
A 1.1-Mb Transcript Map of the Hereditary Hemochromatosis Locus
- David A. Ruddy1,
- Gregory S. Kronmal1,3,
- Vincent K. Lee1,3,
- Gabriel A. Mintier1,
- Leah Quintana1,
- Rodolfo Domingo, Jr.1,
- Nicole C. Meyer1,
- Alivelu Irrinki1,
- Erin E. McClelland1,
- Amy Fullan1,
- Felipa A. Mapa1,
- Theodore Moore1,
- Winston Thomas1,
- Deborah B. Loeb1,
- Cyrus Harmon2,
- Zenta Tsuchihashi1,
- Roger K. Wolff1,
- Randall C. Schatzman1, and
- John N. Feder1,4
Abstract
In the process of positionally cloning a candidate gene responsible for hereditary hemochromatosis (HH), we constructed a 1.1-Mb transcript map of the region of human chromosome 6p that lies 4.5 Mb telomeric to HLA-A. A combination of three gene-finding techniques, direct cDNA selection, exon trapping, and sample sequencing, were used initially for a saturation screening of the 1.1-Mb region for expressed sequence fragments. As genetic analysis further narrowed the HH candidate locus, we sequenced completely 0.25 Mb of genomic DNA as a final measure to identify all genes. Besides the novel MHC class 1-like HH candidate gene HLA-H, we identified a family of five butyrophilin-related sequences, two genes with structural similarity to a type 1 sodium phosphate transporter, 12 novel histone genes, and a gene we named RoRet based on its strong similarity to the 52-kD Ro/SSA lupus and Sjogren’s syndrome auto-antigen and the RET finger protein. Several members of the butyrophilin family and the RoRet gene share an exon of common evolutionary origin called B30-2. The B30-2 exon was originally isolated from the HLA class 1 region, yet has apparently “shuffled” into several genes along the chromosome telomeric to the MHC. The conservation of the B30-2 exon in several novel genes and the previously described amino acid homology of HLA-H to MHC class 1 molecules provide further support that this gene-rich region of 6p21.3 is related to the MHC. Finally, we performed an analysis of the four approaches for gene finding and conclude that direct selection provides the most effective probes for cDNA screening, and that as much as 30% of ESTs in this 1.1-Mb region may be derived from noncoding genomic DNA.
[The sequence data described in this paper have been submitted to GenBank under accession nos. U90543–U90548,U90550–U90552, and U91328.]
Transcript maps have been influential in the positional cloning of numerous genes, including those responsible for cystic fibrosis (Rommens et al. 1989), ataxia telangiectasia (Savitsky et al. 1995), and familial Alzheimer’s disease (Sherrington et al. 1995). Two techniques are used primarily to isolate expressed sequence fragments (ESFs) for the construction of transcript maps. The direct selection approach (Lovett et al. 1991) involves the hybridization of cDNA fragments to genomic DNA. It is extremely sensitive and capable of isolating portions of rare transcripts. Exon-trapping (Buckler et al. 1991; Church et al. 1994) recovers spliced exons from in vivo-expressed genomic DNA clones and produces candidate exons without any prior knowledge of the target gene’s expression.
Two additional approaches, sample sequencing and complete genomic sequencing, use the high-throughput capabilities of genomic DNA sequencing and subsequent comparisons of these sequence data to public databases of expressed sequences. Sample sequencing attempts to generate a 1× sequence coverage across a genomic region by random end sequencing of subclones from large-insert bacterial clones. This compares to 3–10× coverage needed (depending on the sequencing strategy) to obtain complete genomic sequence across the same region. Thus, sample sequencing offers a high-speed, low-resolution screening method for expressed sequence tags (ESTs) across large regions of genomic DNA. Complete genomic sequencing in conjunction with sequence analysis software, like GRAIL, is a high-resolution method for screening of expressed sequences. However, it is expensive and labor intensive. Nevertheless, genomic sequencing as a tool for gene finding is beginning to make an impact in positional cloning, for example, the Werner’s syndrome gene (Yu et al. 1996), and cloning by homology, for example, the Alzheimer’s disease gene on chromosome 1 (Levy-Lahad et al. 1995).
With a combination of direct selection, exon trapping, sample sequencing, and complete genomic sequencing, we performed a saturation screen for ESFs in a 1.1-Mb region genetically defined to contain the hereditary hemochromatosis (HH) gene (Feder et al. 1996). In this report, we describe the resulting transcript map. We identified 12 histone genes and cloned 19 cDNAs, including the major histocompatilibity complex (MHC) class 1-like HH candidate geneHLA-H. These cDNAs encode a broad range of proteins with diverse predicted structures and functions, including homlogs for butyrophilin and a sodium phosphate transporter (NPT). We performed preliminary expression experiments and amino acid comparisons with several gene families from this 1.1-Mb region. Structural comparisons of these genes revealed the conservation of an amino acid domain, the B30-2 exon, in several of the newly discovered genes.
We used this transcript map, and the accumulated genomic sequence of this region, to assess the efficacy of the four methods for future gene-finding projects. We analyzed the individual methods for quality of probes and types of background. We also examined the issues of genomic resolution and ESF identification between the sample-sequencing and complete genomic sequencing approaches. We further demonstrated that a sampling strategy combined exclusively with searches of the EST database remains an insufficient technique by itself for gene finding because of the high background of genomic DNA in this database.
RESULTS
Identification of Transcribed Sequences
To clone positionally a candidate gene for HH, initially we performed direct cDNA selection, exon trapping, and sample sequencing on chromosome 6p21.3 between the genetic markers D6S2230 and D6S2237 (Fig. 1). The starting material for these experiments was a 0.9-Mb yeast artificial chromosome (YAC) y899G1 and a 1.1-Mb large-insert bacterial clone contig (Lauer et al., this issue).
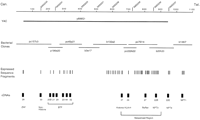
Combination genetic, physical, and transcription map of the HH candidate gene region. The first line shows the relative positions of selected genetic markers that define the HH region. The bold line below represents the YAC clone used in the direct selection experiments. The order and positions of the bacterial clones used in the exon trapping and sample sequencing are indicated under the YAC. The thin bars under the bacterial clones represents the approximate locations of the subset of ESFs that mapped to the contig. The thicker bars show the location of a subset of the cDNAs cloned. Two regions are bracketed: the butyrophilin family of genes (BTF), and the region where complete genomic sequencing was carried out.
Direct cDNA selection (DS) experiments were carried out as previously described (Morgan et al. 1992) by using pooled cDNA made from fetal brain, liver, small intestine poly(A)+ RNA, and hybridized to gel-purified y899G1 DNA. Four hundred fifty-six clones were sequenced and the resulting data searched by BLAST (Altschul et al. 1990). Those clones representing repetitive, bacterial, yeast, mitochondrial, and histone sequences were eliminated from further study as potential HH candidate genes. The remaining sequences were searched for overlaps and assembled into 108 unique DS contigs. The number of clones per DS contig varied from 1 to 22, and the length of each contig ranged from 250 to 850 bp. Small sequence-tagged site (STS) PCR assays were developed for each DS contig. Each STS was mapped to the bacterial clone contig and tested for its presence in cDNA libraries. Overall, 80% of the DS contigs mapped to the region and were found in cDNA libraries. The 20% mapping failure rate is probably an overestimate, as PCR assays that cross exon–intron boundaries would be expected to fail or give larger size products.
Exon-trapping experiments were carried out as described previously (Buckler et al. 1991; Church et al. 1994) with minor modifications on the large-insert bacterial clone contig. Preliminary experiments suggested that to detect rare splice events, 96 trapped products per large-insert bacterial clone needed to be sequenced (data not shown). Therefore, 768 potential exons from the 8 large-insert bacterial clones trapped were sequenced and the resulting data analyzed by BLAST. In addition, each potential exon was searched against a database of the DS contigs to eliminate redundant sequences. PCR assays were developed for each of the potential exons and they were tested for their presence in cDNA libraries. A total of 48 potential exons (or 6% of the total trapped) remained after these screening steps.
The identical set of large-insert bacterial clones were sample sequenced to generate a representative portion of genomic sequence for the 1.1-Mb region. A total of 3794 end sequence reactions were run to achieve the theoretical 1× coverage (see Methods). These sequences were screened against all available public databases. Eighty-five percent of these sequences contained inserts with no sequence similarity to vector or any known Escherichia coli sequence. An additional 1060 end sequence reactions were run from the opposite end of the cloning vector to augment the sequence coverage and prepare for complete genomic sequencing across selected regions. BLAST searches of all publicly available databases identified portions of 14 histone genes (2 representing pseudogenes) and 74 unique ESTs. The ESTs were cross-referenced against the DS and exon-trapped databases to eliminate redundancies. Fifty-eight unique ESTs, representing 39 distinct clones, remained (Table 1).
ESTs Found by Sample Sequencing Large Insert Bacterial Clones
Isolation of Full-Length cDNAs and the Transcript Map Surrounding the HLA-H Gene
A compilation of 195 ESFs from the three approaches led to the construction of a transcript map that served as the framework for the isolation of full-length cDNAs (Fig. 1). Probes that appeared to be derived from the coding portions of cDNAs, based on BLASTX searches, were developed for 82 ESFs and the appropriate cDNA libraries were screened. Nineteen cDNAs were isolated, 17 representing previously uncloned sequences.
A list of the cloned cDNAs and a comparison of the methods used to find them are presented in Table 2. Direct selection identified portions of 14 of the 18 cDNAs sequences contained within the boundaries of the template used in the selection, YAC y899G1. Exon trapping found exons in 15 of the 19 cDNAs contained within the boundaries of the large-insert bacterial clone contig. Sample sequencing identified 11 cDNAs that had corresponding ESTs in the public databases at the time of this analysis. The only combination of the three techniques that cloned all 19 cDNAs was direct selection and exon trapping.
Comparison of the Three Gene-Finding Methods Used to Construct the Hemochromatosis Region Transcript Map
0.25 Mb of Sequence Surrounding the HLA-H Gene
A detailed genetic analysis of the HH gene region delineated a 0.25-Mb subregion flanked by the markers D6S2238 and D6S2241 as the probable location of the HH gene (Feder et al. 1996). This subregion is within the 1.1 Mb for which we built the transcript map, and contains four novel genes—cDNA 37 (histone 2A), cDNA 24 (HLA-H), cDNA 27 (RoRet), and cDNA 22B (NPT3) (Fig. 1).
As a final measure to identify all of the genes, we performed complete genomic sequencing across the 0.25-Mb region. We identified a tiling path with overlapping end sequences from the sample sequence database. We then sequenced each 3-kb clone within the path using randomly selected transposable elements as platforms for dual-end sequencing. These individual clones were assembled in conjunction with the sample sequences from all bacterial clones in the region. An additional 2500 sequencing reactions were required to construct the complete genomic sequence across the 0.25-Mb region. The resulting sequence (Fig. 2) was analyzed systematically with BLASTX searches and the GRAIL 1.2 program to identify novel open reading frames (ORFs). The BLASTX searches did not produce any novel ESTs or potential coding regions that had not already been identified by sample sequencing. However, we were able to map accurately all histones genes within the 0.25-Mb region. GRAIL 1.2 identified 29 candidate exons with the designation “excellent,” the program’s highest degree of confidence. Of 29 candidate exons, 20 were associated with the genes in the 0.25-Mb region. GRAIL found at least one exon for each of the genes in this region. It was an excellent tool for elucidating the structures of the histone genes and the histone-associated cDNA 37 clone, but it had difficulties predicting the actual gene structure of the remaining three cDNAs. Figure 2 displays the positions, the exon and intron arrangement, and the relative orientation of transcription of the four novel genes within the 0.25-Mb region. The positions and transcriptional orientations of the histone genes and ESTs are also shown.

Schematic of the 0.25 Mb of genomic sequence carried out in the HH critical region. Those genes located on top of the line are transcribed from left to right (centromere to telomere), those below the line are transcribed from right to left (telomere to centromere). The positions and exon–intron structure of all four novel genes, cDNA 37, cDNA 24, cDNA 27 (cDNA25 is a smaller variant of cDNA27), and cDNA 22B, are denoted. Two versions of the gene structure are shown. The longer version represents both the coding and noncoding exons and the shorter version shows only the coding exons. The positions of the histone genes and the ESTs are also shown. The asterik denotes a pseudogene. The 0.25 Mb of genomic sequence has been deposited in GenBank (accession no.U91328). The sequence of the individual cDNAs have also been deposited in GenBank (accession nos: cDNA 37, U90551; cDNA 24, U60319; cDNA 27,U90547; cDNA 22B, U90544.
ESTs in the Large-Insert Bacterial Contig
In an effort to account for ESTs that did not associate with those cDNAs that we had already characterized in the 1.1-Mb region, we analyzed each 3′ EST for two characteristics: (1) a recognizable polyadenylation signal (Birnstiel et al. 1985); and (2) a stretch of genomic encoded poly(A) within 10–30 bp 3′ of the end of the EST. Of the 39 clones found in this 1.1-Mb region, 3′ end sequence was available for 30. Of these 30, 14 had recognizable poly(A)+ addition signals, 15 did not, and 1 could not be determined because of poor sequence quality. Of the 15 that did not have poly(A)+ addition signals, 9 had stretches of genomic encoded poly(A) within 10–30 bp from the end of the 3′ EST sequence. Five of these 9 clones contained Alu sequences within their sequence. Therefore, 30% (9 of 30) of the clones in this region probably arose from oligo(dT) priming of Alu repeat containing genomic DNA or unprocessed heterogeneous nuclear RNA transcripts. Of the 15 that did not have poly(A)+ signals, 5 did not associate with genomic poly(A) stretches. Two of these five were associated with the 3′ ends of cloned cDNAs and probably arose from internal priming of a mRNA molecule. Of the 14 that did have poly(A)+ addition signals, 8 were accounted for in the cDNAs cloned from the region, 2 clones showed homology to histone genes, and the remaining 4 were mapped outside of the 0.25-Mb HH critical region and were not pursued. An example of three ESTs clones that were found to be associated with or near cDNA 37, a histone 2A gene (Fig. 2), and their partial alignments to their corresponding genomic sequence (U91328) are shown in Figure3. These clones represent three classes of ESTs: in Figure 3A, an EST clone that corresponds to the actual end of the cloned cDNA and has a poly(A)+ addition signal; in Figure 3B, an EST that is associated with a cloned cDNA but appears to have originated by internal priming ∼180 bp 5′ of the authentic 3′ end; and in Figure 3C, an EST that is neither associated with coding DNA, nor has a poly(A)+ addition signal, but aligns 5′ to a genomic encoded stretch of poly(A) (within an Alu repeat). The latter clone was found to be a spliced product and to align with the intron of cDNA 37 (see Fig. 2).
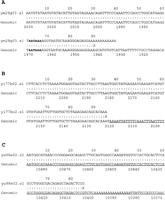
A FASTA alignment (Pearson and Lipman 1988) between three EST clones associated with cDNA 37 and their corresponding genomic DNA (only 90 bp of the 3′ end of each EST are shown). (A) An authentic EST corresponding to the end of cDNA 37 that possesses a poly(A)+addition signal (AATAAA). Note that the poly(A) tail has not been completely trimmed. (B) An EST that is associated with the end of cDNA 37, but does not have a poly(A)+ addition signal and appears to have arisen by internal priming. The sequence starts 180 bp 5′ of the true end of the clone. Analysis of the sequence trace does not suggest that this sequence read was excessively trimmed to produce the 180-bp difference (data not shown). The underlined sequence immediately 3′ to the end of the alignment is an Alu sequence. (C) An EST that aligns to the intron of cDNA 37 and contains no poly(A)+addition signal (this EST sequence was edited from the available sequence traces files) The alignment begins 5 bp 5′ of a stretch of poly(A) (in italics). The underlined sequence represents anAlu sequence.
Butyrophilin Gene Family
We cloned the human homolog of the bovine butyrophilin gene (BT) and mapped this gene ∼0.48 Mb centromeric toHLA-H (see Fig. 1). BT is a transmembrane protein that constitutes 40% of the total protein associated with the fat globule of bovine milk (Jack and Mather 1990). The human homolog of BT has also been cloned by others using a different approach (Taylor et al. 1996). BT is a member of a gene family with at least five other members residing in this region (see Fig. 1). A comparison of these proteins is shown in Figure 4. Each of the five proteins display varying degrees of homology to BT—BTF1 (cDNA 21), BTF2 (cDNA 32), BTF5 (cDNA 44), and BTF3 (cDNA 29)—are 45%, 48%, 46%, and 49% identical to BT, respectively, whereas BTF4 (cDNA 23), which is more similar to BTF3 (cDNA 29), is only 26% identical. This low degree of identity to BT is largely attributable to a truncation at the carboxyl terminus of the protein. The BTF family falls into two groups: BTF1 and BTF2, which are more related to each other than to BT or the other BTF members, and BTF3, BTF4, and BTF5, which appear different enough from BT or BTF1 and BTF2 to suggest a common evolutionary origin.

Alignment of the predicted amino acid sequence of the BTF proteins. Sequences were aligned in a pair-wise fashion using CLUSTAL to deduce the most parsimonious arrangement and are presented as such. The “stars” under the alignment represent those amino acids conserved in all six proteins, the “dots” represent conservative amino acid substitutions. Boxed are the regions within the proteins that correspond to three conserved motifs: (1) the B-G domain, (2) the transmembrane (TM) domain, and (3) the B30-2 exon domain. The sequences for all BTF cDNAs have been deposited in GenBank (accession nos: BTF1,U90543; BTF2, U90550; BTF3, U90548; BTF4, U90546; and BTF5, U90552).
There are three major components of BT—a B-G immunoglobulin superfamily domain containing the V consensus sequence (Miller et al. 1991), a transmembrane region, and a B30-2 exon. These motifs are found in all of these proteins with the exception of BTF4 (cDNA 23), which lacks the B30-2 exon by virtue of a carboxy-terminal truncation. The B30-2 exon is a previously noted feature of the MHC class 1 region found ∼0.2 Mb centromeric to the HLA-A gene (Vernet et al. 1993). This exon is also found in several genes of diverse functions telomeric to HLA-A, namely the myelin oligodendrocyte glycoprotein (MOG) gene (∼0.2 Mb telomeric to HLA-A) and the RET finger protein (RFP) gene (∼1 Mb telomeric toHLA-A) (Amadou et al. 1995).
To determine the size of the BTF transcripts and their expression level in a variety of tissues, we performed Northern blot analysis (Fig. 5A). BTF1 and BTF2, which are 90% identical, are expressed as a single major transcript of 2.9 kb. A minor transcript of 5.0 kb was present in some tissues. (The results for BTF1 are shown in Fig. 5A.) By Northern blot analysis, BTF1 was observed to be expressed at high levels in all the tissues tested with the exception of the lung, liver, and kidney where the expression level was lower. To expand the number of tissues examined, RT–PCR experiments were carried out (Fig. 5B). Amplification was observed in all tissues, including those observed by Northern blotting to be expressed at lower amounts, kidney and liver. A larger size fragment, not observed in the positive control lane, was presented in all lanes, indicating possible alternative splicing. Identical results were obtained with primers for BTF2 (data not shown).
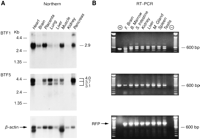
Expression analysis of the BTF family. (A) Northern blot analysis of representative members of the two groups of BTF proteins, BTF1 and BTF5. BTF1 hybridized to all tissues on the blot as a major transcript at 2.9 kb. BTF5 hybridizes to several transcripts ranging between 4.0 and 3.1 kb and displays as a similar expression profile to BTF1, with the exception of less expression in the brain. Autoradiography was for 24 hr. The β-actin hybridization shows the variation in poly(A)+ RNA between the lanes. Autoradiography was for 1 hr. (B) RT–PCR analysis confirms that the expression of both genes is widespread. Included in the (+) lane are BTF1 and BTF5 cDNAs as positive controls, the (−) lane represents the no-DNA control. Amplification using primers for the RFP gene (Isomura et al. 1992) controls for the integrity of the cDNA. All first strand cDNAs were checked for contaminating genomic DNA amplification by carrying out an identical experiment excluding the reverse transcriptase. In all cases, no amplification was obtained (data not shown).
BTF3, BTF4, and BTF5 were expressed as three transcripts ranging in size from 3.3 to 4.0 kb. (The results forBTF5 are shown in Fig. 5A.) BTF5 was expressed at moderate levels in all tissues tested, with the exception of the brain and kidney, where the expression level was less. RT–PCR experiments indicated that mRNA from the BTF5 gene could be found in all tissues tested, including fetal brain and kidney (Fig. 5B). Identical results were obtained with primers from the other genes in this group (data not shown).
RoRet, a Gene with Similarity to 52 kD Ro/SSA Autoantigen
Located ∼0.12 Mb telomeric to the HLA-H gene (see Figs. 1 and 2) is a gene that has 58% amino acid identity to the 52-kD Ro/SSA protein, an auto-antigen that is frequently recognized by antibodies in patients with systemic lupus erythematosus and Sjogren’s syndrome (Anderson et al. 1961; Clark et al. 1969). This novel sequence also has 29% amino acid identity to RFP (Isomura et al. 1992). On the basis of these two homologies we propose that this novel gene be calledRoRet. Alignment of the RoRet amino acid sequence to the 52-kD Ro/SSA demonstrated that a putative DNA-binding cysteine-rich motif [C-X-(I,V)-C-X(11-30)-C-X-H-X-(F,I,L)-C-X(2)-C-(I,L,M)-X(10-18)-C-P-X-C] found at the amino terminus (Freemont et al. 1991) and the B30-2 exon located near the carboxyl terminus of the 52-kD Ro/SSA protein are both conserved in RoRet (Fig. 6A).


The predicted RoRet and NPT3 and NPT4 proteins are similar to the 52-kD Ro/SSA auto-antigen and sodium phosphate transporter protein NPT1, respectively. (A) Alignment of the predicted amino acid sequence of the RoRet gene to the 52-kD Ro/SSA auto-antigen protein. The asterisks (*) under the alignment represent conserved amino acids, the dots represent conservative amino acid substitutions. The putative DNA-binding cysteine-rich domain and the B30-2 exon domain are boxed. The sequence of the RoRet cDNA has been deposited in GenBank, accession no. U90547. (B) Alignment of the predicted amino acid sequence of the two novel putative sodium phosphate transport proteins to that of the NPT1. Boxed are four potential transmembrane domains known to be conserved between human NPT1 and its rabbit homolog (Chong et al. 1993). The sequences of the NPT3 and NPT4 cDNAs has been deposited in GenBank, accession nos. U90544 and U90545, respectively.
Using Northern blots, the RoRet gene was found to be expressed as two major transcripts of 2.8 and 2.2 kb and two minor transcripts of 7.5 and 4.4 kb. Expression was observed in all of the tissues examined at levels reflective of the RNA amounts as determined by β-actin probing. One notable exception was the lung, where the expression appears to be less (Fig. 7A). Low-level expression was also detected in small intestine, kidney, liver, and spleen by RT–PCR (Fig. 7B).
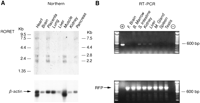
Expression analysis of the RoRet gene. (A) Northern blot analysis of the RoRet gene. The RoRet cDNA hybridizes to four different transcripts, ranging from 7.5 to 2.2 kb. Autoradiography was performed for 4 days. The rehybridization of the blot with a β-actin probe shows the variation in poly(A)+ RNA between the lanes. Autoradiography was for 1 hr. (B) RT–PCR analysis of the RoRet gene. Included in the (+) lane is a RoRet cDNA positive control. Weak amplification of the correct size is observed in the small intestine, kidney, and liver. The other tissues are negative as is the no-DNA control lane (−). The RFP primers show the integrity of the DNA.
Two Novel Genes with Homology to a Sodium Phosphate Transporter
A cDNA for a type 1 sodium phosphate transport (NPT1) protein has been cloned previously and mapped to 6p21.3 by using a somatic cell hybrid panel (Chong et al. 1993). We isolated this gene in our study by exon trapping and sample sequencing, and mapped it 0.32 Mb telomeric to HLA-H (see Figs. 1 and 2). Two additional cDNAs were cloned that show appreciable homology to NPT1 (see Fig. 6B). These genes, NPT3 and NPT4, mapped 0.15 and 0.1 Mb centromeric to the NPT1 gene (see Fig. 1). The predicted gene products of NPT3 and NPT4 are extremely hydrophobic, like NPT1, and reflect the probable location of these proteins in the plasma membrane. Both proteins also gave hydrophilicity profiles that were indistinguishable from NPT1 (data not shown).
Northern blot analysis revealed that NPT3 and NPT4have dramatically different patterns of expression (Fig.8A). NPT3 was expressed at high levels as a 7.2-kb transcript predominately in muscle and heart. Lesser amount of the mRNA were also found in brain, placenta, lung, liver, and pancreas. RT–PCR analysis revealed that expression of the proper size PCR fragment for NPT3 was clearly present in the small intestine, kidney, spleen, and testis, but was absent in fetal brain, bone marrow, and mammary gland (Fig. 8B). A smaller size fragment that may have arisen because of alternative splicing was detectable in all tissues with the exception of the liver. Although expression was apparently absent from the kidney by Northern blot analysis, it was detectable by RT–PCR. NPT4, on the other hand, was expressed only in the liver and kidney as a group of transcripts ranging from 2.3 to 1.7 kb (Fig. 8A). RT–PCR confirmed these results, yet a small amount of the proper size PCR fragment was also found in other tissues, notably the small intestine and testis (Fig. 8B). Other tissues showed amplification, but the fragments were of larger and smaller sizes than that produced by the cDNA positive control.
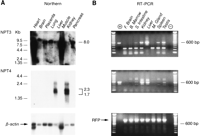
Expression analysis of the NPT3 and NPT4 genes. (A) Northern blot analysis of NPT3 and NPT4. NPT3 is expressed at high abundance in the heart and muscle as a single 8.0-kb transcript. Lesser amounts are found in the other tissues. The expression pattern of NPT4 is more restricted, being found only in the liver and kidney as a mixture of transcripts ranging from 2.3 to 1.7 kb. (B) RT–PCR analysis of theNPT3 and NPT4 genes. Included in the (+) lane are the NPT3 and NPT4 cDNA positive controls. TheNPT3 gene is expressed as the proper size PCR fragment in the small intestine, kidney, liver, spleen, and testis. A smaller fragment is detected in all tissues with the exception of the liver. The no-DNA control lane (−) is negative. NPT4 is expressed as the proper size fragment in the small intestine, kidney, liver, and testis. Larger and smaller size fragments are found in all other tissues with the exception of the brain. For both genes, these different size fragments may indicate alternative splice events. The no-DNA control lane (−) is negative. The RFP primers show the integrity of the cDNA.
DISCUSSION
Gene-Finding Methods
We used four methods for gene finding that depend on separate sets of criteria and resources to ensure the cloning of all HH candidate genes. We discovered that a combination of three techniques were needed to identify the 19 cDNAs and 12 histones genes in the 1.1-Mb region. Future gene-finding projects will require the scanning of multiple genomic regions for genes of diverse function. It would be difficult and expensive to use saturation screening across multiple loci with all four methods. Therefore, we examined the strengths and weaknesses of the individual techniques to determine the optimal approach for future gene-finding projects.
Direct cDNA selection provided the greatest amount of raw material from which the transcript map was constructed. We sequenced the fewest clones of the four techniques (456 DS clones compared to 768 exon-trapped clones and 4854 sample-sequence reactions. An additional 10,000 sequencing reactions would have been required to complete the genomic sequence across the 1.1-Mb region), yet we still accounted for 14 of the 19 cDNAs. The resulting cloned fragments were also more representative because the input cDNA was created by random priming, and the coding portions of cDNAs were more often recognizable by BLASTX searches. These data regarding the potential type of gene in the region allowed for a more efficient prioritization of ESFs for cDNA screening, and a rapid classification of cDNAs in the 1.1-Mb region. Although we did not determine the actual degree of enrichment obtained with the direct selection experiment, subsequent analysis of HLA-H cloning effort indicates that the enrichment was sufficient to clone very rare transcripts (Feder et al. 1996). The sensitivity of the technique led to the isolation of DNA fragments (∼15%) that map back to the region but are not represented in cDNA libraries. These clones appear to be either contaminating genomic DNA in the cDNA pool or portions of cDNA made from heterogeneous nuclear RNA transcripts. In addition, one 3.4-kb cDNA clone (cDNA 36) was isolated using a direct selection probe that appears to represent genomic DNA based on the fact that its poly(A) tail is genomic encoded and it does not contain an ORF. Direct selection failed to isolate a representative of the BT gene (cDNA 20). The most likely explanation is that this gene is mainly expressed in a tissue (breast) not used in the direct selection experiment. In spite of these problems, we found the probes produced by direct selection to be optimal in three regards: (1) high percentage of clones having ORFs, leading to more efficient prioritization; (2) the distribution of the selected products throughout the cDNAs, with sequences located near the 5′ end of messages facilitating the isolation of full-length clones upon cDNA library screening; and (3) highest ratio of transcripts to number of costly sequencing reactions.
Exon trapping is also a powerful method for isolating candidate exons from genomic DNA. It allows for the isolation of potential exons from genomic DNA without any knowledge of what tissues the particular gene target may be expressed in. Nonetheless, the method is technically demanding and extra steps must be taken to eliminate background splice products from further consideration. We used 32P-labeled oligonucleotides and colony hybridization to remove exons representing the common background splice products (i.e., vector–vector and vector–cryptic splice-site splicing). The result was that >90% of the candidate exons sequenced were of genomic DNA origin. By sequencing 96 clones per large-insert bacterial clone for a total of 768 sequencing reaction, exons were trapped for 15 of the 19 cDNAs isolated from the 1.1-Mb region, including one not cloned by direct selection, cDNA 20 (BT). However, in the case of cDNA 24, the HH candidate gene, and the ultimate target for this project, the two exons trapped for this gene were represented only once out of the 96 sequenced for the bacterial clone 75l14. Therefore, for this particular project, the number of candidate exon clones sequenced per large-insert bacterial clone appears to represent the minimum number.
The number of exons cloned per cDNA ranged from one to as many as nine. In many instances (30%), the exon trapped from a cDNA represented a noncoding portion of the gene. Therefore, exons could not be evaluated solely on the presence of ORFs. Many trapped exons (25%) that failed to test positive in cDNA libraries and thus, were eliminated from screening presumably arose from cryptic genomic splice events. However, three exons that are present in cDNA libraries gave rise to clones (cDNAs 41, 43, and 39) with no ORFs and appear to represent genomic DNA, based on the fact that their poly(A) tails are genomic encoded. Thus, both direct selection and exon trapping produce sequences that can lead to the cloning of putative noncoding cDNAs present in cDNA libraries. Nevertheless, the only combination of methods to clone all 19 cDNAs from the 1.1-Mb region was direct selection and exon trapping.
A sample sequencing strategy scans quickly and effectively large regions of genomic DNA for ESFs without the added cost and labor involved in a complete genomic sequencing effort. On the basis of our study, we estimate that sample sequencing produced sequence data five times faster at one third the cost of a complete genomic sequencing effort. Sample sequencing alone generated ∼67% sequence coverage across the 1.1 Mb, identifying 12 of the 19 cDNAs eventually cloned. This sequence information can also assist with probe development, exon–intron boundaries, marker identification, and physical mapping. In addition, we were able to identify pieces of all 12 histone genes (and the two pseudogenes) and ESTs representing three of the four cDNAs in the 0.25-Mb HH critical region. The RoRet gene did not have a corresponding EST at the time when the comparisons were done, yet we developed probes to this gene because of recognizable homology between end sequences and the Ro/SSA gene. Thus, increasing the resolution from sample sequencing to complete genomic sequencing did not affect the identification of ESFs in this region.
At present, an exclusive utilization of a genomic sequencing strategy combined with screening of the EST database is an inadequate method for gene finding because of the incomplete and inaccurate nature of the data in the EST database. In our study, 11 of the 19 cDNAs had corresponding ESTs at the time of analysis, and only 11 of 34 EST clones (32%) with 5′ end sequence (Table 1) appeared to represent coding portions of cDNAs. In our efforts to clone all of the genes in the 0.25-Mb critical region, we screened six EST probes that were not associated with cloned cDNAs against cDNA libraries made from the same tissues from which the ESTs were derived, and all six failed to produce clones. Upon completion of the genomic sequencing in this region and aligning the EST sequences to the genomic sequence, it was determined that all six ESTs were derived from noncoding DNA. When this analysis was extended to all the ESTs in the 1.1-Mb region, a total of nine ESTs appeared (9 of 30) to be derived from noncoding DNA. The majority of the noncoding ESTs (7 of 9) are listed as single clones in UNIGENE, and thus, do not fall into clusters. One clone is in a cluster of two ESTs, and the other is not listed in UNIGENE, but is a single clone in dbEST. Six of the nine noncoding ESTs originate from the same library (Soares fetal liver spleen). Although the number of ESTs we have analyzed is relatively small and restricted to a 1.1-Mb region of the genome, these data suggest that the percentage of ESTs associated with noncoding DNA could be higher than previous estimates of 2%–3% (Aaronson et al. 1996) and that the majority of these clones may reside as single isolates in dbEST.
Although the majority of noncoding ESTs in this region have repetitive DNA at their 3′ ends, we were reluctant to remove them from consideration based on this factor alone, for many cDNAs, including cDNA 24 the eventual HH candidate gene, have repetitive sequence elements at their 3′ ends. We observed that the presence of a poly(A)+ addition signal remains the best predictor of an EST clone’s authenticity. A recent study has shown that between 14% and 34% of ESTs in dbEST are of low complexity sequence (Aaronson et al. 1996). We suspect that most of the noncoding ESTs we have encountered might be removed in the process of building a higher quality EST database (Aaronson et al. 1996), and we expect that the percentage of 5′ ESTs with coding sequence should increase with the development and implementation of new normalized cDNA libraries constructed to improve cDNA 5′ end representation (Bonalda et al. 1996).
Genomic sequencing approaches are inherently problematic for finding expressed products because of the low percent of coding sequence in human genomic DNA. In the 0.25-Mb HH critical region, we estimate that 10% of the genomic DNA is coding. In the 1.1-Mb region, nearly 5000 sampling reactions were required to achieve adequate sequence coverage. An additional 2500 reactions were required to complete the sequence across the 0.25-Mb region. Both approaches incur high costs primarily attributable to sequencing reagents.
Genes in the Hemochromatosis Region
The bovine butyrophilin protein is one of the major proteins in the membrane layer surrounding the secreted milk droplet (Franke et al. 1981). We have isolated five novel cDNAs that show substantial sequence similarity to the BT gene. The expression pattern of theBTF family members differs from BT in that they are expressed in other tissues in addition to being present in the breast. The five cDNAs fall into two classes, suggesting that they arose from an early duplication event followed by additional gene duplications and divergence. Four of the five cDNAs share motifs found in theBT gene—a signal sequence, a domain related to the chicken MHC B blood group system (B-G) antigens, a transmembrane region, and the B30-2 exon. BTF4 contains the first three motifs, yet lacks the B30-2 exon because of a truncation of the carboxyl terminus. This exon was isolated originally from the HLA class 1 region and has appeared to “shuffled” into many genes distal to the MHC, including MOG, RFP, and BT (Vernet et al. 1993). The role that this exon plays in these genes and its relationship to the MHC remains unclear. However, a functional comparison of the B30-2 deficient BTF4 with BTF1,BTF2, BTF3, and BTF5 may be illustrative in this regard.
The B30-2 exon is also found at the carboxyl terminus of a completely different gene located 0.5 Mb telomeric to the BTF cluster, theRoRet gene. The RoRet protein is 58% identical to the 52-kDRo/SSA lupus erythematosus and Sjogren’s syndrome autoantigen. The 52-kD Ro/SSA gene is located on chromosome 11p15.5 (Frank and Mattei 1994). The function of the 52-kD Ro/SSA protein, which is part of a large complex of at least two other proteins, is unknown. However, the protein has been shown to bind DNA (Frank et al. 1995), a function that is presumably imparted by the novel cysteine-rich sequence (Freemont et al. 1991) found at the amino terminus of the protein. This cysteine-rich motif is also conserved in the RoRet gene. Of obvious interest will be if patients with lupus erythematosus or Sjogren’s syndrome who have autoantibodies to the 52-kD Ro/SSA protein also have auto-antibodies to the RoRet gene product and if such antibodies contribute to the cause of these diseases.
NPT1, located at the telomeric end of our contig, is a member of a gene family with two highly similar genes, NPT3 andNPT4. NPT4 has an expression pattern similar toNPT1, and therefore, may act like NPT1 in the renal reabsorption of phosphate. However, NPT3 is expressed at high levels in muscle and heart, as well as in the kidney. Therefore, these two genes are under the control of different regulatory elements giving rise to differential patterns of expression. At present there are two autosomal inherited disorders whose genes have not been mapped that are thought to be caused by defective reabsorption of phosphate in the proximal tubule of the kidney (Rasmussen and Tenenhouse 1989). What role either of these two newly discovered genes may play in these particular hypophosphatemias is awaiting determination.
In summary, we have used four gene-finding techniques to construct a 1.1-Mb transcript map 4.5 Mb telomeric to HLA-A. We compared the four techniques used in the process of making the map and conclude that direct cDNA selection is currently the most efficient procedure for generating raw material for transcript maps in terms of the quality of probes produced per number of clones sequenced. By adding exon trapping and sample sequencing, the efficiency of gene isolation increases notably. Using the transcript map, we identified and cloned a candidate gene for hereditary hemochromatosis, HLA-H, as well as 18 other cDNAs, most representing novel genes. The data presented here suggest that there have been duplication events in this region, resulting in a family of butyrophilin-like genes and a homolog of the 52-kD Ro/SSA gene that include a MHC-related domain B30-2. Further study into the function of these genes should provide insight into the evolutionary relationship between genes in this 1.1-Mb region of chromosome 6p21.3 and the MHC.
METHODS
Direct Selection
Poly(A)+ RNA from fetal brain, liver, and small intestine (Clontech, Palo Alto, CA) were converted into cDNA using random primers and a Superscript cDNA synthesis kit (Life Technologies, Gaithersburg, MD). The cDNA was digested with MboI and ligated to cDNAMboI linker adapters (Morgan et al. 1992). Unligated linker adaptors were removed by passage through cDNA spun columns (Pharmacia, Piscataway, NJ). Five nanograms of each of the ligated cDNAs were amplified using the cDNA MboI S primer (5′-CCTGATGCTCGAGTGAATTC-3′). The amplified products were purified on S-400 spun columns (Pharmacia, Piscataway, NJ), ethanol precipitated, and resuspended at 1 μg/μl in TE buffer (10 mm Tris, 1 mm EDTA). Gel-purified YAC y899G1 DNA (a gift from Dr. A. Gnirke, Mercator Genetics, Menlo Park, CA) was processed as described (Morgan et al. 1992). The cDNAs were mixed in equal molar amounts for a total of 3 μg, and blocked with a mixture of 4 μg Cot-1 DNA (Life Technologies, Gaithersburg, MD), and a cocktail of Sau3A-digested ribosomal and five different histone plasmid DNAs. The blocked cDNAs were hybridized to biotinylated YAC y899G1 DNA and streptavidin capture was carried out as previously described (Morgan et al. 1992). After the second round of selection, the eluted cDNAs were amplified using the cDNA MboI S primer, which included a [CUA]4 repeat at the 5′ end to facilitate cloning into a version of pSP72 (Promega, Madison, WI) constructed for use with uracil–DNA glycosylase cloning (Life Technologies, Gaithersburg, MD). Recombinants were transformed into DH5α, 960 clones picked into a 96-well format, and clones prepped for DNA sequencing using AGTC boiling 96-well mini-prep system (Advance Genetic Technologies, Gaithersburg, MD).
Exon Trapping
CsCl-purified genomic P1, BAC, and PAC DNAs were digested withBamHI, BglII, PstI, SacI, andXhoI, and 125 ng of each digest ligated into 500 ng of pSPL3 (Church et al. 1994) (Life Technologies, Gaithersburg, MD) digested with the appropriate restriction enzyme and phosphatased with CIAP (U.S. Biochemical, Cleveland, OH). One-tenth of the ligation was used to transform XL1-Blue MRF′ cells (Stratagene, La Jolla, CA) by electroporation. Nine-tenths of the electroporation were used to inoculate 10 ml of LB + 100 μg/ml of carbenicillin and after overnight growth, DNA was prepared using Qiagen Q-20 tips (Qiagen GmbH, Hilden, Germany). The remaining one-tenth was plated on LB + 50 μg/ml carbenicillen plates to evaluate the efficiency on cloning and to test individual clones for the presence of single inserts. COS-7 cells were seeded at a density of 1.4 × 105/well in six-well dishes and grown overnight. One microgram of DNA was transfected using 6 μl of Lipofect-Ace (Life Technologies, Gaithersburg, MD). Cytoplasmic RNA was isolated 48 hr after transfection. RT–PCR was carried out as described (Church et al. 1994) using commercially available reagents (Life Technologies, Gaithersburg, MD). The resulting CUA-tailed PCR fragments for each restriction-digested bacterial clone were pooled and UDG cloned into pSP72-U. The DNA was transformed into DH5α and the cells plated onto nylon membranes. After overnight growth, duplicates were made and the DNA hybridized to 32P end-labeled oligonucleotides designed to detect various background products associated with the pSPL3 vector. One set of filters was hybridized overnight with the following gel-purified oligonucleotides in 6× SSC aqueous hybridization solution at 42°C: vector–vector splicing, 5′-CGACCCAGCAACCTGGAGAT-3′; cryptic donor-1021, 5′-AGCTCGAGCGGCCGCTGCAG-3′; and cryptic donor-1134, 5′-AGACCCCAACCCACAAGAAG-3′. The filters were washed in 6× SSC, 10 mm NaPPi at 60°C, 30 min, 2×.
After overnight autoradiography, nonhybridizing clones were picked and grown in 250 μl of LB + 100 μg/ml of carbenicillin in 96-well minirack tubes. The samples were analyzed by PCR using the secondary PCR primers supplied in the kit (Life Technologies, Gaithersburg, MD) and those clones with inserts >200 bp were selected for sequencing.
Sample Sequencing
The minimal set of bacterial clones across YAC y899G1 was prepped with the Qiagen Maxi-Prep system (Qiagen GmbH, Hilden, Germany) and CsCl banded. Ten micrograms of DNA from each bacterial clone was sonicated in a Heat Systems Sonicator XL and end-repaired with Klenow and T4 polymerase (U.S. Biochemical, Cleveland, OH). The sheared fragments were size selected between 3 and 4 kb on a 0.7% agarose gel and then ligated to BstXI linkers (Invitrogen, San Diego, CA). The ligations were gel purified on a 0.7% agarose gel and cloned into a pSP72 derivative plasmid vector. The resulting plasmids were transformed into electrocompetent DH5α cells and plated on LB–carbenicillin plates. A sufficient number of colonies were picked to achieve 15× clone coverage. The appropriate number of colonies were selected by the following equation to generate a 1× sequence coverage: Number of colonies = size of bacterial clone (in kilobase)/average sequence read length (0.4 kb). These colonies were prepped in the 96-well AGCT system and end sequenced with a mitogen-associated protein 1 (MAP1) oligonucleotide using standard ABI Dye Terminator protocols. The oligonucleotide sequence is 5‘-CGTTAGAACGCGGCTACAAT-3‘. The MAP1 sequences were screened with BLAST against all available public databases. All sequence identities were cataloged and cross-referenced to the direct selection and exon-trapped databases.
cDNA Library Screening
Superscript plasmid cDNA libraries (brain, liver, and testis) were purchased from Life Technologies, Gaithersburg, MD. Colonies were plated on Hybond N filters (Amersham, Arlington Heights, IL) using standard techniques. Insert probes from direct selection products, exons, and ESTs (IMAGE clones) were all isolated by PCR followed by purification in low-melting point agarose gels (FMC, Rockland, ME). The DNAs were labeled in gel using the Prime-it II kit (Stratagene, La Jolla, CA). Small exon probes were labeled using their respective STS PCR primers instead of random primers. Up to five different probes were pooled in a hybridization. Filters were hybridized in duplicate using standard techniques. Putative positives were screened by PCR using the probe’s STSs to identify clones. Inserts from positive clones were subcloned into pSP72 and sequenced.
Northern Blots and RT–PCR Analysis
Multiple tissue Northern blots were purchased from Clontech and hybridized according the manufacturer’s instructions. RT–PCR was carried out on random primed first strand cDNA made from poly(A)+RNA (Clontech, Palo Alto, CA) using AmpliTaq Gold (Perkin Elmer, Foster City, CA). Control reactions were performed on RNA samples processed in the absence of reverse transcriptase to control for genomic DNA contamination.
Genomic Sequencing
The MAP1 sequences from the bacterial clones b132a2, 222K22, and 75L14 were assembled into contigs with the Staden software package. A minimal set of 3-kb clones were selected for sequencing by using the MAP2 oligonucleotide that sits on the opposite end of the plasmid vector. The sequence of MAP2 is 5‘-GCCGATTCATTAATGCAGGT-3‘. The MAP2 sequences were entered into the Staden database in conjunction with the MAP1 sequences to generate a tiling path of 3-kb clones across the region. These sequences were also screened by BLAST and all novel sequence identities were noted. The plasmid 3-kb libraries were transformed concurrently in 96-well format into pox38UR. The transformants were mated subsequently with JGM in 96-well format (Strathmann et al. 1991). All matings of the 3-kb clones within the tiling path were streaked on LB–carbenicillin–kanamycin plates and a random selection of 12 colonies per 3-kb clone were prepped in the AGCT system. We sequenced off both ends of the transposon with the −21 oligonucleotide (5‘-CTGTAAAACGACGGCCAGTC-3‘, and the REV oligonucleotide, 5‘-GCAGGAAACAGCTATGACC-3‘). Each 3-kb clone was assembled in conjunction with the end sequence information from all bacterial clones to generate complete sequence across the region. The genomic sequence was analyzed with BLAST and the GRAIL 1.2 to identify novel ORFs for gene finding.
Acknowledgments
We thank A. Gnirke and P. Lauer for supplying us with the YAC and large-insert bacterial clone contigs used in our experiments. We also thank R. Myers and M. Ellis for their comments on the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.








