
CAGGG Repeats and the Pericentromeric Duplication of the Hominoid Genome
Abstract
Gene duplication is one of the primary forces of evolutionary change. We present data from three different pericentromeric regions of human chromosomes, which indicate that such regions of the genome have been sites of recent genomic duplication. This form of duplication has involved the evolutionary movement of segments of genomic material, including both intronic and exonic sequence, from diverse regions of the genome toward the pericentromeric regions. Sequence analyses of the target sites of duplication have identified a novel class of interspersed GC-rich repeats located precisely at the boundaries of duplication. Estimates of the evolutionary age of these duplications indicate that they have occurred between 10 and 25 mya. In contrast, comparative analyses confirm that the GC-rich pericentromeric repeats have existed within the pericentromeric regions of primate chromosomes before the divergence of the cercopithecoid and hominoid lineages (∼30 mya). These data provide molecular evidence for considerable interchromosomal duplication of genic segments during the evolution of the hominoid genome and strongly implicate GC-rich repeat elements as playing a direct role in the pericentromeric localization of these events
Genome evolution is dependent on the processes of single-base-pair mutation and gene duplication. Duplication of a gene followed by mutation is responsible for the emergence of new genes with specialized functions in an evolving species. Two distinct molecular mechanisms of gene duplication are generally recognized. Tandem duplication of an ancestral gene, vis-à-vis processes of unequal crossover, produces a clustered family of related genes (Smith 1976). In contrast, whole-genome duplication (polyploidy) followed by chromosomal rearrangement and the re-establishment of the disomic state has been proposed as a mechanism for the interchromosomal duplication of large segments of a genome (Ohno 1970). The latter model, put forward originally by Susumu Ohno, explains observations of conserved genic synteny among nonhomologous chromosomes. Because of the genetic consequences of polyploidy, such events, although important in the early expansion of eukaryotic genomes, are rare and ancient (Ohno 1993;Wolfe and Shields 1997). Within the vertebrate lineage, for example, the last tetraploidization event leading to genome duplication is estimated to have occurred >400 mya (Lundin 1993). If polyploidy were the only model by which entire genes could be duplicated among nonhomologous chromosomes, it would then follow that interchromosomal paralogs of recent origin would be unexpected.
Recent analyses of data from the Human Genome Project suggest a third form of genomic duplication, which is mechanistically distinct from polyploidy and tandem duplication models of genome evolution. Several independent reports indicate that genic segments, ranging in length from 5 kb to 30 kb, possess duplicate copies within the pericentromeric regions of human autosomes (Borden et al. 1990; Wong et al. 1990;Eichler et al. 1996, 1997; Regnier et al. 1997; Zimonjic et al. 1997;Ritchie et al. 1998). (Pericentromeric DNA, for our purposes, refers to the chromosomal region that begins immediately distal to the α-satellite repeat and extends into the first distinguishable cytogenetic Giemsa-stained band on either side of the centromere.) The estimated evolutionary age for a few of these duplications has precluded tetraploidization as a possible mechanism for these duplication events (Eichler et al. 1996, 1997; Regnier et al. 1997). During our analysis of one such recent (∼10 mya) duplication of the creatine transporter locus, we identified an interspersed CAGGG repeat sequence located at the junction of the duplicated Xq28 genomic segment within 16p11.2. (Eichler et al. 1996) (Fig. 1). This association and the similarity of these repeats to switch recombination signal sequences (Dunnick et al. 1993) led us to propose that the presence of such repeat elements within these regions might mediate the integration of duplicated segments. If the presence of CAGGG repeats, at least in part, were responsible for the reported pericentromeric bias of duplications, we had three expectations: (1) These repeat sequences should occur exclusively within the pericentromeric region, (2) the repeats should have evolved prior to the arrival of the genomic segments within these regions, and (3) the repeat sequence should delineate the boundaries of other recently duplicated genomic segments. The following experiments were designed to test these different aspects of our model.
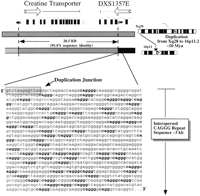
Pericentromeric CAGGG repeats flanking a duplicated genic segment in 16p11.2 A schematic diagram summarizing the duplication of 26.5-kb genomic fragment from Xq28 to 16p11.2. The intron-exon structures of both the DXS1357E and the creatine transporter genes are depicted. Filled and open arrows indicate the position of SINE and LINE repetitive elements, respectively. The sequence of one of the paralogy boundaries within 16p11.2 as well as its proximity to the flanking 3.0-kb CAGGG repeat is shown. The data are based on a large-scale sequence comparison of cosmid subclones from 16p11.2 and Xq28 (Eichler et al. 1996).
RESULTS
Genomic Distribution of the CAGGG Repeat
To assess the genomic distribution of the CAGGG repeat, FISH was performed against a human metaphase spread using as probe a 1.9-kb CAGGG repeat construct (196.3.12). The probe consisted entirely of interspersed CAGGG repeat sequence derived from a previously sequenced 16p11.2 cosmid (HSU41302). A highly nonrandom genomic distribution of these repeat sequences was observed. A total of nine distinct hybridization signals were detected in all metaphases examined, corresponding to cytogenetic band locations 1q12, 2p11, 9q12, 9p11, 10p11, 14q11, 15q11, 16p11, and 22q11 (Fig.2a). Under less stringent hybridization conditions, signals were also detected on chromosomes 7p11, 18p11, and 21q11. All signals were exclusively pericentromeric in location. With the exception of chromosome 9, FISH analysis indicated that the repeats demonstrate an unusual polarity in their map location with respect to the primary point of constriction. Among metacentric and submetacentric chromosomes (2, 7, 10, 16, and 18), the repeats map to the short arm (p11) chromosomal positions, whereas among the acrocentric chromosomes (14, 15, 21, and 22), the repeat hybridizes only to the long arm (q11) of the chromosomes. Only one homolog on chromosome 9 showed signals (9p11.2 and 9q12) on both sides of the centromere.
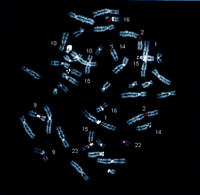
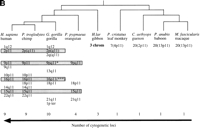
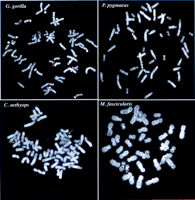
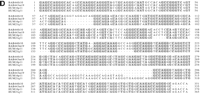
Evolutionary conservation and distribution of the CAGGG repeat. (a) A 1.9-kb subclone (p196.3.12) was constructed within the CAGGG interspersed repeat sequences that flank the CTR-CDM (creatine-transporter/DXS1357E) duplicated locus on 16p11.2 (Fig. 1). FISH analysis performed on a human metaphase chromosomal spread identified nine pericentromeric cytogenetic localizations (1q12, 2p11, 9q11, 9q12, 10p11, 14q11, 15q11, 16q11, and 22q11). (b) A summary of comparative FISH analysis using the same probe on metaphase chromosomal spreads from nine different primate species. The locations of these signals with respect to human phylogenetic group assignment are presented in the context of a generally accepted phylogeny of these species. Note that hominoid species show a general increase in the number of chromosomal sites that hybridize with this probe. The human phylogenetic assignments among the cercopithecoids were confirmed by using whole or partial human chromosome painting probes (species-specific band assignments are shown in parentheses). (*) The correspondence to human 9p11 or 9q11 cannot be unambiguously resolved because of recent pericentric inversion. (***) Three distinct hybridization signals could be distinguished on GGO chromosome 17q11 (HSA 16p11 equivalent). (c) Comparative FISH analysis of CAGGG repeat probe, 196.3.12. Representative hybridizations of chromosomal metaphases are shown for two hominoids (gorilla and orangutan, G. gorilla and P. pygmaeus) and two Old World monkey species (guenon and macaque,C. aethyops and M. fascicularis). (d) A sequence alignment of a portion (∼350 bp) of the CAGGG repeat structure from two baboon loci (denoted BAB followed by BAC coordinate; GenBank accession nos. AF144085 and AF144086) and two human (HUM; GenBank accession nos. AC004527 and AC002041) loci. Regions of sequence identity are shaded.
We approximated the haploid copy number of the CAGGG repeat elements by hybridizing the insert from 196.3.12 against various, high-density arrayed genomic libraries. Based on the total number of clones identified after screening two different human BAC insert libraries (RPCI-11 and CIT-HSP), we estimated a total of 42–44 copies of the repeat element per haploid genomic equivalent (Table1). This estimate may be considered a minimum as it assumes a single copy of the CAGGG repeat element per 150-kb BAC insert. Complete sequence analyses of BAC clones that harbor this repetitive sequence confirm this distribution in the genome (see below). Analysis of four chromosome-specific cosmid libraries revealed a nonuniform distribution of the repeat, ranging from 2 to 16 copies among the different chromosomes. As an independent validation of copy number, Southern analysis was performed using the 0.9-kb subclone of 196.2.1 as probe against nylon-transferred PstI-digested DNA from a monochromosomal hybrid panel of all human chromosomes (ONCOR). Based on the CAGGG repeat consensus sequence of this 900-bp region,PstI does not restrict within the repeat structure. A total of 32 distinct PstI hybridizing fragments were identified among all chromosomes (1, 2, 9, 10, 14, 15, 16, 18, 20, 21, 22, and Y), suggesting a similar copy number estimate of this pericentromeric repeat.
Genomic Distribution of the CAGGG Repeat
Evolution of the CAGGG Repeat
To evaluate the evolutionary conservation of the CAGGG repeat, four additional hominoid (P. troglodytes, G. gorilla, P. pygmaeus,and H. lar) and four cercopithecoid species(M. fascicularis, P. cristatus, C. aethyops, and P. anubis) were examined by FISH. Our analysis revealed strong hybridization signals localized almost exclusively to the pericentromeric regions of primate chromosomes (Fig. 2b,c). Among all hominoid species examined, cross-hybridization was detected among multiple chromosomes. Considerable variation in the distribution of these repeats was observed even among closely related primate species. In contrast to the hominoids, each of the metaphases from four representative Old World monkeys showed pericentromeric hybridization to a single chromosome, suggesting that the repeat element underwent extensive amplification early in the evolution of the hominoid lineage (Fig. 2b). To confirm the molecular basis for the cross-hybridization, genomic subclones of the CAGGG repeat element were identified and isolated from one representative Old World monkey species, P. anubis. A degenerate PCR assay was designed to amplify a portion of the CAGGG repeat structure, and the products were directly sequenced (see Methods). Alignment of the baboon sequences with copies of the CAGGG repeat from human chromosomes 21q11 and 16p11 revealed an average pairwise sequence similarity of 88.2 ± 1.7% (Fig. 2d). The comparative data indicate that both the sequence of the repeat and its nonrandom distribution within the pericentromeric region have been conserved properties of primate genome since the radiation of the catarrhine primates (∼30 mya). (Li 1997; Takahata and Satta 1997)
CAGGG Repeat Breakpoint Analysis
GenBank searches of data generated recently as part of the Human Genome Project were used to determine the organization of the full-length CAGGG repeat structure. Using the CAGGG repeat segment of HSU41302 as query, BLASTN sequence similarity searches identified four highly significant hits that corresponded to BAC/cosmid clones that had been mapped to human chromosomes 21q11 (AC004527), 22q11 (D87003/018), and 16p11 (AC002041 and AC002042). As expected, all chromosomal map assignments were in the vicinity of the centromeric markers confirming that the repeats are largely restricted to pericentromeric regions. Multiple pairwise sequence alignments (CLUSTAL W) delineated the size and the structure of the repeat (Fig.3a). From these four pericentromeric sequences, a tripartite organization was deduced, composed of (1) an interspersed CAGGG repeat block, (2) a short spacer region, and (3) a region of subtelomeric-like repeat sequences (HSREP522, HSREP271, TAR, HSREP282) (Fig. 3b). The overall length of these four repeat structures ranged from 4.3 kb (16p11.2, AC002041) to 9.9 kb (22q11, D87003/018). The pairwise sequence similarity (GAP alignment) among these four repeat elements varied from 87.3 ± 0.6% to 95.4 ± 0.2% (5072 bp on average aligned between any two sequences). The 21q11 repeat copy showed the lowest average pairwise sequence similarity (88.02 ± 0.51%).
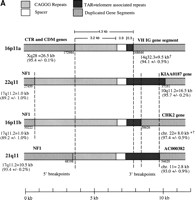
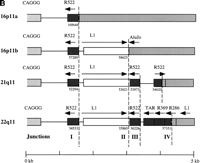
Duplicated genomic segments flank the CAGGG repeat structure. (A) Four human CAGGG repeat elements [21q11, 22q11, and two from 16p11 (a and b)] were identified based on BLAST sequence similarity searches of htgs and nr divisions of GenBank (accession nos.AC004527, D87003, AC002041, and AC002402, respectively). Multiple pairwise sequence alignments (CLUSTA W) were used to determine the extent of the repeat structure. The diagram shows the structure of the repeat element, composed of (1) an interspersed CAGGG repeat block (mean length = 4490 bp; range, 2937–5915 bp), (2) a short spacer region (mean length = 881 bp; range, 851–943), and (3) a variety of subtelomeric-like repeat sequences (HSREP522, HSREP271, TAR, HSREP282) (mean length = 1899 bp; range, 793–3018 bp). Broken lines represent breakpoints within the aligned repeat units. Analysis of sequences flanking the repeat structure identified paralogous gene structures located near or at the boundaries of each repeat. The duplicated genic segments are shown as hatched bars. Numbers below these boundaries refer to the breakpoint positions within the GenBank accession. The percent sequence similarity, the location of the ancestral sequence, and the estimated size of the duplication are summarized below each bar. (*) Calculation of sequence similarity was based on sim4 comparisons of cDNA and genomic segment. (†)Minimal size of duplicated region was based on the genomic distance between first and last paralogous exons. All interchromosomal duplication boundaries were confirmed by PCR amplification and sequencing of products from a monochromosomal somatic cell hybrid panel. (B) The location of the 3′ boundary of the repeat element with respect to various telomere-like repeat elements is shown in greater detail. Note the preponderance of breakpoints near or within the HSREP522 (R522) element.
Once the extent of the CAGGG repeat structure had been determined, the sequences located proximal and distal of the repeat were systematically examined for the presence of recently duplicated segments. Sequences were masked for common primate repeat elements (RepeatMasker v2.0), and BLASTN nucleotide sequence similarity searches were performed against nr, dbEST, htgs, and monthly GenBank databases. The size of the duplicated segments and the degree of sequence similarity were determined by a combination of dot-matrix (DOTTER), Miropeat, and GAP alignment analyses between target and query sequences. All junctions were validated by PCR and sequencing from total genomic human DNA. Examination of the eight boundaries indicated the presence of recently duplicated segments that had originated from elsewhere in the genome (Fig. 3; Table 2).
Pairwise Sequence Comparisons of Duplicated Sequences
Duplication Junction PCR Oligonucleotides
Five known genes from various locations in the genome showed specificity in their duplication and transposition to the defined CAGGG repeat structure. These included the creatine-transporter gene and DXS1357E genes from Xq28 (26.5 kb); two different portions of the neurofibromatosis locus in 17q11.2 (10-and 1-kb segments); a 16-kb segment (eight exons) of a full-length cDNA, KIAA0187, from 10q11.2; a 9-kb segment including two immunoglobulin variable heavy chain exons from 14q32.3, and the first six introns and exons of a human protein kinase gene, CHK2, assigned to chromosome 22. In addition, sequence similarity to an uncharacterized cDNA (96% sequence similarity) (AC306657) with nonprocessed gene structure (three exons over 4 kb) was identified along the 3′ flank of the 21q11 CAGGG repeat. All sequence alignments showed a remarkably high degree of sequence similarity (89%–97%) over substantial tracts of genomic DNA (1–27 kb). Based on calculated substitution distances (Kimura two-parameter model) among noncoding portions of the aligned sequences and estimates of rates of substitution for pseudogenes (2.2 × 10−9substitutions/bp per year), we have determined that duplications probably occurred at different times (10–25 mya) during hominoid evolution (Table 2).
DISCUSSION
Three properties of the CAGGG repeats characterized in this study support their involvement in the process of pericentromeric duplication. First, the repeat elements are distributed almost exclusively within the pericentromeric regions of primate chromosomes (Fig. 2). Several of the chromosomal locations identified in this study (10q11, 15q11, 16p11, and 22q11) have been described previously as particular hot spots for recent interchromosomal gene duplication (Eichler 1998; Jackson et al. 1999). Second, comparative analyses indicate that these repeat elements have existed within the pericentromeric regions prior to the divergence of the Old World and hominoid primate lineages (Fig. 2b), generally estimated to have occurred 30–35 mya (Li 1997; Takahata and Satta 1997). In contrast, many of the duplications associated with these elements, such as the creatine transporter and neurofibromatosis loci, appear to have originated more recently (10–25 mya) with no evidence of duplicated loci found among Old World primates (Eichler et al. 1996; Regnier et al. 1997). A greater evolutionary age would be expected if the CAGGG repeats were functioning as putative transposition integration signals. In other words, if the repeats were responsible for attracting duplicated segments toward the pericentromeric regions, the presence of such repeat structures should predate the evolutionary arrival of the duplicons. Perhaps the strongest evidence supporting the involvement of these repeats in the pericentromeric-directed mechanism of gene duplication was the finding that these structures delineate the boundaries of several recent and apparently independent genomic duplication events (Fig. 3). We identified five genic segments originating from diverse regions of the human genome (Xq28, 14q32, 10q11, 17q11) that have duplicatively transposed precisely to the pericentromeric CAGGG repeat structures described in this study. We propose that such an association is unlikely to be the result of stochastic events but, rather, is indicative of a functional role of these repeats in this evolutionary process.
With the exception of the creatine-transporter cassette, all gene duplications were incomplete. In most cases only segments or portions of the original gene structure have become duplicated to the pericentromeric region. The ancestral copy and the direction of the interchromosomal transposition were inferred based on the location of the expressed full-length copy of each gene. The evolutionary origin of two of these duplications has been determined previously (Eichler et al. 1996; Regnier et al. 1997) and coincides with the normally expressed copy of the gene. Only one of the eight duplicates, in this study, involved the transposition of a complete genic region (all introns and exons as well as putative promoter region of the creatine transporter gene from Xq28). Transcribed cDNAs have been identified and characterized for both the Xq28 and 16p11.2 paralogs (SCL6A8 and SCL6A10) (Iyer et al. 1996; Sandoval et al. 1996). Interestingly, the ancestral Xq28 copy (Eichler et al. 1996) is expressed in all tissue types, whereas transcription of the chromosome 16 paralogous creatine transporter gene is restricted in humans to the testis. Although the function of SCL6A10 is unknown, the data suggest that transcriptional potency can be maintained within the pericentromeric region—albeit in a tissue-specific fashion.
Sufficient genomic sequence was available to determine the precise duplication junction with respect to the CAGGG element in six of eight boundaries. Three out of the four paralogous segments situated 5′ of the CAGGG repeat (sense strand) terminated with the sequence motif CCAGGG, whereas the fourth was situated 12 bp upstream from the first motif of this sequence. Among those duplicated sequences situated 3′ of the CAGGG repeat structures, comparative sequence analysis indicated that the duplication boundaries occurred in close proximity (<100 bp) to various subtelomeric repeat sequences (TTAGGG degenerate sequences). The repeat structures that demarcate the boundaries of the duplications are unusually GC rich. Similar GC-rich sequences have been reported at the boundaries of other recently duplicated genomic segments including duplications of adrenoleukodystrophy and immunoglobulin variable κ-chain locus (Borden et al. 1990; Eichler et al. 1997). The telomere-like elements (HSREP522, TAR, and HSREP286) that define the opposite of end of these repeat structures are likewise composed of GC-rich sequences. All of these duplication boundary elements share, as a common sequence motif, runs of at least three guanine nucleotides (GGG). Several lines of experimental evidence indicate that repetitive tracts of three or more G's are required to drive the formation of G4 DNA (Williamson 1994). Since its initial discovery (Sen and Gilbert 1988), G4 DNA has been implicated in biological processes such as homologous chromosomal pairing and recombination (Dunnick et al. 1993; Frantz and Gilbert 1995; Liu et al. 1995). The CAGGG repeats we have described in this study bear a striking resemblance both in sequence and organization to switch recombination signals (Mills et al. 1990) that have been shown to mediate genomic rearrangement events that juxtapose immunoglobulin heavy chain variable regions [V(D)J] and constant regions (Snapper et al. 1997). The unusual Hoogsteen pairing potential of such guanine-rich sequences has been thought to underlie this phenomenon.
Biologically, G-rich DNA have been identified in three distinct regions of the genome (telomeres, rDNA, and switch-recombination regions) (Dempsey et al. 1999). Our findings suggest a fourth region, the pericentromeric regions of human chromosomes, where these sequences define the boundaries of recently duplicated genomic segments. We propose that the evolution of such sequences in these regions effectively serves as preferred sites of integration, perhaps because of their potential to promote pairing and ectopic recombination. Duplicated segments may have accrued in these regions as a result of the potential hyperrecombinogenic properties of the CAGGG repeat structure. Of course, such a model remains speculative and will require validation that such sequences are recombinogenic and promote transpositional integration. In this regard, it may be noteworthy that subtelomeric regions of the genome have also been shown to be hot spots for recent gene duplication (Trask et al. 1998a,b; Brand-Arpon et al. 1999; Grewal et al. 1999). Degenerate telomeric repeats (TTAGGG) are common in such regions of the genome and have been found in close proximity to interchromosomally and intrachromosomally duplicated segments (Amann et al. 1996; Chute et al. 1997). The presence of G-rich DNA sequences in both of these genomic microenvironments would provide a unifying theory to explain the accumulation of duplicated segments in these particular regions of the human genome.
Examination of sequence organization within the pericentromeric region of human autosomes provides molecular evidence for a mechanism of gene duplication that is distinct from tandem (regional) and tetraploidization models (Ohno 1970; Smith 1976). This form of duplication involves the apparent movement of genomic material, including both intronic and exonic sequence, from one location of the genome to a second region without the apparent loss of the ancestral locus (Borden et al. 1990; Eichler et al. 1996; Regnier et al. 1997). Unlike tetraploidy models of genome duplication, this mechanism has occurred recently within the lineage of human evolution (10–25 mya by our estimates). Unlike models of regional (tandem) duplication, this process creates paralogous copies that are unlinked and therefore liberated from the homogenizing influences of gene conversion or the disruptions of unequal crossing-over associated with tandemly arrayed multigene families (Dover 1982; Charlesworth et al. 1994). The pericentromeric-directed mechanism of gene duplication may be likened to “duplicative transposition.” There is, however, no evidence for the production of short direct repeats at the site of integration—suggesting that the process is mechanistically distinct from classically defined transposition (Borden et al. 1990; Eichler et al. 1996, 1997). Whether the process of pericentromeric-directed gene duplication is an anomaly of primate genome evolution remains to be determined. To our knowledge, in no species other than human and its closest relatives has there been a demonstration of the duplication of gene “cassettes” within the pericentromeric regions of chromosomes. The juxtaposition of different modular components of genes that originate from diverse regions of the genome could, in theory, lead to the formation of new genes with new functions. Such alternative molecular pathways for gene duplication may have evolved to compensate for the genetic restrictions of ploidy cycles of genome duplication. Furthermore, the recent evolutionary nature of these events suggests that such processes may be important in the rapid genomic diversification of chromosome structure among closely related primate species.
METHODS
Probe Construction
Two synthesized oligonucleotide primers 59336 (GGAAAGGGCAATATCAGGACAAG) and 59339 (5′-CGGCGTAACATGGCTCTGCATTGG) were designed to amplify a 1901-bp fragment of CAGGG repeats from chromosome 16p11.2 (GenBank accession no. HSU41302 corresponding to positions 29639–31517). PCR products were subcloned into the PGEMT TA cloning vector system (Promega), and the transformants were screened and sequence verified as described previously (Eichler et al. 1997). One subclone, p196.3.12, was selected for subsequent analysis. P196.2.1 was similarly constructed using oligonucleotide primers 59338 (5′-GGCCACAACTAGGTCTGTGTTATG) and 59339.
Fluorescence in situ Hybridization
Chromosome metaphase spreads were prepared from lymphoblastoid cell lines representative of five hominoid species (H. sapiens, P. troglodytes, G. gorilla, P. pygmaeus, and H. lar) and three cercopithecoids (M. fascicularis, P. cristatus, andP. anubis). Probes were nick-translated with either biotin-16-dUTP or digoxigenin-11-dUTP, and hybridized to chromosomal preparations as previously described (Lichter et al. 1990). A Zeiss Axioskop epifluorescence microscope with a cooled charge-coupled device (CCD) camera was used to generate digital images. Hybridizations were performed in conjunction with human whole-chromosome painting probes to determine orthologous band intervals relative to the human phylogenetic group assignments. As a test of FISH specificity, reciprocal hybridizations were performed against human and baboon metaphases using CAGGG repeat subclones from each species.
Library Hybridization
Four chromosome-specific cosmid libraries (LL02NC02, LA10NC02, LA15NC01, and LA16NC02) and four total-genomic libraries (RPCI-11, CIT-HSP, RPCI-43, and RPCI-41) were probed with a PCR-amplified insert of the CAGGG repeat subclone p196.3.12. Probes were radiolabeled and hybridized as described previously (Eichler et al. 1997) with the exception that the probe was blocked with Cot1 DNA for 1 hr at 65°C prior to hybridization. Southern analysis was performed using a monochromosomal somatic cell hybrid panel, PstI-restricted DNA (ONCOR), per manufacturer's suggested protocol. Filters were washed with a solution of 0.05 m NaPO4, 0.5% SDS, and 1 mm EDTA solution using high-stringency conditions: three washes, 45 min each at 65°C. Filters were exposed overnight to autoradiographic film, and the positives scored.
PCR
PCR amplification reactions were performed in a final volume of 20 μl containing 0.20 mm dNTPs (Pharmacia), 20 pmoles of each primer, and 0.35 units of Taq polymerase in standard 1X reaction buffer (Boehringer Mannheim). PCR conditions were identical for all primers used in this study: an initial denaturation of 5 min at 95°C, followed by 35 cycles of 30 sec at 95°C, 30 sec at 55°C, and a 45-sec extension at 72°C. A final extension of 5 min was performed at 72°C. Primer pair CAGGG-9 (5′-AGGGCTGGGTCCAGGGCCAGAATC) and CAGGG-11 [5′-CTTTCC(CT)TGGCCCTGAGCTGGCAG] corresponding to a 394-bp conserved portion of the CAGGG repeat tract (31383-31777, GenBank accession no. 41302) was used to amplify the corresponding sequence from both human and baboon clones. Synthesized oligonucleotide primers used to confirm the CAGGG paralogy boundaries are summarized in table 3. Junctions were amplified and sequenced from both clone and human control DNA templates. All cycling conditions were optimized for use in a PE 9600 Thermocycler (Perkin-Elmer Applied Biosystems).
Sequencing
Amplified PCR products were directly sequenced using both forward and reverse PCR primers and dichlororhodamine sequencing dye-terminator sequencing chemistry (Perkin-Elmer Applied Biosystems). Reactions were performed per manufacturers protocol with the following modifications. Prior to sequencing, 8 μl of each PCR product were treated with 1.5 units of exonuclease I and 0.30 unit of shrimp alkaline phosphatase to remove excess-single strand DNA and deoxynucleotide triphosphates. Reactions were incubated at 37°C for 5 min and then heat inactivated at 72°C for 15 min. Cycle sequencing reactions were performed in a total reaction volume of 10 μl: 4 μl (30–90 ng) of Exo/SAP treated product, 20 pmoles of sequencing primer, and 3 μl of dichlororhodamine dye-terminator mix. All fluorescent traces were analyzed using the Applied Biosystems Model 377 DNA Sequencing System (Perkin-Elmer Applied Biosystems), and the quality of sequence data was assessed with PHRED/PHRAP/CONSED software (http://genome.wustl.edu).
Sequence Analysis
BLASTN sequence similarity searches were performed against nr and htgs divisions of GenBank using as query, repeat-masked CAGGG repeat sequence corresponding to coordinates 29833–32504 from GenBank accession number 41302 (Eichler et al. 1997). Four significant hits (HSP value < e − 25) were identified: (AC002041,AC004527, D87003, and AC002042). AC002041 was found to completely overlap with the query sequence (HSU41302). The degree of sequence similarity, however, is relatively low for an allelic variant (98.7%), suggesting that this may represent a paralogous copy of this sequence within 16p11.2. CLUSTAL W v.1.7 (gap opening penalty, 10.0; gap extension penalty, 5.0) (Thompson et al. 1994) and Miropeats software (Parsons 1995) were used to determine the extent of the CAGGG repeat structure. The composition of the four CAGGG repeat elements was very similar, consisting of a CAGGG repeat flank, a spacer region, and a cluster of telomeric repeats. It was, therefore, possible to effectively align all four the CAGGG repeat elements (the differences in length were largely due to deletions or insertions in the basic organization). Once these were aligned, points of transition from CAGGG repeat element to non-CAGGG repeat sequence could be easily identified. These flanking segments were then individually searched for the presence of duplicated sequence. Repeat-masked versions (http://ftp/genome/washington.edu/cgi-bin/RepeatMasker) of sequences flanking each of the repeats were systematically searched for the presence of duplicated segments. Two criteria were used when identifying tentative regions of paralogy: (1) The sequence shows >89% and <98% nucleotide identity to previously sequenced regions of >400 bp (with the additional requirement of a nonprocessed exon/intron structure when screening dbEST), and (2) mapping data exists to place the paralogous sequence in a another nonhomologous chromosomal location. Pairwise genomic sequence alignments were performed with GAP optimal alignment software (http://genome.cs.mitu/edu/align/align.html). Sim4 software, which optimizes alignment based on known structural properties of exon/intron structure, was used for cDNA to genomic comparisons. Percent similarity (Fig. 2) was calculated as L (number of matched bases) / L + number of gaps × 100%. Standard error was estimated as the square root of the binomial distribution. The number of substitutions per 100 bp and its associated variance was determined using Kimura's two-parameter method (Kimura 1980). Deletions and insertions were excluded in this analysis.
Molecular Clock
Published rates of nucleotide substitution among pseudogenic sequences vary dramatically. Estimates ranging from 12.6 × 10−9 substitutions per site per year for α-globin pseudogenes to 1.0 × 10−9 substitutions per site per year for η-globin sequences have been reported (Miyata and Yasunaga 1981; Miyamoto et al. 1987). Based on available data for two of the duplications in this study (the creatine transporter and neurofibromatosis loci), we estimated the rate of nucleotide substitution to be 2.2 × 10−9 substitutions per site per year. For example, molecular and cytogenetic data indicate that duplications of the creatine transporter locus occurred during the hominid line of evolution after the separation of orangutan from the greater apes. Using generally accepted estimates for the divergence of these two lineages (11.9 mya) and the number of substitutions per 100 bp (4.43 ± 0.13 ) we calculated a substitution rate (r = K / 2T) of 2.2 × 10−9 substitutions per site per year. Similarly, Regnier et al. showed that no duplications of the NF1 locus could be identified among Old World Monkeys with the first evidence of duplication being found among the gibbon (25.3 mya). Using a value ofK as 10.92 (Table 2), we calculate a substitution rate of 2.1 × 10−9. Because these values estimate the maximum divergence (the duplications occurred probably after these dates), our estimates are conservative and provide a lower boundary of the true rate of nucleotide substitution. The timing of all other duplication events (T = K / 2r) were calibrated using this molecular clock that is slightly higher than the silent substitution rate (1.5 × 10−9) (Li 1997).
Acknowledgments
We thank Drs. Larry Deaven and Norman Doggett for providing chromosome-specific libraries (LA10NC02, LA15NC01, and LA16NC02) and Jeff Bailey for computational assistance. This work was supported, in part, by grants from the National Institutes of Health/National Institute of General Medical Sciences (HG58815-01) and the National Science Foundation (DEB9806913) and by a Basil O'Connor Scholar award to E.E.E. (FY99-0519) from the March of Dimes Birth Defects Foundation. The chromosome specific gene libraries were constructed under the auspices of the National Laboratory Gene Library Project sponsored by the U.S. Department of Energy.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL eee{at}po.cwru.edu; FAX (216) 368-3432.
-
- Received July 7, 1999.
- Accepted September 22, 1999.
- Cold Spring Harbor Laboratory Press








