative molecular studies have been useful for identifying and analyzing animal models of human disease. Mouse models have been particularly relevant to the genetic analysis of Hirschsprung disease (HSCR, OMIM no. 142623). HSCR is recognized clinically as aganglionic megacolon, the absence of intrinsic ganglion cells in the myenteric (Auerbach’s) and submucosal (Meissner’s) plexuses of the distal gastrointestinal tract with subsequent failure of peristalsis. In association with sensorineural deafness or melanocyte deficiencies, congenital aganglionosis is categorized as Waardenburg-Shah syndrome (also called WS4, OMIM no. 277580). The multigenic nature of this disorder is reflected by the number of spontaneous mouse mutants with phenotypes similar to WS4: piebald(Ednrbs )/piebald lethal(Ednrbs-1 ), lethal spotting (Edn3ls ), and Dominant megacolon(Sox10Dom ). Comparative genetic analyses led to the identification of EDNRB mutations ins/sl mice and HSCR families (Hosoda et al. 1994; Puffenberger et al. 1994). Similarly EDN3 mutations have been identified in ls/ls mice and HSCR patients (Baynash et al. 1994; Edery et al. 1996; Hofstra et al. 1996). More recently, the elucidation of a Sox10 mutation in Dominant megacolon mice (Southard-Smith et al. 1998) has led to identification of mutations of the human SOX10 ortholog in WS4 families (Pingault et al. 1998; this report).
Murine models are also useful for dissecting the genetic factors that contribute to the severity of disease phenotypes. Genetic analysis of the WS4 mouse model Ednrbs was useful in identifying loci that modify the hypopigmentation defect (Pavan et al. 1995); however, these crosses did not exhibit significant variation in the aganglionosis defect (W.J. Pavan, unpubl.). In contrast, theSox10Dom mouse is a particularly relevant model for WS4 because it mimics the variable penetrance and expressivity of the aganglionosis observed in HSCR patients. The phenotype ofSox10Dom mice is comprised of hypopigmentation that appears as a belly spot, white feet and white forelock (head spot) accompanied by aganglionic megacolon. Initial characterization of these mice revealed variation in the severity of the megacolon phenotype and indicated that the aganglionosis was affected by backcrosses to either parental strain, C57BL/6J (B6) or C3HeBFeJLe-a/a (C3H) (Lane and Liu 1984; Kapur et al. 1996).
Molecular genetic analysis of Sox10Dom mice led to identification of a single base insertion in the B6 allele of theSox10-coding region (Herbarth et al. 1998; Southard-Smith et al. 1998). The Sox gene family is defined by sequence similarity of its members to the HMG DNA-binding domain (SRY box) present in the mammalian sex-determining gene, SRY (Prior and Walter 1996; Pevny and Lovell-Badge 1997). Members of this gene family control developmental decisions that dictate cell fate in sex determination (SRY andSox9), neuroepithelial lineages (Sox1 andSox2), and hematopoiesis (Sox4). By association of mutations in Sox10 with the presentation of neural crest defects, an essential role for Sox10 in neural crest development has been established in mouse (Herbarth et al. 1998;Southard-Smith et al. 1998) and man (Pingault et al. 1998). In vitro studies have confirmed that the same mutant forms of SOX10 observed in WS4 patients fail to transactivate expression of reporter constructs in transient transfections (Kuhlbrodt et al. 1998b).
This study describes the construction of Sox10Dom /+ congenic lines and demonstrates the utility of this mouse model for dissection of genetic determinants that contribute to the variable hypopigmentation observed in WS4 patients. We also present comparative genomic analyses of SOX10 in mouse and man, the identification of two new mutations in SOX10 of individuals with WS4, and a structural analysis of the SOX10 HMG DNA-binding motif.
RESULTS
Sox10Dom Congenic Lines Segregate Modifiers of Hypopigmentation
The Sox10Dom mutation originally arose in the C57BL/6J allele of an F1 hybrid cross and has been maintained on a C57BL/6J × C3HeB/FeJLe-a/a (B6C3F1/J) hybrid background (Lane and Liu 1984). To identify the genetic determinants responsible for the variation in the Sox10Dom phenotype, we established congenic lines of this mutant on the B6 and C3H parental strain backgrounds. As described previously (Lane and Liu 1984), survival is markedly affected by the strain background. We observed only 30% ofSox10Dom /+ animals surviving to weaning on the B6 background in contrast to 82% survival on the C3H background. However, in addition to the variation in survival, we demonstrate that the degree of hypopigmentation (spotting) also varies greatly between congenic lines as early as N2. In the B6.Sox10Dom /+ pedigree, 3% (1/28) ofSox10Dom /+ mice demonstrate head spotting, which appears as only a few hairs (Fig. 1). In contrast, 50% of Sox10Dom /+ mice at N4 of the C3H pedigree display visible white forelocks (head spots that are significantly larger than those seen in B6.Sox10Dom /+mice). The absence of head-spotted individuals in the B6. Sox10Dom /+ congenic line was not a consequence of preweaning mortality of severely affected animals in this pedigree. We were able to assess head spotting in almost all the mice as lethality usually did not occur prior to pigmentation (to 7–10 days postnatum).
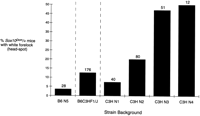
Effect of parental strain on frequency of white forelock inSox10Dom mice. The frequency of white forelock inSox10Dom /+ mice on either the B6, B6C3HF1, or C3H backgrounds is shown. The total number of animals assessed for head spotting in a particular strain background is indicated above each bar on the plot. The increase in head spotting through successive generations of breeding onto the C3H background suggests a recessive C3H modifier.
Modifier loci that influence hypopigmentation have been localized to mouse chromosomes 2, 5, 8, and 10 in crosses segregating theEdnrbs locus (Pavan et al. 1995). Additional analyses that characterized the influence of the chromosome 10 modifier on dorsal spotting colocalized this locus with two microsatellite markers D10Mit178 and D10Mit96 (H. Rhim and W.J. Pavan, in prep.). To determine whether the same or a closely linked locus could account for the variation in hypopigmentation observed in the Sox10Dom congenic strains, we performed linkage analysis on a pedigree of Sox10Dom /+ mice that had been maintained by crosses to B6C3F1/J mice. One hundred and seventy-six Sox10Dom /+ mice were analyzed from this pedigree and genotypes determined for D10Mit178 andD10Mit96. Of the twenty animals with white forelocks, all were homozygous for the C3H allele at the chromosome 10 locus. Two additional animals had small head spots consisting of only a few hairs similar to that seen infrequently in the B6.Sox10Dom /+ pedigree. Both animals were homozygous B6 at these markers. There were also nineteen Sox10Dom /+ animals that were homozygous C3H at the chromosome 10 locus, but did not exhibit white forelocks. This is consistent with the 50% penetrance of the white forelock phenotype observed forSox10Dom /+ mice in the C3H pedigree at N4.
Parametric linkage analysis was performed assuming a 50% penetrance with FASTLINK version 4.0P (Cottingham et al. 1993; Schaffer et al. 1994) in which the head-spot trait was modeled as recessive with 50% penetrance and a phenocopy rate was based on the observation that two mice out of 22 were false positives for the trait. This analysis indicated significant linkage for D10Mit178 (lod score of 5.63 with P value of 2.3 × 10−6) at no recombination (υ = 0.0). Nonparametric linkage analysis with the program SimIBD and 100,000 replicates, 500 simulated null distribution replicates, and 1000 bootstraps gave a P value of 0.009992. The higher P value achieved from the nonparametric analysis is consistent with previous observations that nonparametric linkage methods have lower power than parametric linkage methods when the trait model is correctly specified (Goldin and Weeks 1993). Haplotype inspection revealed only one recombination event betweenD10Mit96 and D10Mit178 in an animal (C3H/C3H and B6/C3H respectively) that lacked a white forelock. In addition all 183Sox10Dom /+ mice displaying white forelocks from the C3H pedigree were homozygous for the C3H allele on chromosome 10. Homozygosity at these markers would be expected for subsequent generations in this pedigree. We propose that the chromosome 10 C3H locus acts as a recessive modifier of white forelock inSox10Dom /+ mice with reduced penetrance. The increased penetrance observed in subsequent generations of the C3H pedigree suggests that additional modifier loci are inherited from the C3H background to account for the increasing penetrance of the white forelock trait. Further linkage analyses will be needed to determine the location of the additional loci that influence hypopigmentation.
Comparative Genomic Analysis: Conservation, Expression, Genomic Structure
The neurocristopathies in Sox10Dom mice, the expression pattern of Sox10, and the altered expression of other neural crest marker genes in these mutant mice indicate a key role for SOX10 in neural crest development (Southard-Smith et al. 1998). To evaluate the conservation of Sox10 in vertebrate evolution and assess the presence of orthologous genes in additional species, a cDNA fragment 3′ to the HMG box of mouse Sox10was hybridized to zoo blots (Fig. 2). Significant hybridizing bands were observed in DNA from all mammals analyzed and from fish. The presence of a Sox10 ortholog in these species is consistent with an essential role for this gene in development (Southard-Smith et al. 1998). Inconsistent hybridization to genomic DNA samples of chicken and yeast between the two blots was observed and thought to be a consequence of either incomplete DNA digestion or strain differences between the samples analyzed.
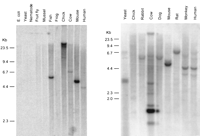
Cross-species conservation of Sox10. Southern blot autoradiographs from hybridization of EcoRI-digested genomic DNAs with the 3′ end of a mouse Sox10 cDNA probe (left, Quantum Biotechnologies; right, Clontech). Organism names are indicated above each lane. Specific species DNAs applied to the blot at left include Escherichia coli,Saccharomyces cerevisiae, Caenorhabditis elegans,Drosophila melanogaster, Tautoga onitis, Mytilus edulis, Bovis domesticus, Xenopus laevis,Gallus domesticus, Mus musculus, and Homo sapiens. (Left) Positions of molecular size standards in kb. Longer exposures of the filter in B revealed several weakly hybridizing bands within the chicken DNA sample that are not visible in this reproduction. Hybridization of a probe for a second gene to the filter in A produced a similar high molecular weight band and streaking in the chicken lane as seen here with theSox10 3′ cDNA probe.
Northern blot hybridization of mouse and human poly(A)+ RNA was performed to compare the expression pattern of Sox10 between mouse and human (Fig. 3). Expression patterns, if comparable, were expected to further validate theSox10Dom mouse as a human disease model and potentially identify additional tissues that should be evaluated for effects of SOX10 mutations in patients. Northern blot analysis demonstrated that Sox10 expression is not detectable in 7-d.p.c. mouse embryos but is expressed in older embryos through to adulthood. This pattern is consistent with the initiation of expression at day 8.5 d.p.c. observed by in situ hybridization studies in mouse and rat (E.M. Southard-Smith unpubl.; Herbarth et al. 1998; Kuhlbrodt et al. 1998a). In adult mouse tissues, Sox10 mRNA is detected in heart, brain, lung, skeletal muscle, and testes. Expression in human tissue poly(A)+ RNA samples was similarly observed in these tissues. Additional sites of Sox10 expression were observed in human pancreas, prostate, ovary, stomach, spinal cord, trachea, and adrenal gland. The high levels of hybridization to human brain and spinal cord RNAs are consistent with Sox10 expression in glial cells and astrocytes (Kuhlbrodt et al. 1998a). SOX10 expression was observed throughout the human digestive tract (stomach, small intestine, and colon) as well, consistent with our previous analyses in the mouse (Southard-Smith et al. 1998). Hybridization signals forSox10 mRNA were consistently absent in kidney and spleen of both mouse and human. The faint signal observed in mouse lung, consistent with expression observed in the developing lung bud of 12.5-d.p.c. embryos (Southard-Smith et al. 1998), was notably absent in hybridizations to human lung poly(A)+ RNA.
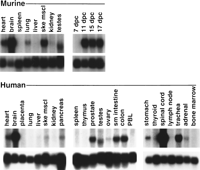
Expression profile of Sox10 in mouse and human total tissue RNA samples. Northern blot hybridization of a Sox10 cDNA probe to multiple mouse and human poly(A)+ RNA samples (10 μg/lane, Clontech). (Top) Hybridizations with 1.3- and 1.7-kb 3′ end Sox10 cDNA probes that exclude the HMG box for mouse and human samples, respectively; (bottom) hybridizations with the RNA loading control L-32 (Dudov and Perry 1984).
Comparative sequence analysis of Sox10 genomic structure between mouse and human was performed to assess the conservation of exon–intron boundaries and facilitate mutation detection in the human ortholog. Mouse exon–intron boundaries were identified by comparison of the mouse cDNA sequences with genomic sequences obtained from single pass sequencing of BAC43P19 (Southard-Smith et al. 1998; E.M. Southard-Smith, J.E. Collins, J.S. Ellison, K.J. Smith, A.D. Baxevainis, J.W. Touchman, E.D. Green, I. Dunham, and W.J. Pavan, in prep.). Five exons and four introns were identified. Each of the identified splice junctions possesses the consensus for splice donor and acceptor sites. Although the majority of Sox genes exhibit a monoexonic structure, Sox10 is analogous to Sox9,SOX5, Sox17, and SOX20, which possess multiple exons that partition the ORF within the HMG box. By use of the junction sequences from mouse Sox10, the human exon boundaries were identified within cosmid J81I2 and confirmed by sequencing of additional human BAC clones (Table 1). Exon–intron organization and splice-site positions appear conserved between mouse and human; however, some variation in exon size is apparent. Exons 2, 3, and 5 differ by 4, 6, and 170 bases, respectively. Comparison of intron sequence flanking splice junctions demonstrates a considerable lack of conservation outside exonic regions. This information was used to establish a set of primers that amplify human exons for mutation detection analysis.
Comparison of Sox10 Splice Sites in Mouse and Human
Our sequence analysis of Sox10 mouse cDNA additionally revealed two potential initiator methionines (Fig. 4) the most 5′, MetALT, residing within a representative Kozak consensus sequence (Kozak 1996) and the second residing 309 bp more 3′, Met1, within a less representative Kozak region. Because no stop codons are apparent in the mouse between these two positions, either methionine could serve as the initiator residue. To investigate which methionine acts as initiator for the Sox10ORF, we extended the human SOX10 cDNA sequence using clones derived from three independent human brain cDNA clones and compared this to cDNA sequences of mouse (Southard-Smith et al. 1998) and rat (Kuhlbrodt et al. 1998a) as well as to mouse genomic sequences (E.M. Southard-Smith and W.J. Pavan, unpubl.). Repeated attempts to generate sequence information for the human SOX10 exon 1 that might contain a MetALT analogous to that in the mouse by cDNA isolation, 5′ RACE, and genomic sequencing of human BACs with mouse primers were unsuccessful. However, comparative alignment of the available human sequence with mouse and rat sequences for exons 2 and 3 revealed 90% sequence identity between the murine and human sequences, implying structural relevance. Despite this high degree of conservation, which exceeds the average aligned identity (67%) of human and mouse 5′ UTRs (Makalowski et al. 1996), our sequence analysis of the human SOX10 cDNA clones identified a single base deletion in comparison to the mouse and rat that would frameshift the human ORF. This deletion was observed in three independent human cDNA clones our human genomic clones and has been seen by Pusch and colleagues (Pusch et al. 1998). The single base deletion would preclude the use of an orthologous MetALT and supports initiation of the ORF from Met1 in exon 3.
Comparison of mouse, human, and rat sequences 5′ of the initiation methionine (Met1, boxed) proposed by Kuhlbrodt and colleagues (Kuhlbrodt et al. 1998a). Coding sequences derive from cDNA (human; E.M. Southard-Smith, unpubl.; mouse GenBank accession no. AF017182; rat GenBank accession no. AJ001029) or genomic sequences (M. Angrist, unpubl.). An alternative start codon MetALT found in the mouse Sox10 5′ UTR is also boxed.
Mutation Detection in Waardenburg-Hirschsprung’s Disease Patients
On the basis of the definition of exon–intron boundaries provided by cross-species comparisons, PCR assays were developed to assess the integrity of SOX10-coding regions in patients that had biopsy-proven HSCR in addition to one or more major stigmata associated with Waardenburg syndrome (sensorineural hearing loss, pigmentary anomalies, and/or bicolored irides). Direct sequencing of the threeSOX10-coding exons in nine patients with HSCR and Waardenburg-associated phenotypes revealed two nonsense mutations. The patient from family 140 (Fig. 5) has short segment HSCR, profound sensorineural hearing loss, hypopigmentation on his abdomen and neck, and is heterozygous for a Y207X mutation. Both parents are phenotypically normal and neither carries the mutation. Y207X occurs in exon 4, 27 residues downstream of the carboxyl end of the HMG box and 14 residues downstream of the corresponding site in the mouse where the insertion responsible for theSox10Dom phenotype is located (Fig. 6;Southard-Smith et al. 1998).

Pedigrees of WS4/Sox10 families. (Left) Pedigree of family 140. The Proband (arrow) was found to be heterozygous for theSOX10 mutation Y207X. (−) Mutant allele; (+) wild-type allele. This patient has short segment HSCR, profound deafness, and multiple areas of hypopigmentation on his neck and abdomen. DNA was not available from the proband’s phenotypically normal sister. (Right) Pedigree of family 192. The Proband and his sister were both found to be heterozygous for the SOX10 mutation Q377X. Both siblings have nystagmus and ataxic cerebral palsy and are profoundly deaf. Only the proband has an abnormal enteric neuronal phenotype. Sequence analysis revealed a faint mutant band in the father corresponding to the T nucleotide in the TAG stop codon in addition to the wild-type band of normal intensity, suggesting possible germ-line mosaicism for Q377X.
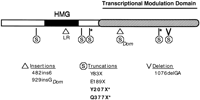
Summary of Sox10 mutations detected in mouse and human HSCR. The effect of the Sox10Dom mutation and multiple independent human mutations [this study (*); Pingault et al. 1998] on SOX10 are diagrammed beneath the protein domains. The category of mutation within the Sox10 coding sequence is indicated below. Note that both 929insGDom and 1076delGA produce frameshifts that introduce heterologous amino acid sequence, 99 and 37 residues, respectively, before terminating the protein at the indicated positions on the diagram.
The second mutation was found in the proband of family 192 (Fig. 5), which has sensorineural deafness and variable diagnoses of enteric function ranging from hypoganglionosis to long segment HSCR. The mutation (Q377X) truncates the SOX10 protein within the transcription modulation domain (Fig. 5). In addition to the two mutations described above, Pingault and colleagues (1998) have described four distinct mutations in SOX10. Two of these truncate the protein 3′ of the HMG-binding domain—similar to the mutations we detected—while a third truncates the protein 5′ of the HMG-binding domain. The fourth mutation, a Leu–Arg duplication within the HMG box (482ins6), alters the third helix of the SOX10 DNA-binding domain. To examine further the effect of the 482ins6 mutation on SOX10 function, the predicted structure of the SOX10 HMG domain was evaluated. Model structures were generated for the wild-type SOX10 sequences from human, rat, and mouse, as well as for the human SOX10482ins6 mutant. Each query sequence was threaded through the NMR coordinates of the second HMG-1 box from rat (rHMG1.2) as described previously in a structural study of the HMG-1 box family of proteins (Baxevanis et al. 1995). For each sequence, all possible placements of the sequence within the structure were considered, with a conformational energy (ΔG R‖M) being calculated for each placement. Threads with the most favorable conformational energies (i.e., those with the lowest ΔG R‖M) were selected for further study.
The final placement of each sequence with respect to the NMR structure of rHMG1.2 is shown in the multiple sequence alignment in Figure7. The structural alignment is not vastly different from what would be produced by a traditional, sequence-based alignment method in this particular case. However, because the sequences are being aligned to the HMG-1 box structure, each of the core regions (roughly corresponding to the three α helices) is necessarily constrained to being ungapped. This constraint has a significant effect on the alignment of the 482ins6 mutant, in that the insertion occurs within the third α helix. By virtue of this location, the best thermodynamic solution results in all residues carboxy-terminal to the Leu–Arg insertion being shifted out of position by two amino acids.
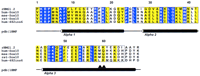
Structural alignment of the HMG-1 box domains of human, mouse, and rat Sox10 proteins. The sequence of HMG-1 box 2 from rat whose NMR structure was used as the basis for the threading experiments (Weir et al. 1993) is shown in the first line of the alignment (rHMG1.2). (Blue) Positions exhibiting absolute identity; (yellow) conserved positions. (▴) The positions of the 2-residue Leu–Arg insertion in human 482ins6. Positions of the secondary structural elements found in the HMG-1 box NMR study are shown below the alignment. Core segments defined for the threading algorithm (Bryant and Lawrence 1993) are boxed. ALSCRIPT V. 2.0 (Barton 1993) was used to format the alignment.
To illustrate the network of pairwise interactions responsible for maintaining the SOX10 structure, as well as to examine the effect of the 482ins6 mutation, a series of energy scaffolds was generated (Fig.8). The view presented focuses on the interactions between core 1 and core 3, as the interactions between core 2 and cores 1 and 3 are, for the most part, identical. Simple visual inspection of the colored bars connecting core 1 and core 3 for both the human SOX10 wild type and 482ins6 mutant immediately shows that there is a definite change in how the inward-facing residues located between the core regions interact with one another. The most significant change is the shift in a large, favorable interaction seen between Val-1 and Asp-64 in wild-type SOX10. In the 482ins6 mutant, this interaction is missing and is instead replaced by a positive interaction between Val-1 and Arg-60, in essence moving the major interaction between Val-1 and core 3 one helical turn amino-terminal on core 3 (from position 64 to 60). The previously favorable interaction between positions 1 and 64 is now replaced by a small, yet negative, one. Two additional unfavorable interhelical interactions are also seen in 482ins6, between Lys-2 and Lys-64, and between Arg-3 and Lys-64. Two significant changes are seen within helix 3: the addition of a favorable interaction between Glu-53 and Arg-60, as well as an unfavorable interaction between Arg-56 and Lys-64. The net effect of these two new intrahelical interactions is the destabilization of helix 3, as their sum produces a positive ΔG.
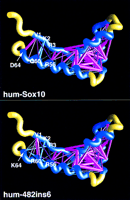
Energy scaffolds for wild-type and mutant Sox10 sequences containing the HMG-1 box domain. The α carbon backbone of the protein is depicted as a curving worm. Within the backbone, segments of the HMG-1 box domain comprising the core folding motif are shown in blue, while the intervening loop regions are shown in yellow. Pairwise residue interaction energies between core residues (Bryant and Lawrence 1993) are indicated by the thickness and coloring of the connecting α carbon positions in the protein backbone. Thick, magenta-colored cylinders are the most favorable interactions; thick, cyan-colored cylinders indicate the least favorable interactions. Intermediate colors and cylinder thicknesses represent interactions falling between these extremes. Numbering corresponds to that in the multiple sequence alignment in Fig. 6 and in Baxevanis et al. (1995). Scaffolds were generated by use of the graphics program GRASP (Nicholls et al. 1991). (Top) Human SOX10; (bottom) human 482ins6 mutant, yielding a Leu–Arg insertion at position 59.
Despite the fact that most of the favorable interactions seen in SOX10 still exist in 482ins6, the chance occurrence probability (E R‖M changes from 0.02 in the wild type to 0.13 in the mutant. Because its E R‖M > 0.05, the 482ins6 mutant does not produce a statistically significant match of sequence to structure. It appears that the change in the interaction pattern is moving α helices 1 and 3 out of position with respect to one another, and this net change in structure may be responsible for the observed phenotype in 482ins6 mutants.
DISCUSSION
We present data from a series of molecular and genomic studies aimed at understanding the role of SOX10 in HSCR and associated phenotypes (WS4). In WS4, both hypopigmentation and aganglionosis demonstrate variable penetrance and expressivity, as illustrated by hypopigmentation documented in only four of the six SOX10mutations identified to date (Fig. 6; Pingault et al. 1998). This variable penetrance observed in WS4 could arise from effects of modifier loci acting in conjunction with a primary genetic defect inSOX10. Identity-by-descent mapping in HSCR families has already documented the effects of a presumptive chromosome 21 locus that modifies the extra-enteric phenotypes (white forelock, bicolored irides, and hearing loss) associated with HSCR (Puffenberger et al. 1994).
Sox10Dom /+ mice model the variable penetrance and expressivity of hypopigmentation and aganglionosis observed in HSCR families. In this study, we used congenic lines ofSox10Dom mice to analyze one of the variable aspects of the WS4 phenotype, hypopigmentation. During construction of our congenic lines, we documented that only 30% of theSox10Dom /+ animals survived to weaning in the B6 pedigree in contrast with 82% survival in the C3H pedigree. This finding is consistent with the initial report of decreased survival in B6. Sox10Dom /+ lines (Lane and Liu 1984) and the observation by Kapur et al. (1996) that aganglionosis is increased inSox10Dom /+ first generation progeny derived from matings to B6. Further analysis is needed to confirm that increased lethality in our B6. Sox10Dom /+ line stems from increased severity of aganglionosis. The B6.Sox10Dom /+ and C3H. Sox10Dom /+ congenic lines will be invaluable in crosses to segregate the modifier locus(i) responsible for variations in neural crest defects. The orthologous gene(s) in humans for this modifier(s) will be an excellent candidate for interactions with HSCR and WS4 disease loci.
Segregation of loci that influence the penetrance of white forelock hypopigmentation of Sox10Dom /+ mice was also observed with the congenic lines. Linkage analysis demonstrated that penetrance of the white forelock in Sox10Dom /+ mice segregates with markers D10Mit96 and D10Mit178. This region of mouse chromosome 10 contains a modifier locus that results in variation of white spotting in another mouse model of WS4,Ednrbs (H. Rhim and W.J. Pavan, in prep.). Although it is not possible to determine whether the same locus accounts for the white forelock phenotype in both Ednrbs andSox10Dom mice, the inheritance of the C3H allele is associated with an increased frequency of white forelocks in both mutants. The identity of the chromosome 10 modifier gene has yet to be established; however, the locus cosegregates with mast cell growth factor (MGF). MGF, the ligand for the Kit tyrosine kinase receptor, is essential for melanocyte development and consequently is an excellent candidate for modifying hypopigmentation.
To evaluate further the role of SOX10 in neural crest development and disease, we performed comparative genomic analyses. Zoo blot hybridizations indicate that SOX10 is conserved in all mammals examined. The gene is conserved in fish as well, a model organism relevant to the study of neural crest development. Evidence ofSOX10 gene conservation in fish and the availability of neural crest mutants in Danio rerio are likely to lead to rapid identification of a SOX10 ortholog in this species. In particular, the colorless (cls) mutant, which lacks pigmentation, exhibits ear and otolith defects (Kelsh et al. 1996), and displays an absence of enteric neurons and a reduction in sensory neuron numbers (R.N. Kelsh and J.S. Eisen, unpubl.), is a primary candidate for mutation analysis of the SOX10 gene. However, conservation of the SOX10 gene in other organisms, particularly avians and yeast is questionable. While one zoo blot showed hybridization of high molecular weight bands in chicken genomic DNA, no hybridization was detected on the other blot. These differences could be due to variation in the genomic DNA quality, partial digestion, or strain selected for genomic DNA preparation. Similar inconsistent hybridization to yeast genomic DNA was also observed for the zoo blots. BLASTn analysis of the probe used for hybridization against the yeast genome database did not detect any homologies to ORFs, suggesting that the bands observed in the yeast lane on the one zoo blot are irrelevant. Further evaluation of Sox10 conservation in chickens will be of interest as this is one of the principal model organisms for study of neural crest development.
Extensive sequence conservation is observed on comparison of theSOX10 coding region between mouse, man, and rat (Pingault et al. 1998) although assignment of the initiatior methionine has not been definitively determined. An in-frame stop codon preceeding a single initiator methionine (Met1) in the rat (Kuhlbrodt et al. 1998a), absence of alternate 5′ methionines in the rat, and conservation of the coding region around Met1 between rat, human, and mouse suggest this residue is used for initiation of the ORF. We investigated a second methionine upstream of Met1 in the mouse, MetALT, by comparative alignment of sequences in exons 2 and 3 between mouse, human, and rat. Although we cannot exclude the possibility that MetALT functions as an initiator codon in the mouse, our analysis of the human SOX10 cDNA sequence identified a single base deletion that rules out usage of an orthologous MetALT in the human. These analyses together with the characterization of the rat 5′ UTR supports use of Met1 as the initiator methionine and, at the same time, suggests a functional role for the highly conserved 5′ UTR sequence, perhaps in message translation or stability. Although analysis of translated products is needed to determine the functional protein(s) encode by the Sox10 locus, these comparisons have identified conserved regions that may be relevant for mutational analysis in WS-4 individuals.
Several SOX10 mutations have been identified in individuals with WS4 phenotypes (Fig. 6). These patients exhibit sensorineural deafness and hypopigmentation in addition to bowel dysfunction (this study; Pingault et al. 1998). Given the highly variable phenotypes observed in theSox10Dom /+ mouse (Lane and Liu 1984; Kapur et al. 1996) and the identification of hypoganglionic patients possessingSOX10 mutations (this study; Pingault et al. 1998), this gene should be evaluated in patients who have chronic bowel dysfunction, but lack biopsy results pointing to a definitive diagnosis of aganglionosis. One of the families reported here illustrates this point. In family 192 (Fig. 5), the proband (found to carry the Q377X mutation) was biopsied multiple times and given diagnoses ranging from hypoganglionosis to long segment HSCR. In addition to congenital sensorineural hearing loss, the proband also has nystagmus and ataxic cerebral palsy. His sister shares all of his phenotypic features except aganglionosis/hypoganglionosis. Interestingly, although brother and sister are discordant for their enteric neuronal phenotypes, both siblings are heterozygous for Q377X. These patient phenotypes emphasize the variation observed in HSCR disease phenotypes and expand the potential systems affected by SOX10 mutations. The additional effects documented in these patients are consistent with SOX10 being essential for development of multiple aspects of the peripheral nervous system (PNS) as well as enteric neurons and melanocytes (Southard-Smith et al. 1998).
We used structural modeling to analyze one SOX10 defect identified within the HMG DNA-binding domain (Pingault et al. 1998) of a WS4 patient. Our results reveal by both structural alignments and energy scaffolds that the third α helix of the HMG domain is shifted out of its normal conformation by this 482ins6 mutation. Because the HMG-1 box is L shaped, with an angle of ∼80° between the two arms (Weir et al. 1993; Jones et al. 1994), the model presented here predicts that the mutant does not possess the canonical structure believed to facilitate the binding of HMG-1 box proteins to DNA. Our analysis is consistent with recent studies that demonstrate by gel mobility-shift assay the inability of this mutant form of SOX10 to bind the consensus sequence for SOX proteins (Kuhlbrodt et al. 1998b).
In summary, we established congenic lines ofSox10Dom mice and demonstrated the utility of these lines for identification of modifiers of neural crest defects by analysis of hypopigmentation. We investigated conservation ofSOX10 in multiple species and found similarity of expression for SOX10 in mouse and man. On the basis of the conserved role of SOX10 in neural crest development of mouse and human, we propose that this gene will be relevant to neural crest lineages in other species as well. These studies include the description of two new mutations of the SOX10 gene in WS4 patients. Our identification of WS4 patients with defects in peripheral nervous system components other than the enteric nervous system and variability in aganglionosis indicates the need to evaluate patients with peripheral nervous system defects that may or may not be accompanied by a diagnosis of aganglionosis for mutations in SOX10. Lastly, our structural modeling to evaluate the effects of human mutation on the HMG DNA-binding domain verifies the susceptibility of this region to mutagenesis and is consistent with in vitro DNA-binding assay results.
METHODS
Mice
Mice segregating for the Sox10Dom mutation (Lane and Liu 1984) on a B6C3F1/J hybrid background (C57BL/6JLe × C3HeB/FeJLe-a/a)F1 were obtained from the Jackson Laboratory (Bar Harbor, ME). Congenic strains for identification of loci that modify the phenotype ofSox10Dom mice were generated by crossingSox10Dom /+ animals in successive generations to either C57BL/6J or to C3HeB/FeJLe-a/a and selecting for animals that exhibited two of the three phenotypic features (belly spot, white feet, and head spot). Subsequent to the identification of the causative mutation, genotype was determined by a PCR-based assay (Southard-Smith et al. 1998). Animal care was in accordance with NIH guidelines. Tail DNAs were isolated by the salting out method (Thomas et al. 1992).
Genetic Mapping
Simple Sequence Length Polymorphism of Microsatellite Marker Loci
Primers and PCR fragment sizes for the D10Mit178 andD10Mit96 microsatellite markers were as described (Dietrich et al. 1992; the Whitehead Institute/MIT Center for Genome Research athttp://www.genome.wi.mit.edu/cgi-bin/mouse/index). PCRs forD10Mit markers were performed in 20 μl, consisting of 1× buffer, 0.2 μm dNTPs, 0.25 μmprimers, and 0.5 units of Taq DNA polymerase. Thermocycling parameters for the above markers consisted of a denaturation step at 94°C for 5 min, 35 cycles of 94°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec followed by 72°C extension for 10 min. The resulting PCR products were analyzed by electrophoresis on 10% native polyacrylamide gels (Pavan and Tilghman 1994) and visualized with ethidium bromide stain.
Statistical Analysis
Parametric linkage analysis calculations were done with FASTLINK, version 4.0P (Cottingham et al. 1993; Schaffer et al. 1994), whereas nonparametric analysis was performed with SimIBD, version 2.1 (Davis et al. 1996; Ott 1989, 1991) and a Sun Sparc workstation. Head spotting was modeled as a recessive trait with 50% penetrance. On the basis of this model, a penetrance function of 0.5 for C3H/C3H genotypes, 0.0125 for C3H/B6 genotypes, and 0.025 for B6/B6 genotypes was employed. The penetrances for C3H/B6 and B6/B6 genotypes were assigned to account for the phenocopy rate (Ott 1989), presumably equivalent for C3H/B6 and B6/B6 haplotypes, and for the observation of two false positives in 22 affecteds. The frequency for the affected allele was taken as 0.5 on the basis of the pedigree structure.
Northern Blot Analysis
Mouse and human multiple-tissue Northern blots were purchased from Clontech. A 1.3-kb mouse Sox10 cDNA fragment (dcgs10-1/PvuII–MluI) that excludes the HMG domain was gel purified and labeled with [α-32P]dCTP by random priming. Hybridization to human RNA samples was similarly performed with a 1.7-kb gel-purified cDNA fragment (HSox10-1/PstI–MluI).
Mutation Analysis in Human Patients
Patient Samples
We chose patients (9 from a total of ∼200) that had biopsy-proven HSCR or hypoganglionosis in addition to one or more major stigmata associated with Waardenburg syndrome, namely: sensorineural hearing loss, pigmentary anomalies and/or bicolored irides (Read and Newton 1997). DNA samples from six unrelated HSCR patients, as well as three related patients from a large Mennonite kindred (Puffenberger et al. 1994), were screened for mutations in the three coding exons of the human SOX10 gene. Of the Mennonite patients, two were found previously to be heterozygous and one homozygous for the W276C mutation in EDNRB (Puffenberger et al. 1994). Informed consent was obtained in all cases according to a protocol approved by the Case Western Reserve University Institutional Review Board (11-93-364). The University Hospitals of Cleveland’s Institutional Review Board operates under the U.S. Department of Health and Human Services Multiple Project Assurance of Compliance (M 1521 02).
PCR Amplification
Primer sequences (see Table 2) were derived from a publicly available cosmid spanning the entire humanSOX10-coding region (GenBank accession no. AF006501). PCR amplification was carried out in 50-μl reaction volumes containing 0.25 μm dNTPs, 1.5 μm Mg2+, 20 pmoles of each forward and reverse primer, 1 m betaine (Sigma), 1× reaction buffer and 2.5 units of Taq polymerase (both from Boehringer-Mannheim).
Primers Used for Genomic Amplification of Human SOX10 Coding Exons
Sequencing Primers
Internal primers used for sequencing each coding exon (in addition to the PCR primers above) were as follows: Exon 3, 3201R, 5′-CACGGGGAACTTGTCATC-3′; 3189F, 5′-CGGCGAGGCGGACGATGAC-3′; dcg110h2midFa, 5′-GGTGCTCAGCGGCTACGACTG-3′; 3048F, 5′-CCCCGTGGGCTCGGAGGAG-3′. Exon 4, 8614F, 5′-GCCCCTCTGCTGTCCTCTC-3′; 8932R, 5′-GCCCCACCCTCAGCTCTGTCAT-3′. Exon 5, 4BF, 5′-CGCCCACCTGCCTCTAACCTG-3′; 12892R, 5′-ACGCCTGGTGGCTTGGAGATC-3′; 12815F, 5′-GGCCACGTGAGCAGCTACTC-3′; 12950F, 5′-AAAGCCCAGGTGAAGACAGAGAC-3′; 13053R, 5′-GAGGGGAAGGCTGAGCCATAGT-3′; 13079F, 5′-TCCCGCCCCCAGTTTGACTAC-3′.
Sequence Analysis
Residual primers and dNTPs were removed from PCR products with Qiaquick PCR purification columns (Qiagen). Then 2–10 μl was included in cycle sequencing reactions. Cycle sequencing was carried out for 34 cycles with the ThermoSequenase radiolabeled terminator cycle sequencing kit (Amersham) according to the manufacturer’s protocol. Extensions were performed at 60°C for the first 17 cycles and 70°C thereafter. A 5-μl sample of each reaction was loaded onto 6% polyacrylamide gels prepared with Sequagel-6 (National Diagnostics, Atlanta, GA) and run for ∼2 hr at 90 W.
Homology Model Building
Threading experiments were performed by the method of Bryant and Lawrence (1993), with detailed derivations and methodology provided therein. All possible alignments of the query sequences in the Sox10 data set with the structure of the second HMG-1 box from rat (Weir et al. 1993) were examined as described in the text. Core segments used in the threading experiments were defined previously (Baxevanis et al. 1995). For each possible alignment, individual pairwise residue interactions were determined on the basis of chemical type and distance intervals with no use of arbitrary gap penalties (Bryant and Lawrence 1993). With these values, a conformational energy (ΔG R‖M), defined as the expected work for substitution of a specific sequence R for a random sequence with the same composition in the context of folding motif M, was then calculated for each alignment. Z-scores and chance occurrence probabilities (E R‖M) were calculated to compare conformational energies for different alignments. Chance occurrence probabilities give the odds that a random sequence of the same length and amino acid composition would yield a threading energy as low as that for the query sequence R. A significant match of sequence to structure results when E R‖M for that thread is <0.05 (5%). Calculation of energies and statistical values were performed with C and S-PLUS subroutines. All energy scaffolds were visualized by use of the GRASP software package (Nicholls et al. 1991).
Acknowledgments
We thank Drs. Leslie Biesecker, Alejandro Schaffer, and Robert Nussbaum for scientific discussions; Dr. Stacie Loftus for critical reading of the manuscript; Lisa Garrett and Amy Chen for assistance in derivation of congenic lines; and Darryl Leja for assistance with graphics. We thank Dr. Robert Kelsh for providing unpublished observations. M.A. and A.C. were supported by National Institutes of Health grant HD28088.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL bpavan{at}nhgri.nih.gov; FAX (301) 402-2170.
-
- Received September 28, 1998.
- Accepted January 19, 1999.
- Cold Spring Harbor Laboratory Press








