
Single-nucleus multiomic profiling of the aging mouse substantia nigra reveals conserved gene alterations linked to Parkinson's disease
- Kangli Wang1,
- Weikun Xia1,
- Yingli Gu1,
- Songpeng Zu1,
- Qian Yang2,
- Maria Luisa Amaral1,
- Yaozhi Wang1,
- Allen Wang2,
- Xiang-Dong Fu3,
- William C. Mobley4 and
- Bing Ren1,2,5,6
- 1Department of Cellular and Molecular Medicine, University of California San Diego, School of Medicine, La Jolla, California 92093, USA;
- 2Center for Epigenomics, University of California San Diego, School of Medicine, La Jolla, California 92093, USA;
- 3Westlake Laboratory of Life Sciences and Biomedicine, School of Life Sciences, Westlake University, Hangzhou, Zhejiang 310024, China;
- 4Department of Neuroscience, University of California San Diego, La Jolla, California 92093, USA
Abstract
Parkinson's disease (PD) is a prevalent neurodegenerative disorder predominantly affecting individuals over 60. Its motor symptoms stem from the deterioration of dopaminergic neurons within the substantia nigra. Despite aging being a significant risk factor, the specific mechanisms linking aging and PD pathology remain unclear. Leveraging advancements in single-cell genomics, this study utilizes single-nucleus multiome sequencing to capture transcriptomic and epigenetic profiles from 40,125 cells across the lifespan of the mouse substantia nigra. Our analysis pinpoints age-associated changes at a cell type–specific level, revealing a subset of genes that increasingly express with age and are enriched in PD-related pathways, notably in oligodendrocytes at late aging stages. Integration with five public PD single-cell RNA-seq data sets highlights 85 genes consistently differentially expressed with aging and PD. Key genes such as Hsp90aa1 and Hsp90ab1 are upregulated at late aging stages in oligodendrocytes, microglia, and glutamatergic neurons. Additionally, Apoe in microglia and genes related to protein folding in oligodendrocytes are upregulated at late aging stages, whereas genes involved in myelination are downregulated at early aging stages in oligodendrocyte. Our multiomic atlas underscores the substantial regulatory network changes during aging that may predispose to PD, providing valuable insights for furthering understanding of PD pathogenesis and potential therapeutic targets.
Parkinson's disease (PD) is a common neurodegenerative disease with an average onset age of 60 years (Pagano et al. 2016; McGregor and Nelson 2019; Aarsland et al. 2021; Bloem et al. 2021). The characteristic motor symptoms of PD, including tremor, rigidity, and postural instability, are largely attributed to the degeneration of dopaminergic neurons, particularly within the substantia nigra region of the midbrain (Dauer and Przedborski 2003; McGregor and Nelson 2019). This progressive loss of dopaminergic neurons in the substantial nigra, a hallmark of PD, underscores the disease's complexity and points to the involvement of various cell types in its pathogenesis. Aging is the predominant risk factor for PD, as disease prevalence increases substantially in later life (Hou et al. 2019). Notably, several cellular alterations that accompany normal aging—including mitochondrial dysfunction, chronic inflammatory activation, and oxidative stress—mirror key pathological processes observed in PD (Hindle 2010; Reeve et al. 2014; Collier et al. 2017). In addition, genetic susceptibility and environmental exposures are believed to interact with age-related cellular decline to influence disease onset and progression (Pang et al. 2019). Despite aging being the greatest risk factor for developing PD, the mechanisms by which aging promotes PD are not fully understood. High-resolution studies of cellular and molecular changes during aging could yield essential insights into the disease's pathophysiology.
Recent advances in single-cell genomics offer new opportunities to unravel the cellular processes that link aging and PD pathology. Single-cell multiome sequencing (Vandereyken et al. 2023) is a powerful technique that enables the simultaneous capture of both gene expression (RNA modality) and chromatin accessibility (ATAC modality) within individual cells, allowing for high-resolution insights into the regulatory mechanisms underlying aging and PD. Previous studies on midbrain aging primarily employed RNA sequencing (Jin et al. 2025), which, although informative, does not fully elucidate the complex regulatory mechanisms underlying transcriptional programs. Gene expression is regulated by cis-regulatory elements (CREs) in a spatiotemporal manner through precise gene regulatory networks (Lee and Young 2013; Levine et al. 2014; Long et al. 2016). Understanding these regulatory mechanisms beyond transcription may reveal critical aspects of how cellular changes during aging contribute to PD pathology.
A pivotal study employing the multiome technique in human aging and PD samples highlighted the utility of such analyses, identifying extensive changes not only in gene expression but also in the connections between genes and chromatin regions in glia populations during aging and PD (Adams et al. 2024). However, human samples often suffer from inherent genetic variability, limited age representation, and low neuron capture rates, complicating the interpretation of age-related effects. The laboratory mouse provides a genetically homogeneous model and allows for longitudinal studies across multiple age stages, offering a more controlled investigation into aging mechanisms. Whereas traditional aging studies often compare just two age groups, a more nuanced approach incorporating multiple age points can detect molecular changes that may occur at different stages of aging, enhancing our understanding of aging processes. Mice, for example, progress from a suckling phase to adulthood within weeks and reach a state akin to human elderly past 18 months, providing a practical model for studying aging-related diseases (Dutta and Sengupta 2016; Wang et al. 2020b).
In this study, we performed 10x Genomics multiomic sequencing on eight mouse substantia nigra samples at four distinct age stages. This approach allowed us to assess cell type–specific transcriptional and epigenetic changes across aging in an unbiased way at single-cell resolution.
Results
Multiomic atlas of mouse substantia nigra across age
To investigate cell type–specific transcriptomic and epigenetic changes associated with aging in the substantia nigra, we profiled this brain region in mice at four different age stages: 2 months (adolescent), 6 months (mature adult), 12 months (middle-aged), and 18 months (old) using 10x Genomics Chromium Multiome (Fig. 1A). We conducted two biological replicates for each stage, resulting in a total of eight samples for both single-nucleus ATAC-seq (snATAC-seq) and single-nucleus RNA-seq (snRNA-seq).
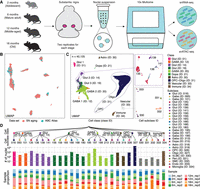
Single-nucleus multiomic analysis of the mouse substantia nigra across aging. (A) Schematic of the mouse substantia nigra samples analyzed, with 10x multiome data sets generated at four age stages: 2, 6, 12, and 18 months, with two replicates for each stage. (B) Coembedding UMAP (McInnes et al. 2018) of cells from the Allen Brain Cell (ABC) Atlas (Yao et al. 2023) scRNA-seq data and snRNA-seq data from this study, colored by data set. SN, substantia nigra. (C) UMAP embedding and clustering analysis of snRNA-seq data (n = 40,125 nuclei) from this study, with cells colored based on cell classes. The panels show examples where cell classes are separated into subclasses, and cells are colored based on cell subclass IDs. Cell class and subclass are annotated according to the ABC Atlas (Yao et al. 2023). Detailed information is provided in Supplemental Table S1. (Astro) astrocyte; (OPC) oligodendrocyte precursor cells; (Oligo) oligodendrocytes; (Glut) glutamatergic; (Gaba) GABAergic; (Dopa) dopaminergic; (VLMC) vascular leptomeningeal cells; (Peri) pericytes; (SMC) smooth muscle cells; (Endo) endothelial cells. (D) Top: Taxonomy tree of the 27 cell subclasses organized in a dendrogram, with cell class and subclass IDs marked. Middle: Bar plots representing the number of nuclei analyzed per subclass, colored by cell class. Bottom: Bar plots showing the proportion of nuclei in each sample for each subclass. 2 m: age 2 months; 6 m: age 6 months; 12 m: age 12 months; 18 m: age 18 months; rep1: replicate 1; rep2: replicate 2.
After sequencing, we performed rigorous quality control, filtering out low-quality cells based on criteria such as the number of detected features and mitochondria content in the RNA modality using Seurat (Hao et al. 2021), and transcription start site (TSS) enrichment score and fragment count in the ATAC modality using SnapATAC2 (Zhang et al. 2024a; Methods; Supplemental Fig. S1). Potential doublets were also excluded, resulting in 40,125 high-quality cells retained for further analysis.
Next, we performed iterative clustering using the RNA modality and integrated the data with the single-cell RNA-seq data from the Allen Brain Cell Atlas (Yao et al. 2023) to annotate cell types using Seurat (Supplemental Fig. S2; Fig. 1B; Methods). This analysis identified 10 cell classes and 27 cell subclasses (Fig. 1C; Supplemental Table S1), including nine subclasses within four nonneurons (NN) classes, six subclasses within two GABAergic (GABA) classes, eleven subclasses within three glutamatergic (Glut) classes, as well as one specific subclass of dopaminergic (Dopa) class. We validated our annotations by examining the expression of representative marker genes (Supplemental Fig. S3). The number of cells per subclass varied, ranging from a few dozen in some vascular subclasses to >10,000 in oligodendrocyte (Fig. 1D). In the subsequent analysis, we focused on the subclasses, which represent the most informative layer for cell type analysis. Therefore, subclasses are referred to as cell types in the following sections.
Age-associated differential gene expression in subclasses
We first assessed whether cell type proportions change with age by analyzing the cell subclass composition in each sample (Fig. 2A; Supplemental Fig. S4A; Methods). To do this, we calculated the cell subclass proportion using speckle (Phipson et al. 2022), estimating the variability of each subclass proportion at different age stages by leveraging biological replicates. To assess whether cell subclasses increased or decreased with age, we applied a linear model with age as the explanatory variable using limma (Ritchie et al. 2015). Statistical modeling showed no significant compositional changes with age after correcting for multiple tests.
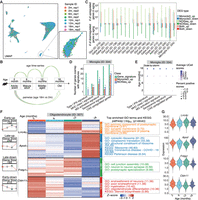
Profiling of age-associated gene expression variations across substantia nigra cell subclasses. (A) UMAP representation of all nuclei, colored by sample IDs. (B) The two comparison models used for age-associated differential expression analysis. (C) Summary of the number of age-associated DEGs identified within each subclass, categorized by upregulated or downregulated effect size and colored by comparison models (Monocle3 [Trapnell et al. 2014] linear model, FDR < 0.05; NOISeq [Tarazona et al. 2015] pseudobulk, probability > 0.95; and overlap in both methods). up, upregulated DGEs in aging; down, downregulated DEGs in aging. (D) Recurrence of age-related signatures in age-associated DEGs within the microglia subclass. Asterisks represent significant enrichment (FDR < 0.05) by hypergeometric test. IFN, interferon; MHC, major histocompatibility complex; MG, microglia; CAS, common aging score. (E) Dot plot representing the aggregated expression of age-related signatures across ages in the microglia subclass. Dot color and size represent the scaled average aggregated signature score calculated by UCell and scaled percent of positive scores of all nuclei, respectively. (F) Groups of age-associated DEGs within the oligodendrocytes subclass. Left: Box plots representing gene expression across ages for four DEG groups. Middle: Heat map of pseudobulk gene expression for each DEG group across ages. Right: Top significant GO and KEGG enrichments for genes in each DEG group in oligodendrocytes subclass. Early-up, early upregulated DEGs in aging; Late-up, late upregulated DEGs in aging; Early-down, early downregulated DEGs in aging; Late-down, late downregulated DEGs in aging. (G) Expression distribution of all nuclei in the oligodendrocytes subclass across ages for representative genes from the four age-associated DEG groups in panel F.
Next, we performed age-associated differential expression analysis for each cell subclass, fitting a single-cell linear model to identify differentially expressed genes (DEGs) associated with aging. In this analysis, we treated age as a continuous variable and accounted for the logarithm of total counts and biological replicates as covariates using Monocle3 (Trapnell et al. 2014; Fig. 2B; Methods). We also applied a nonparametric pseudobulk method using NOISeq (Tarazona et al. 2015) as a complementary and comparative approach, given that linear regression can be sensitive to cell number variation across subclasses. Certain subclasses were excluded from the differential analysis due to insufficient cell numbers or representation in any samples (Supplemental Fig. S4B,C). Overall, we identified age-associated DEGs in 19 subclasses. We observed strong concordance between age-associated expression estimates derived from the Monocle3 linear model and log2 fold-changes obtained from the pseudobulk NOISeq analysis. For example, in oligodendrocytes, the two methods showed a correlation of 0.89 (Supplemental Fig. S4D). Using the linear model, we found that most age-associated DEGs were specific to one or two subclasses, whereas only a small subset showed broad changes across many subclasses. Notably, genes such as Malat1 and Rpsa were significantly upregulated in 10 subclasses (Supplemental Fig. S4E). Using the linear model, the largest number of age-associated DEGs (FDR < 0.05) was found in oligodendrocytes, the most abundant cell type in the substantia nigra. Oligodendrocytes, the myelinating cells of the brain, are known to undergo changes during aging and have been implicated in the onset and progression of PD (Errea and Rodriguez-Oroz 2021; Sams 2021; Han et al. 2022). The pseudobulk method generally detected more DEGs (probability > 0.95) than the linear model, and the DEGs identified by pseudobulk analysis largely encompassed those detected by the linear model (Fig. 2C). The higher number of DEGs detected by the pseudobulk method may be attributed to its insensitivity to cell numbers within subclasses and its approach of comparing only two age points without considering other stages. Here, we focus on DEGs identified by the linear model, which accounts for expression trends across all age stages (Supplemental Table S2).
In the aged brain, microglia, as the major innate immune cell, particularly exhibit an activated state that expresses neuroinflammatory phenotype and undergoes senescence (Mattson and Arumugam 2018; Matsudaira et al. 2023; Han et al. 2024; Rim et al. 2024; Zhang et al. 2024b). Given that immunoreactive pathways and cellular senescence are key drivers of aging, we compiled five gene signature groups related to these pathways and evaluated their expression in the microglia data set. We observed a significant enrichment of upregulated DEGs in these signatures (Fig. 2D), and signature scores displayed a clear trend of increasing expression with age (Fig. 2E).
Functional enrichment analysis of age-associated DEGs revealed that upregulated DEGs in several subclasses were significantly enriched in the Parkinson's disease KEGG pathway (Kanehisa et al. 2023). Specifically, the linear model identified enrichment in seven out of 19 cell subclasses, and the pseudobulk method identified enrichment in 17 out of 19 cell subclasses (Supplemental Fig. S5A,B). Notably, age-associated DEGs within GABAergic neurons (ID 197), glutamatergic neurons (ID 166), and oligodendrocytes (ID 327) showed the most significant enrichment across both methods. Key PD-related KEGG pathway genes altered in these subclasses included Uqcrh, Cox4i1, Slc25a4, Psmb4, Ubc, and others. To further examine the link between DEGs and PD, we compared age-associated DEGs with PD-associated genes curated from the MalaCards (Rappaport et al. 2017) and Gene4PD (Li et al. 2021a) databases. Age-associated DEGs in GABAergic neurons (ID 197), oligodendrocytes (ID 327), glutamatergic neurons (ID 135), and glutamatergic neurons (ID 166) showed the strongest enrichment across both methods and databases (Supplemental Fig. S5C–F). Among the most significantly altered PD-associated genes in these subclasses were Gstm1, Apoe, Cox4i1, Eef1a1, Gpx4, Ubb, and Fth1. Amyotrophic lateral sclerosis (ALS) is another major neurodegenerative disorder that primarily affects motor neurons and shares several molecular and cellular pathways with PD (Spencer 2022; Lualdi et al. 2023). Consistent with this overlap, we found that upregulated DEGs in oligodendrocytes and dopaminergic neurons were also enriched in the ALS KEGG pathway (Supplemental Table S3; Supplemental Fig. S5G). As an additional control analysis, we further examined whether age-associated DEGs were enriched in ALS-associated genes curated in the MalaCards database. Only DEGs in dopaminergic neurons (ID 215) analyzed using the pseudobulk model exhibited slight enrichment (FDR = 0.04), suggesting a more specific association between aging-related transcriptional changes and PD pathology.
In dopaminergic neurons, which are specifically vulnerable in PD, we detected relatively few DEGs using the linear model, likely due to small cell numbers limiting the ability to detect aging effects (Fig. 2C). Therefore, we performed functional enrichment on DEGs identified by the pseudobulk method. Upregulated DEGs in aging dopaminergic neurons were enriched in neurodegenerative disease pathways, including PD, ALS, and Huntington's disease (Supplemental Fig. S5G), whereas downregulated DEGs were enriched in cell adhesion molecules pathways (Supplemental Fig. S5H).
To explore the dynamics of gene expression changes with age, we performed clustering analysis of DEGs, identifying distinct groups with varied functions. In oligodendrocytes, for example, we identified four DEG groups: early-upregulated, late-upregulated, early-downregulated, and late-downregulated (Fig. 2F,G). Some genes, such as Cldn11, showed decreased expression at early age stages, whereas others, like Fnbp1, exhibited reduced expression at later stages. Cldn11 is a major component of central nervous system myelin (Gow et al. 1999), and its decreased expression has been linked to neurodegenerative disorders (Sobue et al. 2021; Xie et al. 2022; Depp et al. 2023). Fnbp1 is recognized as a risk gene with reduced expression in amyotrophic lateral sclerosis (Andrés-Benito et al. 2017; Nakamura et al. 2020; Alonso et al. 2021), and its decreased expression has also been associated with other neurodegenerative disorders (Chen et al. 2020; Cai et al. 2022; Smajić et al. 2022; Gonzalez-Rodriguez et al. 2023). Notably, DEGs upregulated in late stages of aging were significantly enriched in the PD pathway (Fig. 2F; Supplemental Table S3; Supplemental Fig. S5I).
Recognizing that cellular activities often result from coordinated gene actions within pathways, we further analyzed pathway-level dynamics within each subclass using a pathway scoring method and a linear model, identifying differentially expressed pathways associated with age (Supplemental Fig. S6). We observed consistent upregulation of the PD pathway score across 10 cell subclasses. Additionally, clustering analysis of age-associated differentially expressed pathways in dopaminergic neurons identified two groups: early-upregulated and late-upregulated, with the dopaminergic synapse pathway in the former and the PD pathway in the latter (Supplemental Fig. S7).
Age-associated differential chromatin accessibility in subclasses
To investigate epigenomic changes in the substantia nigra with aging, we visualized scATAC-seq data using UMAP at a 500-base pair (bp) resolution for genomic bin features, revealing strong consistency with RNA-based cell annotations (Supplemental Fig. S8A). We validated chromatin accessibility at marker genes for cell subclasses (Fig. 3A). Next, we performed peak calling on each subclass to identify reproducible peaks following a previous established procedure (Zu et al. 2023) using MACS2 (Zhang et al. 2008; Methods). Reproducible peak calling analysis identified tens of thousands of candidate cis-regulatory elements (cCREs) for each subclass (Supplemental Fig. S8B), with a total of 134,369 cCREs, each 500 bp in length, identified across the mouse substantia nigra. We applied a strategy similar to gene expression analysis to identify age-associated differentially accessible cCREs (DACs) (Methods). The highest number of age-associated DACs was found in oligodendrocytes, with the pseudobulk method identifying a larger number of DACs (probability > 0.95) (Fig. 3B). Unless otherwise specified, we focus on DACs identified by the linear model (FDR < 0.05), as it captures accessibility trends across all age stages. An illustrative example of an age-associated DAC adjacent to the Lrig1 gene in microglia is shown in Figure 3C.
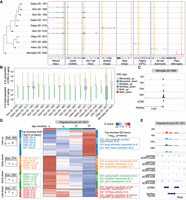
Age-associated differentially accessible cCREs across cell subclasses in the mouse substantia nigra. (A) Genome Browser tracks of aggregate chromatin accessibility profiles for representative subclasses at selected marker gene loci, with yellow blocks highlighting the representative regions. (B) Summary of the number of age-associated DACs (FDR < 0.05 for the Monocle3 model; probability > 0.95 for the NOISeq method) identified within each subclass, categorized by upregulated or downregulated effect size and colored by comparison models. up, upregulated DACs in aging; down, downregulated DACs in aging. (C) Genome Browser tracks of aggregate chromatin accessibility profiles for the microglia subclass across ages at a representative DAC locus. (D) Groups of age-associated DACs within oligodendrocytes subclass. From left to right: Box plots representing chromatin accessibility across ages for DAC groups. Top significant motif enrichments for cCREs of each DAC group in oligodendrocytes subclass. Heat map of pseudobulk chromatin accessibility for each DAC group across ages. Top significant GO and KEGG enrichments for cCREs of each DAC group in oligodendrocytes subclass. Late-up, late upregulated DACs in aging; Early-down, early downregulated DACs in aging; Late-down, late downregulated DACs in aging. (E) Genome Browser tracks of aggregate chromatin accessibility profiles for the oligodendrocytes subclass across ages at a representative DAC locus of the Late-down group in panel D. The predicated motif of the SOX family in the JASPAR database is shown at the bottom.
We further performed clustering analysis to group DACs, followed by motif and functional enrichment analysis. Notably, a DAC group downregulated during late aging in oligodendrocyte was enriched for motifs of the SOX family (Fig. 3D; Supplemental Table S4), consistent with the reduced expression of Sox10 (FDR = 2.04 × 10−9) in oligodendrocyte during aging (Fig. 2F; Supplemental Fig. S8C). Sox10 is a critical transcription factor involved in oligodendrocyte differentiation and maturation, directly regulating genes that encode the major myelin proteins (Stolt et al. 2002; Schlierf et al. 2006; Pozniak et al. 2010; Hornig et al. 2013), and its regulatory networks may play a role in PD progression (Corradini et al. 2014). Two examples of DACs in this DAC group, near the Ptprg and Ptk2 genes, contained SOX family motifs (Fig. 3E; Supplemental Fig. S8D). In addition, motif enrichment analysis of up- and downregulated DACs identified by the pseudobulk method in dopaminergic neurons revealed an enrichment for motif families such as ZF, HTH, and FOX, including CTCF, RFX, and FOXA1 (Supplemental Fig. S8E,F). Foxa1 is a member of the FOX family of winged-helix transcription factors and plays a crucial role in the development and maintenance of midbrain dopaminergic neurons (Ferri et al. 2007; Stott et al. 2013; Pristerà et al. 2015). Dysregulation of Foxa1 has been associated with motor impairments in PD models (Domanskyi et al. 2014).
Cell subclass–specific cCRE–gene links in substantia nigra
Utilizing the multiomic capabilities within individual cells, we identified potential enhancer-gene links at the cell subclass level using the SCENT workflow (Sakaue et al. 2024; Fig. 4A; Methods). In brief, we identified cCRE–gene pairs by linking a cCRE within 500 kb of a gene promoter and used a Poisson model to test the association between chromatin accessibility and gene expression for each distal cCRE–gene pair within each cell subclass. We discovered a range of significant cCRE–gene links, from 74 in microglia to 6193 in a GABA (ID 199) subclass (Fig. 4B; Supplemental Table S5), totaling 40,913 cCRE–gene links, comprising 21,396 cCREs and 10,431 putative target genes across the mouse substantia nigra. By applying reciprocal liftOver to map mouse cCREs to the human genome and identifying orthologous gene pairs, we found that 3632 cCRE–gene links (Supplemental Table S6) were conserved in humans based on comparison with cCRE–gene correlations reported in a previous human brain study (Adams et al. 2024). We observed moderate overlap between cCRE-gene links, age-associated DEGs, and DACs. For instance, in oligodendrocytes, 116 age-associated DEGs overlapped with genes linked by cCREs and 35 DACs overlapped with cCREs linked to genes (Fig. 4C,D). Noteworthy examples of oligodendrocyte-specific cCRE–gene links associated with DEGs and DACs include the Apod, Opalin, Septin4, Il33, Ccp110 gene loci (Fig. 4E; Supplemental Fig. S9A). Apod, a member of the lipocalin family, is elevated in brains of aging and neurodegenerative disorders, and its increased levels in glial cells surrounding dopaminergic neurons in the brains of PD might be linked to myelin degradation (Muffat et al. 2008; Dassati et al. 2014; Zucca et al. 2018; Dai et al. 2024).
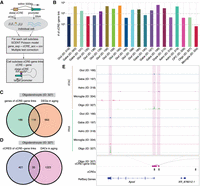
Inference of subclass-specific cCRE–gene links in the mouse substantia nigra. (A) Schematic of the computational strategy for linking chromatin accessibility of cCREs to expression of target genes for each cell subclass. (B) Bar plot showing the number of significantly (FDR < 0.1) correlated cCRE–gene pairs identified in each cell subclass. (C) Venn diagram showing the overlap of genes linked by cCREs and age-associated DEGs in oligodendrocytes. (D) Venn diagram showing the overlap of cCREs linked to genes and age-associated DACs in oligodendrocytes. (E) Genome Browser tracks of aggregate chromatin accessibility profiles and RNA signals for representative subclasses at selected age-associated DAC and DEG pairs.
Our findings, along with previous studies, highlight neuroinflammation driven by the cGAS-STING pathway, which is activated during aging (Fig. 2D,E; Gulen et al. 2023). The cCRE–gene maps provide an opportunity to further explore the regulatory networks and identify putative enhancers and potential transcription factors involved in these pathways. We identified 80 putative enhancers related to these genes, and motif enrichment analysis revealed that transcription factors such as CTCF, BORIS, TCF3, and PRDM15 may serve as key regulators (Supplemental Fig. S9B). Tcf3 encodes a member of the E protein family of helix-loop-helix transcription factors and plays a role in macrophage activation and inflammatory induction (Benayoun et al. 2019; Zhu et al. 2022; Deng et al. 2024).
Integration with human PD data sets highlights conserved age-related changes
The integration of our data set provides an opportunity to examine how aging may contribute to PD risk at cellular and molecular levels. Differential analysis revealed changes in gene expression during aging related to PD pathways. To pinpoint which PD-associated genes in specific cell types are also affected by aging, we integrated our mouse aging data set with five publicly available human PD single-cell data sets, focusing on conserved cell types between mouse and human substantia nigra (Fig. 5A; Supplemental Fig. S10A–D). We observed strong conservation of glial cells, including oligodendrocytes, astrocytes, OPCs, microglia, and vascular cells, across all data sets. However, due to biological complexity, neuron loss in PD, and technical challenges with human samples, neuronal subclasses were less consistently conserved than glial cells. Only a few neuronal populations—dopaminergic neurons (ID 215), specific GABAergic neurons (ID 213), and glutamatergic neurons (ID 016, 138, and 168)—were recurrently identified in more than two data sets.

Integrative analysis of aging and PD-associated gene expression changes. (A) Schematic of integration analysis between mouse and human single-cell data sets (Smajić et al. 2022) to identify consensus cell types and common age- and PD-associated genes. DaNs, dopaminergic neurons. (B) Heat map showing age-associated differentially expressed genes, with evidence of altered expression in PD across consensus cell types shown in the bottom panel. GO terms of genes with at least three evidence points in the GeneCards (Stelzer et al. 2016) database are displayed at the bottom.
Next, we identified homogeneous genes that were differentially expressed during aging and dysregulated in human PD across conserved cell types in at least two data sets. Notably, Hsp90aa1 and Hsp90ab1 were upregulated at late aging stages, and their homogeneous genes also showed increased expression in human PD in several conserved cell types, including microglia, oligodendrocytes, and glutamatergic neurons (Fig. 5B; Supplemental Fig. S10E). In microglia, Apoe was also upregulated at the late aging stage and showed elevated expression in PD. In oligodendrocytes, early aging stages showed upregulation of genes related to response to iron ion (Bcl2 and Snca) and downregulation of genes involved in myelination (Mbp, Mag, Mal, Ptn, Erbb3, and Pllp) and gliogenesis (Aspa, Mag, Mal, Ptn, Erbb3, and Opalin), whereas late aging stages were marked by upregulation of those involved in protein folding (Hsp90aa1, Hsp90ab1, Hsp90b1, and Cryab) and locomotory behavior (Aplp2, Selenop, and Sez6l2) and downregulation of those related to regulation of cell junction assembly (Clasp2, Ptk2, Srcin1, and Rap1a). The expression of these genes during aging mirrored patterns seen in PD, highlighting potential molecular links between aging and PD pathogenesis.
Discussion
In this study, we generated an atlas for mouse substantia nigra aging, incorporating both transcriptomic and epigenomic data. Our comparative analysis provided valuable insights into the cellular and molecular mechanisms by which aging increases the risk of PD. The cell atlas of the aging substantia nigra that we developed can serve as a crucial resource for future research focused on understanding the cellular changes in PD-related nigrostriatal pathway.
Aging is known to significantly alter transcriptional programs and gene regulatory networks in brain cell types (Mattson and Arumugam 2018; Zhang et al. 2022; Jin et al. 2025). In general, inflammation and cellular senescence are recognized as canonical hallmarks of aging in the mouse brain (Baker and Petersen 2018; Franceschi et al. 2018; Mattson and Arumugam 2018; Tchkonia and Kirkland 2018; Santos et al. 2024). Our study identified an upregulation of cellular senescence markers and the activation of inflammatory transcriptional programs in microglia within the substantia nigra, potentially contributing to the increased vulnerability to neurodegenerative diseases (Chinta et al. 2018; Tansey et al. 2022; Gopinath et al. 2023). Neuroinflammation in aging microglia is a complex process influenced by factors such as oxidative stress, mitochondrial dysfunction, and protein aggregates (Mattson and Magnus 2006; Zhang et al. 2023). Oxidative stress, resulting from the unregulated production of reactive oxygen species (ROS), is both a trigger and a promoter of neuroinflammation (Solleiro-Villavicencio and Rivas-Arancibia 2018; Teleanu et al. 2022; Zafar 2023). It can lead to oxidative posttranslational modifications that affect the function of neuroinflammatory mediators. Furthermore, transcriptional regulators like NF-kB and NRF2, which are activated by ROS, play crucial roles in controlling neuroinflammation. The reciprocal relationship between oxidative stress and neuroinflammation forms a key pathological axis in brain aging and neurodegenerative diseases. Consistent with this, our data revealed increased pathway activity scores for oxidative phosphorylation (P-adjust = 1.25 × 10−11) and ROS (P-adjust = 5.42 × 10−5) during aging in microglia (Supplemental Fig. S6). Both elevated ROS and inflammation are related to cellular senescence and accumulating evidence indicates these changes during aging are involved in the progression of PD (Miller et al. 2022; Russo and Riessland 2022; Sahu et al. 2022). Our results implicate the propagation of oxidative stress and neuroinflammatory processes in substantia nigra aging.
Our analysis also revealed that age-associated DEGs in several cell subclasses were enriched in PD-relevant pathways. This finding aligns with a recent study on the frontal cortex and striatum, which found that genes upregulated with age in neurons are enriched in pathways associated with neurodegenerative disease, including PD (Allen et al. 2023). Notably, in oligodendrocytes, the most abundant cell type in the substantia nigra and a potential contributor to PD etiology (Errea and Rodriguez-Oroz 2021), we observed that many of these genes were upregulated at later stages of aging, suggesting an increased PD risk associated with advanced age. Through a multiomic approach, we uncovered regulatory changes in aging, including late-downregulated DACs in oligodendrocytes that were enriched for the SOX family motifs. The SOX transcription factors are crucial for neurogenesis, and their dysregulation has been linked to PD (Stevanovic et al. 2022). Additionally, by mapping cell type–specific links between age-associated DACs and DEGs, we provided a deeper understanding of cellular regulatory networks in aging.
Our findings highlight genes that may represent age-related risk factors for PD development. By integrating our mouse data with human PD single-cell RNA-seq data sets, we identified a subset of genes that were differentially expressed during aging and also dysregulated in PD. Among these, Hsp90aa1 and Hsp90ab1, both members of heat-shock protein 90 (HSP90) family involved in protein folding and protein stabilization (Chen et al. 2005), were upregulated in multiple conserved cell types. Dysfunction of HSP90 has been implicated in alpha-synuclein aggregation and PD pathways (Hernandez et al. 2020; Hu et al. 2021). In microglia, we found that Apoe, a gene encoding a major apoprotein of the chylomicron, was upregulated during both aging and PD. High Apoe expression indicates microglial reactivation, which is associated with mitochondrial changes, oxidative stress, and neurodegeneration (Lee et al. 2023b; Martirosyan et al. 2024; Peruzzotti-Jametti et al. 2024). In oligodendrocytes, we observed both upregulated and downregulated genes shared between aging and the PD state. Protein folding genes such as Hsp90aa1, Hsp90ab1, and Cryab, along with locomotory behavior genes like Aplp2, Selenop, and Sez6l2, were upregulated. Conversely, myelination genes (e.g., Mbp and Mal) and gliogenesis genes (e.g., Aspa and Opalin) were downregulated. Alterations in myelin have been proposed as a key pathological feature of neurodegeneration (Dean et al. 2016). Furthermore, our analysis revealed distinct temporal patterns of gene expression during aging. For example, myelination-related gene expression decreased in early aging, whereas changes in genes related to cell junction assembly occurred at later stages. These stage-specific changes may reflect both proactive and reactive responses during aging and pathological processes, necessitating further investigation into these distinct temporal dynamics. Overall, these age- and disease-associated genes may reflect molecular changes that predispose the aging brain to neurodegenerative diseases. Given that aging is the primary risk factor for PD, these genes could help elucidate aging-dependent risk factors for neurodegeneration.
Whereas our study utilized mouse substantia nigra to generate a series of time-point data sets and minimize confounding factors, the sample size for several cell subclasses remains underrepresented. This underrepresentation could be attributed to the low abundance of these cells in the substantia nigra or technical limitations in capturing these cells with single-cell methods, resulting in reduced statistical power in the linear model and fewer detected differentially expressed genes in these cell types. Future studies that expand substantia nigra sample sizes and improve cell sorting or capturing techniques could further uncover the molecular and cellular interactions between aging and neurodegenerative diseases. Moreover, our study examined aging in wild-type mice up to 18 months without including comparisons to PD models. Incorporating age-related PD mouse models could reveal additional overlap with patient-derived gene signatures, particularly those related to dopaminergic neuron vulnerability. For example, the AAV–α-synuclein model induces moderate dopaminergic neuron loss and recapitulates key aspects of PD pathology (Björklund and Mattsson 2024). Applying the same experimental and analytical framework to such models would provide a more accurate representation of disease progression in the context of aging. Additionally, increasing the number of age stages, biological replicates, and sex-specific analyses would enhance statistical robustness. Investigating the functional consequences of our findings will further deepen our understanding of PD pathology. Finally, intrinsic species differences were not systematically compared, and biological factors such as aging may contribute to distinct phenotypic outcomes across species. Addressing these variations in future research will be crucial for translating findings to human disease contexts. We observed a relatively low proportion of cCRE–gene links conserved in the human brain data set (Adams et al. 2024). This may reflect the previously reported species specificity of both cCREs and their associated orthologous genes (Li et al. 2023). In addition, our analysis was restricted to cis-regulatory links, which may differ substantially across species due to evolutionary divergence in regulatory architecture. The limited coverage and underrepresentation of certain cell types in both data sets may further constrain the ability to detect conserved regulatory relationships. More comprehensive and higher-resolution data sets from both mouse and human will be essential for a more accurate and systematic comparison of regulatory conservation. Nonetheless, our frameworks offer critical insights and a foundational resource for the aging and PD research community, laying the groundwork for future explorations into the mechanisms of neurodegeneration.
In conclusion, despite minimal changes in cell type proportions with age, we observed significant alterations in gene expression and chromatin accessibility across many cell types during aging. Notably, genes upregulated with aging were enriched in PD-related pathways, highlighting potential mechanisms by which aging may contribute to increased PD susceptibility. Integration with human PD single-cell RNA-seq data sets revealed 85 genes, including Hsp90aa and Hsp90ab1, that were consistently differentially expressed during aging and PD. These findings suggest that age-related changes in the gene regulatory networks within the substantia nigra could play a critical role in PD pathogenesis.
Methods
Tissue preparation and nuclei isolation
All C57BL/6J mice were purchased from the Jackson Laboratory. All procedures were conducted in accordance with the guidelines of the University of California San Diego Institutional Animal Care and Use Committee (protocol no. S99116). Both male and female mice were used in this study. Mice aged 2 months, 6 months, 12 months, and 18 months were selected to collect substantia nigra as representative samples of adolescent, adult, middle-aged, and old brains. No pretests were performed to determine sample sizes.
Brains were extracted from the mice and sectioned into 1-mm coronal sections along the anterior-posterior axis in ice-cold PBS. Midbrain sections were identified by the hippocampus, and substantia nigra regions were dissected according to the Allen Brain Reference Atlas (Wang et al. 2020a). For each sample, brain tissues were pooled from four to six C57BL/6J mice to pile up 800,000 to 1,000,000 nuclei for single-nucleus multiome sequencing. Two biological replicates were performed for each age stage. The brain dissections were flash-frozen in liquid nitrogen (N2) and stored at −80°C until ready to process. Nuclei were isolated and sorted using the SH800S (Sony) for library construction as previously described (Li et al. 2021b; Supplemental Fig. S11).
Single-nucleus multiome
Single-nucleus ATAC-seq and single-nucleus RNA-seq libraries were performed for each sample as previously described (Zemke et al. 2023). For each sample, approximately 18,000 nuclei were loaded onto a 10x Chromium Controller following the Chromium Next GEM Single Cell Multiome ATAC + Gene Expression User Guide (CG000338, 10x Genomics). Libraries underwent shallow sequencing (∼1–5 million reads per library) on a NextSeq 500 (Illumina) for quality control, followed by deep sequencing (∼150–250 million total read pairs per library) on a NovaSeq 6000 (Illumina).
Alignment of sequencing reads
The sequencing reads were aligned to the mm10 (GRCm38) reference genome, downloaded from 10x Genomics (https://www.10xgenomics.com/cn/support/software/cell-ranger/latest/release-notes/cr-reference-release-notes). Feature-barcode matrices and fragments were generated using cellranger-arc (v2.0.0, 10x Genomics) with default parameters.
Preprocessing and nucleus filtering
For snRNA-seq libraries, feature-barcode matrices generated by cellranger-arc were imported into Seurat (v4.3.0) (Hao et al. 2021) within the R environment (v4.2.2) (R Core Team 2022) (https://www.R-project.org/) for further processing. Quality control was performed on a per-sample basis. Low-quality nuclei with fewer than 500 unique feature counts or more than 5% mitochondrial reads were excluded from further analysis. Doublet rates were estimated based on the cell number within each sample, and potential doublets were identified and removed using the DoubletFinder (v2.0.3) (McGinnis et al. 2019) algorithm.
For snATAC-seq libraries, fragments were shifted +4 bp for the positive strand and –5 bp for the negative strand to correct the 9-bp duplication induced by Tn5 transposase (Yan et al. 2020) during processing. We imported fragments into SnapATAC2 (v2.3.2) (Zhang et al. 2024a) for further processing. For each sample, we retained high-quality nuclei with transcription start site enrichment (TSSe) ≥ 10 and uniquely mapped fragments ≥1000 using the function filter_cells in SnapATAC2. Doublets were removed using a modified version of the Scrublet (Wolock et al. 2019) algorithm in SnapATAC2. Briefly, we first added the 500-bp genomic bin features using the add_tile_matrix function, then used the select_features function to select the 500,000 most accessible features across all nuclei. Doublet scores were calculated using the scrublet function, and barcodes with doublet scores > 0.5 were removed from further analysis.
Finally, high-quality nuclei that passed the quality control criteria for both RNA and ATAC modalities were retained for analysis. For snRNA-seq data, all samples were merged together for clustering analysis using the IntegrateData function in Seurat. For snATAC-seq, all SnapATAC2 AnnData objects were merged into one AnnDataSet for further analysis.
Iterative clustering and integration analysis
To annotate cell type for the sequenced nuclei, we performed cell clustering on the RNA modality. Initially, we performed a standard clustering analysis using all nuclei as follow: Principal component analysis (PCA) (Greenacre et al. 2022) was performed on the integrated data set using the function RunPCA in Seurat. The suitable number of PCs was determined with the findPC function in the R package findPC (v1.0) (Zhuang et al. 2022) using the “first derivative” method and confirmed with an elbow plot using ElbowPlot function in Seurat. Uniform Manifold Approximation and Projection (UMAP) (McInnes et al. 2018) was then calculated through RunUMAP function with the parameters a as 1.8956 and b as 0.8006. The k-nearest neighbors (KNN) graph was constructed using FindNeighbors, and Leiden clusters were defined with FindClusters function, selecting the resolution from 0.1 to 2 with a step size of 0.1 based on the Silhouette Coefficient (Rousseeuw 1987) using the silhouette function in the R package cluster (v2.1.4) (https://CRAN.R-project.org/package=cluster). Each clustering result was manually verified on the UMAP plot.
Next, integration analysis was performed with the scRNA-seq data from the Allen Brain Cell Atlas (Yao et al. 2023), focusing on cells from the midbrain ventral tegmental area and substantia nigra (MB - VTA-SN) regions. Public mouse brain single-cell RNA-seq data used for cell integration and annotation are available from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE246717, in accordance with GEO data policies. Using the Allen data set as a reference, the TransferData function in Seurat and canonical component analysis (CCA) method were applied to annotate cell classes. For UMAP visualization, the IntegrateData function was used to create the coembedding space. Consensus scores were calculated as the number of nuclei within a cluster annotated as the corresponding cell class in the scRNA-seq data divided by the number of nuclei in that cluster to annotate clusters. Clusters with a consensus score > 0.8 were assigned to corresponding cell classes. For mixed clusters, additional rounds of clustering and integration were performed (Supplemental Fig. S2).
Further clustering and integration with the Allen data set were conducted for each cell class to annotate cell subclasses. Subsequent analysis focused on the subclass level, providing generalizable insights into gene regulatory elements and a comprehensive characterization of cell types indicated by previous study (Zu et al. 2023).
As both male and female mice were used in this study, we examined the gender distribution within each cell subclass. To do this, we utilized the aforementioned midbrain single-cell RNA seq data set from the ABC Atlas, which contains the gender information, to train a model for gender prediction. Six genes, including Xist and Tsix (located on the X Chromosome) and Ddx3y, Eif2s3y, Uty, and Kdm5d (located on the Y Chromosome), were selected as features for training due to their significant differential expression between genders (Supplemental Fig. S12A). The data were randomly split into 70% for training and 30% for testing. A logistic regression model was trained on the training set and used to predict gender in the test set. The model demonstrated strong predictive performance on the test data (Supplemental Fig. S12B–D). We then applied this model to predict the gender of each cell in our data set. For each cell subclass, we observed a balanced gender distribution, with the proportion of cells in each gender ranging from 31% to 69% (Supplemental Fig. S12E).
Identification of reproducible peaks
We performed reproducible peak calling for each cell subclass using the strategy from a previous study (Zu et al. 2023). Briefly, all fragments were combined to generate a pool of pseudobulk ATAC-seq data set for each cell subclass. We also created pseudobulk data sets consisting of two biological replicates and two pseudoreplicates, each containing half the reads from each biological replicate for each subclass. Peak calling was independently performed on each pseudobulk data set using MACS2 (v2.2.9.1) (Zhang et al. 2008). Peak summits were extended by 250 bp on either side for downstream analysis. To minimize potential false negatives, we did not perform peak calling for pseudobulk data sets of replicates if any individual biological replicates or pseudoreplicates contained fewer than 200 cells. We retained only reproducible peaks, defined as peaks detected in the pooled data set that overlapped ≥50% of their length with peaks in both biological replicates or pseudoreplicates. Reproducible peaks were further filtered with a score per million (SPM) cutoff of 5. Additionally, we excluded peaks overlapping with the ENCODE mm10 blacklist regions (Amemiya et al. 2019) and kept only those located on Chromosomes 1–19 and sex chromosomes. Finally, to filter potential positive peaks, we selected only peaks identified as open chromatin in a significant fraction of the nuclei in each cell subclass. This was achieved by fitting a zero-inflated beta model for the fraction of nuclei showing a signal at the same number of randomly selected non-DHS regions and empirically determining a significant threshold of FDR < 0.01 for peak filtering. All UCSC Genome Browser tracks in this study were displayed using Integrative Genomics Viewer (IGV) (Robinson et al. 2011).
Cell subclass composition across aging
To test for statistically significant compositional changes across age for each cell subclass, we first calculated the cell subclass proportion using the function getTransformedProps in the R package speckle (v1.2.0) (Phipson et al. 2022). For each cell subclass, we estimated the variability of the cell subclass proportion at each age stage by leveraging the biological replicates. To determine whether the cell subclasses were increasing or decreasing with age, we fit a linear model with age as the explanatory variable using the R package limma (v3.58.1) (Ritchie et al. 2015) for each cell subclass. The P-values from limma benefit from empirical Bayes shrinkage of the variances. We obtained adjusted P-values by applying the Benjamini–Hochberg multiple testing correction (Benjamini and Hochberg 1995).
Aging differential gene expression analysis
To identify age-associated genes changing in each cell subclass, we first employed a linear regression model using the function fit_models in Monocle3 (v1.3.4) (Trapnell et al. 2014). We accounted for the logarithm of total counts and biological replicates as covariates in the model to disentangle the true effect of age. We fit the model for cell subclasses with at least 100 nuclei in total and 10 nuclei in any biological replicate of each age stage, focusing on genes expressed in at least 10 nuclei. To avoid sex bias, we only assessed genes on autosomes. The P-value tested whether the effect of age was significantly different from zero, and these P-values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. Age-associated DEGs were defined as those with an FDR < 0.05.
Given that linear regression methods for differential expression analysis are sensitive to the sample size, we also employed a pseudobulk method named NOISeq (Tarazona et al. 2015) as a complementary and comparative approach. NOISeq is a nonparametric method that is more suitable for small sample sizes. We generated the pseudobulk expression for each subclass using the AggregateExpression function in Seurat. Differential expression analysis was then performed between the 2-month and 18- month age groups using the function noiseqbio in NOISeq (v2.46.0). We calculated a posteriori probability of differential expression between groups, corresponding to the 1 minus FDR as defined by the Benjamini–Hochberg method. Genes with a probability > 0.95 (equivalent to FDR < 0.05) were considered significant DEGs.
We then assessed the potential influence of gender on differential gene expression analysis, using oligodendrocytes as an example. The oligodendrocyte data were split into male or female groups based on the gender prediction model described earlier. Differential gene expression analysis was conducted separately for male and female data. Although there was a slightly higher proportion of males in the 12-month age stage, the differential analysis expression results were highly concordant between males and females (Supplemental Fig. S12F–I). The age-associated DEGs showed significant overlap between the male and female data sets (Supplemental Fig. S12G,H), and the age-related differential expression statistics were strongly correlated between two groups (Supplemental Fig. S12I).
We additionally performed a sample size estimation to evaluate the power for detecting age-associated effects in our single-cell data set. Using oligodendrocytes as a pilot cell population, we applied the distribution-free method scPS (v0.5.2) (Hsu et al. 2024). We first characterized key data properties—including gene expression means, cell-cell correlations, and the relationship between gene means and standard deviations—for 1000 candidate genes using the geneCandidate function. Based on the observed data characteristics, power was then estimated across varying cell numbers and effect sizes using sizeCal function. Our analysis indicated that achieving 80% power to detect differentially expressed genes at FDR = 0.05 with a two-fold effect size, consistent with prior mouse brain aging study (Jin et al. 2025), would require approximately 600 cells under the current replicate design (Supplemental Fig. S13).
Aging-related signature in microglia
To compare the reported aging-related signatures to our identified aging DEGs in microglia, we collected five signature gene groups previously reported to show activation in microglia during aging. Type I interferon (IFN) signature, major histocompatibility complex (MHC) class I signature, and microglia (MG) activation signature were reported by Gulen et al. (2023). The senescence signature was reported by Kiss et al. (2020), and the common aging score (CAS) genes were reported by Hahn et al. (2023). The enrichment of microglia aging DEGs in each signature group was assessed using a hypergeometric test. We applied multiple testing correction using the Benjamini–Hochberg method.
Group of differentially expressed genes
We further grouped the aging-associated DEGs based on the pseudobulk expression across age in each subclass. Clustering of aging-associated DEGs was performed using the function clusterData from the R package ClusterGVis (v0.1.1) (https://github.com/junjunlab/ClusterGVis) with the mfuzz (Kumar and Futschik 2007) method. Visualization of the expression patterns for each DEGs group was achieved using the visCluster function in ClusterGVis.
Aging differential pathway expression analysis
Cellular activities often result from the combined actions of genes within pathways. Using a pathway scoring method to aggregate gene effects can enhance statistical power and facilitate a more meaningful biological interpretation. Therefore, we also performed differential pathway analysis to prioritize the most significantly changing pathways across age. We included KEGG pathways containing between five to 300 genes, as smaller gene sets may be overdispersed and larger gene sets may be nonspecific. For each cell subclass, we used UCell (v2.6.2) (Andreatta and Carmona 2021) to aggregate gene scores for each pathway. We then employed limma (Ritchie et al. 2015) to perform age-associated differential analysis, including biological replicates as covariates in the model. The P-values from limma were adjusted using empirical Bayes shrinkage of the variances, and the Benjamini–Hochberg method was applied to correct for multiple testing.
Aging differential chromatin accessibility analysis
We adopted a similar strategy to identify age-associated differentially accessible cCREs as we did for aging differential expression analysis. First, we applied a linear regression model in Monocle3 (Trapnell et al. 2014) to identify age-associated cCREs in each cell subclass. As a complementary approach, we used the nonparametric method NOISeq (Tarazona et al. 2015) for pseudobulk accessibility comparison between the 2-month and 18-month age groups. Age-associated cCREs were defined as those with FDR < 0.05 in Monocle3 or a probability > 0.95 in NOISeq.
Group of differentially accessible cCREs
We further grouped the aging-associated DACs based on the pseudobulk chromatin accessibility across age in each subclass. Clustering and visualization of aging-associated DACs was performed using ClusterGVis, following the same approach as for DEGs.
Gene Ontology and KEGG pathway enrichment
We performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis using the R package clusterProfiler (v4.10.0) (Wu et al. 2021). For age-associated differentially expressed genes, we used the expressed genes in each subclass as the background. For age-associated differentially accessible cCREs, we first annotated the cCREs to genes within 3-kb flanking regions using the R package ChIPseeker (v1.38.0) (Yu et al. 2015). We also filtered out unexpressed genes in each subclass based on RNA modality. The P-values were computed using the Fisher's exact test and adjusted for multiple comparisons using the Benjamini–Hochberg method. An adjusted P-value cutoff of 0.05 was used to determine significant GO terms and KEGG pathways.
PD-associated gene enrichment
Two PD-associated gene lists were obtained from MalaCards (Rappaport et al. 2017) and Gene4PD (Li et al. 2021a), which compile genes with genetic and/or molecular evidence linked to Parkinson's disease. For age-associated differentially expressed genes, the expressed genes within each subclass were used as the background. Gene enrichment analysis was conducted using Fisher's exact test, with multiple comparison adjustments performed using the Benjamini–Hochberg method. A significance threshold of adjusted P-value < 0.05 was applied to identify significantly enriched gene sets.
Motif enrichment
Motif enrichment analysis of age-associated differentially accessible cCREs was performed using Homer (v4.11.1) (Heinz et al. 2010) software with the “given size” parameter.
Inference of cCRE–gene links
We predicted cCRE–gene links for each subclass separately using the SCENT (v1.0.0) (Sakaue et al. 2024) workflow. Gene promoters were defined as genomic regions ±1 kb of transcriptional start sites according to GENCODE vM23 (Harrow et al. 2012). cCREs outside of gene promoters were considered distal cCREs. We identified cCRE–gene pairs as those with a cCRE within 500 kb of a gene promoter. For each subclass, we tested the association between chromatin accessibility and gene expression of each distal cCRE–gene pair using a Poisson model. Covariates in the model included the percentage of mitochondrial reads per nucleus, the logarithm of the number of unique molecular identifiers in the nuclei, and the sample batch. The significance of associations was assessed using bootstrapping procedures in SCENT, and multiple testing correction was applied using the Benjamini–Hochberg method. Statistically significant distal cCRE–gene links were defined by a threshold of FDR < 10%.
We assessed the conservation of cCRE–gene links in humans by comparing our mouse cCRE–gene links to those reported in a previous human brain study (Adams et al. 2024). Mouse cCREs (mm10) were first converted to human genomic coordinates (hg38) using reciprocal liftOver (Hinrichs et al. 2006). Gene symbols were then mapped to their human orthologs using biomaRt (Durinck et al. 2009). A cCRE–gene link was considered conserved if the reciprocally lifted-over cCRE overlapped with a human cCRE and the corresponding orthologous gene symbol matched a gene involved in the human cCRE–gene correlation.
Integration with human PD data sets
To identify consensus cell types and compare age- and PD-associated differentially expressed genes between mouse and human, we performed integration analysis with five publicly available scRNA-seq data sets: Smajić et al. (2022), Adams et al. (2024), Lee et al. (2023a), Martirosyan et al. (2024), and Wang et al. (2024). Five public human brain single-cell RNA-seq data sets used for integration and comparative analyses are available from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE193688, GSE157783, GSE243639, GSE148434, and GSE184950, respectively, and were analyzed in compliance with GEO data policies. Using the human data sets as references, we applied the TransferData function in Seurat along with CCA method to transfer annotations to the mouse data set. For UMAP visualization, the IntegrateData function was used to create a coembedding space. Consensus scores were calculated by dividing the number of nuclei in a mouse subclass annotated as the corresponding cell type in the human data by the total number of nuclei in that subclass. Paired mouse cell subclasses and human cell types with a consensus score > 0.9 were identified as consensus cell types. PD-associated differentially expressed genes from consensus cell types in the human data sets were then compared to age-associated differentially expressed genes in the corresponding mouse cell subclasses.
Statistics
No statistical methods were used to predetermine sample sizes. Randomization of the samples was not performed, and investigators were not blinded to the specimens being investigated. Single-cell differential expression and accessibility analysis was performed using a generalized linear regression model to quantify changes with age, assuming a quasi-Poisson distribution of gene expression counts in the cells. Pseudobulk differential expression and accessibility analysis were performed using the nonparametric and empirical Bayes NOISeqBIO algorithm to compare changes between young and old groups, estimating the null distribution by comparing replicates within the same group. Low-quality nuclei and potential barcode collisions were excluded from the subsequent analysis as outlined above.
Data access
Demultiplexed and processed data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE267831. Custom code and scripts used for analysis are available at GitHub (https://github.com/wkl1990/SN_aging) and as Supplemental Code.
Competing interest statement
B.R. is a cofounder and consultant of Arima Genomics, Inc. and cofounder of Epigenome Technologies, Inc.
Acknowledgments
This work was supported by Aligning Science Across Parkinson's (ASAP-020566) through the Michael J. Fox Foundation for Parkinson's Research (MJFF) (B.R., A.W., X.-D.F., W.C.M.). Work at the Center for Epigenomics was also supported by the UC San Diego School of Medicine. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health Shared Instrument Grant (SIG) (#S10 OD026929). We thank all other members in the Ren laboratory for valuable inputs. We also thank Yoshiaki Tanaka, Malte Spielmann, Inkyung Jung, Matthew G. Holt, and the Bin Zhang lab for their data sharing.
Author contributions: Study supervision: B.R. Contribution to data analysis: K.W., S.Z., and M.L.A. Contribution to data generation and management: K.W., W.X., Y.G., Q.Y., Y.W., A.W., and X.-D.F. Contribution to data interpretation: K.W., B.R., S.Z., M.L.A., and W.C.M. Contribution to writing the manuscript: K.W., B.R., W.X., and Y.G. All authors edited and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.281113.125.
-
Freely available online through the Genome Research Open Access option.
- Received June 23, 2025.
- Accepted March 2, 2026.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








