
Parallel analysis of replication timing, gene expression, and copy number with PARTAGE
- Lakshana Sruthi Sadu Murari1,5,
- Quinn Dickinson1,5,
- Silvia Meyer-Nava1 and
- Juan Carlos Rivera-Mulia1,2,3,4
- 1Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota Medical School, Minneapolis, Minnesota 55455, USA;
- 2Masonic Cancer Center, University of Minnesota Medical School, Minneapolis, Minnesota 55455, USA;
- 3Stem Cell Institute, University of Minnesota Medical School, Minneapolis, Minnesota 55455, USA;
- 4Masonic Institute on the Biology of Aging and Metabolism, University of Minnesota Medical School, Minneapolis, Minnesota 55455, USA
-
↵5 These authors contributed equally to this work.
Abstract
The human genome is partitioned into functional compartments that replicate at specific times during S-phase. This temporal program, referred to as replication timing (RT), is coregulated with the 3D genome organization, is cell type–specific, and changes during development in coordination with gene expression. Moreover, RT alterations are linked to abnormal gene expression, genome instability, and structural variation in multiple diseases, including cancer. However, mechanistic links between RT, large-scale 3D genome architecture, and transcriptional regulation remain poorly understood. A major limitation is that current approaches require the separate profiling of RT and transcriptomes from independent batches of samples, which obscures the complex coregulation between the epigenome and transcriptome. Here, we present PARTAGE, a multiomics approach that enables joint profiling of copy number variation (CNV), RT, and gene expression from the same sample, providing a more accurate integrated view of the complex relationships between RT and gene regulation.
Human cells duplicate their genome by the firing of thousands of origins that are activated in clusters following a precise temporal order (Rivera-Mulia and Gilbert 2016a; Rhind 2022; Vouzas and Gilbert 2023; Hyrien et al. 2025). This replication timing (RT) program partitions the genome into functional units that segregate to distinct nuclear compartments (Rivera-Mulia and Gilbert 2016b; Liu et al. 2025). In fact, RT aligns with the 3D genome organization measured by chromosome conformation methods (Dekker et al. 2002; Lieberman-Aiden et al. 2009), with active and inactive compartments replicating early and late, respectively (Ryba et al. 2010; Dixon et al. 2012; Rivera-Mulia and Gilbert 2016b). Establishment and maintenance of chromatin epigenetic states also require proper RT regulation (Lande-Diner et al. 2009; Klein et al. 2021). RT also changes dynamically during development and is cell type–specific (Hiratani et al. 2008, 2010; Rivera-Mulia et al. 2015, 2019a; Nakatani 2025). Indeed, around half of the human genome changes RT during differentiation into distinct cell fate specification pathways, and these changes occur in coordination with the establishment of cell type–specific transcriptional programs (Rivera-Mulia et al. 2015). Moreover, abnormal RT is linked to altered gene regulation in distinct disease states (Rivera-Mulia et al. 2017; Sasaki et al. 2017; Dixon et al. 2018; Rivera-Mulia et al. 2019b, 2022), highlighting the importance of RT control. The correlation between RT and transcriptional activity has been reported for decades, but the mechanistic bases remain elusive (Gilbert 2002; Hiratani et al. 2009; Rivera-Mulia and Gilbert 2016b; Vouzas and Gilbert 2023). One limitation is the lack of methods enabling the simultaneous measurement of RT and gene expression from the same samples. Although multiple RT profiling methods exist at the ensemble population level (Marchal et al. 2018; Hulke et al. 2020; Rivera-Mulia et al. 2022; Wheeler et al. 2025), as well as at the single-cell level (Dileep and Gilbert 2018; Miura et al. 2020; Bartlett et al. 2022; Gnan et al. 2022; Massey and Koren 2022), links to gene regulation are still being explored by transcriptome analyses on separate sample batches, hindering the complex coregulation of RT and gene expression.
The rapid development of multiomic methods provided tools for the assessment of cell identity and cell-to-cell heterogeneity. Multiomic, single-cell methods also inform on distinct aspects of the epigenome in parallel with gene expression (Cao et al. 2018; Zhu et al. 2019; Flynn et al. 2023). Approaches that map features of 3D genome architecture and transcriptomes have been developed (Cao et al. 2018; Chen et al. 2019; Liu et al. 2023). However, to date, there is no robust method for the joint mapping of RT and gene expression. In addition, distinct challenges remain to align 3D genome features to gene regulation due to data sparsity, and compartment identification is feasible only with data aggregation (Zhang et al. 2022). Here, we developed PARTAGE, a multiomic approach for the simultaneous profiling of RT and gene expression. This approach is based on sample preparation under conditions that enable the preservation of both nucleic acids, enabling joint profiling of RT and transcriptome programs. Because this method is performed on asynchronous cultures of cells maintained in regular media conditions, it eliminates the need for cell cycle synchronization, allowing a more accurate measurement of gene regulation. Moreover, the precise collection of cell populations across distinct stages of the cell cycle allows PARTAGE to also provide accurate measurements of CNV. Overall, PARTAGE is a robust approach for the simultaneous analysis of CNV, RT, and gene expression.
Results
Overview of PARTAGE experimental design
PARTAGE is a multiomics method designed to accurately map CNV, RT, and transcriptome programs from the same samples. It is based on the sample collection for cell cycle analysis under conditions that preserve both nucleic acids. First, cells are labeled with the nucleotide analog BrdU to label nascent DNA and enable downstream purification by immunoprecipitation; then, single-cell suspensions are obtained and intact nuclei purified (Fig. 1). To enable accurate cell cycle analyses and cell sorting into specific populations while preserving both nucleic acids, intact nuclei are stained with cell-permeant DNA dyes. Specifically, we exploited cell-permeant DyeCycle dyes (Invitrogen), which we previously validated for Repli-seq sample preparation (Meyer-Nava et al. 2023). Then, cell cycle analyses are performed based on DNA content, and fractions of nuclei at distinct stages of the cell cycle are collected by fluorescence-activated cell sorting (FACS). Next, genomic DNA and nuclear RNA are copurified using silica-based strategies. Genomic DNA is processed following the optimized Repli-seq method, consisting in enzymatic fragmentation, immunoprecipitation with antibodies against BrdU, and library preparation (Rivera-Mulia et al. 2022). Nuclear RNA is also processed by standard RNA-seq. Because PARTAGE enables the isolation of a pure population of nuclei in G1, an accurate CNV mapping can be performed. DNA isolated from S-phase fractions is exploited to measure RT, and the RNA enables accurate detection of transcriptome control during the cell cycle (Fig. 1).

PARTAGE: Parallel analysis of RT and gene expression. Multiomics approach for the simultaneous profiling of CNV, RT, and transcriptome programs. Target cells are BrdU-labeled (1), dissociated (2), and intact nuclei isolated (3). Then, DNA was stained with cell-permeant dyes (4) and cell cycle analyzed by flow cytometry (5). Nuclei at specific cell cycle stages were collected for nucleic acid copurification (6), and optimized Repli-seq (7) and total RNA-seq (8) were performed. Finally, data analysis and visualization were performed to generate CNV, RT, and gene expression profiles (9).
PARTAGE sample preparation preserves DNA and RNA integrity
To test PARTAGE, we exploited the HepG2 human cell line, a reference cell line used at the ENCODE (The ENCODE Project Consortium et al. 2020) and Roadmap Epigenomics (Roadmap Epigenomics Consortium et al. 2015) consortia with extensive omics data publicly available. Cells were grown up to 60% confluence to avoid contact inhibition, which halts normal cell cycle proliferation, then, dissociated into a single-cell suspension, counted, and assessed for viability (Fig. 2A; Supplemental Fig. S1). Then, intact nuclei were isolated by detergent lysis (Fig. 2B; Supplemental Fig. S1) and DNA stained for cell cycle analysis (Fig. 2C) as previously described (Meyer-Nava et al. 2023). Purified nuclei were analyzed by flow cytometry, and the cell cycle was estimated based on DNA content (Vybrant Violet DyeCycle intensity). Flow cytometry gating strategies, and identification of cell cycle fractions are shown in Figure 2D. Populations of 20 K nuclei for each fraction derived from G1, S-phase (divided into four fractions), and G2/M were sorted into multiwell plates containing a nucleic acid stabilization solution to preserve integrity. Then, high-quality total genomic DNA and RNA were copurified using silica-based column methods. PARTAGE sample preparation generated nucleic acid samples of high quality and integrity adequate for downstream processing by optimized Repli-seq and total nuclear RNA-seq (Fig. 2E–J; Supplemental Figs. S2, S3).
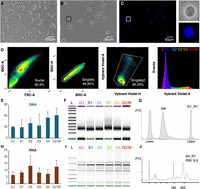
PARTAGE sample preparation. (A) HepG2 cells in culture. (B) Purified intact nuclei. (C) Nuclei staining with Vybrant DyeCycle Violet for accurate DNA content estimation. Magnification of a single nucleus is shown. (D) Flow cytometry and gating strategy. Single nuclei were identified, and gates were established to purify cell populations across the cell cycle: G1, S1, S2, S3, S4, G2/M. Twenty thousand nuclei were collected per fraction, and three independent biological replicates were processed. (E) DNA yields (total ng) obtained from each sorted population. (F) Integrity analysis and size distribution of purified and fragmented genomic DNA. (G) Electropherogram of purified and fragmented genomic DNA. (H) RNA yields copurified with the DNA (total ng) from the sorted cell populations. (I) RNA integrity analysis. (J) Exemplary electropherogram of purified RNA demonstrating RNA integrity (RIN = 0.90).
PARTAGE simultaneously maps CNV, RT, and transcriptomes
To evaluate PARTAGE's capacity to capture the multimodal genomic signals from the same samples, we analyzed HepG2 cells and compared the results against those generated using standard Repli-seq and RNA-seq methods (Fig. 3). Standard Repli-seq was performed on HepG2 nuclei as previously described (Rivera-Mulia et al. 2022). For standard RNA-seq, RNA was isolated from 0.5 million cells, and libraries were prepared for stranded mRNA sequencing. These standard analyses confirmed the close alignment of RT programs with genome compartmentalization as measured by Hi-C, as well as enrichment of active gene expression in active-early replicating genomic compartments (Fig. 3A). However, these data sets were generated independently from separate batches of samples processed by the standard methods of Repli-seq, Hi-C, and RNA-seq (Fig. 3A). Thus, we performed PARTAGE by processing the purified genomic DNA using the optimized Repli-seq method and the RNA using an ultralow input library preparation pipeline. PARTAGE allowed us to simultaneously map CNV, RT, and gene expression from the same sample (Fig. 3B). First, whole-genome sequencing of the cell populations isolated from G1 enabled the accurate mapping of CNV and the detection of multiple amplifications in the HepG2 genome (Fig. 3B). Next, RT was mapped using the multifraction Repli-seq method (Hansen et al. 2010; Zhao et al. 2020). Standard Repli-seq involves the sorting of early and late fractions of S-phase cells/nuclei, and the generation of binary profiles of early versus late replicating values across the genome. In contrast, multifraction Repli-seq involves cell sorting of multiple fractions across the entire S-phase to increase resolution and fine-scale detection of genomic features of the replication domains (Zhao et al. 2020). Thus, PARTAGE multifraction Repli-seq was performed and data visualized as heat maps of normalized sequencing signals per genomic bin. We found that the patterns of early/late replication reproduced the RT timing data obtained with standard Repli-seq (Fig. 3B). Finally, PARTAGE total-nuclear RNA-seq reproduced the transcriptome of the HepG2 cells obtained from separate RNA-seq analysis (Fig. 3). To display global trends in gene expression, we displayed exemplary patterns in three distinct chromosomal regions. To confirm cross-protocol consistency in transcriptomes, we displayed zoomed-in tracks focused on hepatic-specific and housekeeping genes (Fig. 3B).
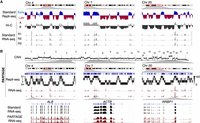
PARTAGE captures CNV, RT, and transcriptomes from the same samples. (A) Standard omics analysis generated from HepG2 cells. Three exemplary chromosomal regions are shown. Standard Repli-seq, chromosome confirmation capture (Hi-C), and RNA-seq were performed from separate sample batches of HepG2 cells. (B) PARTAGE simultaneous mapping of CNV, RT, and transcriptome from HepG2 nuclei. CNV was calculated from the sequencing reads obtained from G1 cell populations. Sequencing reads normalized for depth and per 1-Mb bin are shown. PARTAGE Repli-seq was performed using multifraction RT analysis. Heat map signals of G1-normalized reads are shown per fraction (S1–S4). RNA-seq coverage tracks are shown for the same chromosomal regions. Bottom tracks show a comparison between standard and PARTAGE RNA-seq signals zoomed in on hepatic-specific (ALB) and housekeeping genes (ACTB and RRBP1).
To validate reproducibility and accuracy, we performed in-depth comparisons of PARTAGE multiomic signals against those generated by the separate standard methods. First, because the HepG2 genome contains well-characterized amplifications and deletions, and public CNV data is available; we benchmarked PARTAGE-derived CNVs against ENCODE whole-genome sequencing (WGS) CNV data (The ENCODE Project Consortium 2012). We applied CNVpytor (Suvakov et al. 2021) to the high-coverage ENCODE WGS dataset (60×) to define CNV segments, identifying 25 CNV events with resolution of 1-Mb bins. We then applied the same CNVpytor pipeline to three independent PARTAGE libraries (∼0.5× coverage each) and compared the CNVs detected in PARTAGE to those identified in the ENCODE reference (Fig. 4A). Across the three PARTAGE replicates, we recovered between 92% and 100% of the ENCODE CNV events (Fig. 4B). The statistical significance of the overlap between ENCODE and PARTAGE CNVs was assessed using ProOvErlap (Gualandi et al. 2025), confirming that the concordance is substantially greater than expected by chance (Fig. 4B). Analysis at higher resolution (200-kb bins) resulted in 75 CNVs in the ENCODE reference data, with PARTAGE detecting >85% of these events (Supplemental Fig. S4). Thus, all major genomic amplifications observed in the ENCODE data can be detected using PARTAGE.
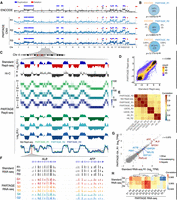
PARTAGE accurately profiles CNV, RT, and transcriptomes. (A) Comparative CNV analysis. CNV profiles derived from ENCODE high-coverage WGS (60× coverage; top track) and from PARTAGE (∼0.5× coverage; three independent replicates) using CNVpytor (see Methods). CNV segments identified in the ENCODE data set (n = 25 events) are shown as duplications (blue) and deletions (red) and were used as a reference. PARTAGE CNV calls are also shown. (B) Overlap of CNV calls between ENCODE and PARTAGE replicates was quantified. PARTAGE correctly detected 92%–100% of the ENCODE CNV events across replicates. The statistical significance of the overlap between ENCODE and PARTAGE CNVs was assessed using ProOvErlap (see Methods). (C) Comparative analysis of standard and PARTAGE Repli-seq data. Standard Repli-seq is shown as log2(E/L) ratio signals. Multifraction Repli-seq is shown in distinct heat map colors per replicate. Signals are normalized counts against G1 per 20-kb bins. Collapsed E/L log2 ratios were calculated from PARTAGE multifraction Repli-seq RT to obtain RT profiles directly comparable to standard E/L Repli-seq. (D) Scatterplot of PARTAGE (y-axis) versus standard (x-axis) Repli-seq data. (E) Genome-wide correlation between a PARTAGE and standard Repli-seq. Genome-wide RT data in 20-kb bins were used. RT from stem cells (H9) and cancer cells (U2OS) were included as a comparison. (F) PARTAGE and standard RNA-seq coverage tracks at the albumin locus showing expression patterns of the hepatic genes ALB and AFP. Raw coverage normalized for sequencing depth was used. (G) Gene-by-gene scatterplots on batch-corrected expression values (log2TPM). Standard RNA-seq (x-axis) versus PARTAGE RNA-seq (y-axis) is shown. Housekeeping genes (blue) and hepatic markers (red) are highlighted; all other genes are gray. (H) Genome-wide cross-protocol concordance between a PARTAGE and standard RNA-seq transcriptomes. Pearson's correlation among all samples, computed on batch-corrected log2TPM values, and clustered by Euclidean distance, is shown. Note that the correlation map is confined to a narrow range near 1.0 reflecting strong similarity across transcriptomes.
Next, we compared PARTAGE RT programs versus standard Repli-seq, and visual inspection confirmed that the multifraction PARTAGE Repli-seq signals faithfully recapitulated the patterns of the standard Repli-seq (Fig. 4C). To enable direct comparison of these different Repli-seq methods, we collapsed the PARTAGE multifraction Repli-seq into log2(Early/Late) ratios. Visual inspection overlaying the RT profiles from the distinct methods and replicates demonstrated the consistency between PARTAGE and standard Repli-seq (Fig. 4C). Moreover, cross-protocol scatterplot comparisons confirmed close concordance of RT values across the genome (Fig. 4D). Furthermore, pairwise/genome-wide correlation analysis confirmed high consistency between PARTAGE and standard Repli-seq RT profiles with r ≥ 0.95 (Fig. 4D,E). Finally, we also validated the concordance between PARTAGE and standard RNA-seq pipelines to ensure that accurate transcriptome profiling was achieved in this method. Visualization of RNA-seq raw coverage tracks (normalized to sequencing depth) demonstrated that PARTAGE recapitulates the transcriptome data obtained from standard methods at specific genes (Figs. 3B, 4F). Because transcriptomes were obtained from distinct cell numbers (0.5 million cells for standard RNA-seq vs. 20,000 nuclei in PARTAGE RNA-seq) and using distinct library preparation pipelines (TruSeq stranded RNA-seq vs. Takara ultra-low input SMARTer Stranded Total RNA-seq), we scaled and normalized the data for proper comparison (Supplemental Fig. S5). Then, log2TPM RNA-seq values were median-centered and batch-corrected (Johnson et al. 2007) to enable proper comparisons. We found strong gene-wise concordance between PARTAGE and standard RNA-seq, and replicate-level scatterplots of gene expression values showed near-isometric agreement, with housekeeping and hepatic-specific marker genes aligning closely (Fig. 4G; Supplemental Fig. S6). Moreover, pairwise Pearson's correlations resulted in a very narrow, high range close to 1.0, indicating a strong agreement between standard RNA-seq and PARTAGE libraries (Fig. 4H). Overall, these results validate the simultaneous and precise mapping of CNV, RT, and gene expression patterns using PARTAGE.
Cell cycle dynamics in RT and gene expression
A long-standing correlation has been observed between early replication and active gene expression (Hiratani et al. 2010; Rivera-Mulia et al. 2015, 2019a; Rivera-Mulia and Gilbert 2016b; Vouzas and Gilbert 2023). However, previous studies were based on RT and gene expression data obtained independently from distinct batches of samples generated in parallel. Thus, we exploited PARTAGE to explore how closely early replication and gene expression are linked, taking advantage of the RT and transcriptome measurements from the same sample that reduces technical noise and batch effects. Genomic tracks of PARTAGE RT profiles and transcriptomic coverage patterns confirmed that active transcription is enriched at early replication chromosomal regions (Fig. 5A). To measure the strength of this correlation, we calculated the percentage of expressed genes in relation to their RT. To do so, we identified all expressed genes and plotted them according to their RT (collapsed E/L ratios). Our results recapitulate the well-established correlation between early replication and active gene expression, which further validates this multiomic method (Fig. 5B). Moreover, a regression analysis was performed and showed a strong correlation between RT and transcriptional activity (Fig. 5B). These results confirm the coregulation between RT and active transcription with the advantage of PARTAGE measuring these modalities within the same sample and cell-cycle–defined populations, minimizing batch and compositional differences.
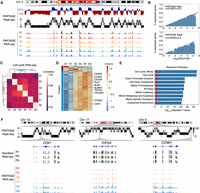
Cell cycle regulation of DNA replication and gene expression. (A) Exemplary genomic locus from Chromosome 4 showing enrichment of gene expression at the early replicating regions. Collapsed E/L ratios from PARTAGE Repli-seq data, PARTAGE multifraction signals, and coverage RNA-seq tracks are shown. (B) Genome-wide correlation between replication timing and gene expression. Expressed genes were identified (threshold of ≥2 log2TPM) and sorted by their RT. The y-axis represents the percentage of expressed genes within each bin. Logistic regression (inner line) and 95% confidence intervals (outer lines) display the correlation strength. Two PARTAGE replicates are shown. (C) Genome-wide correlation of transcriptome programs across the cell cycle. Average TPMs were calculated per cell cycle stage after batch correction (ComBat 2.0), and gene expression values ≥ 0.5 log2TPM in at least three cell cycle stages were used. Pairwise Pearson's correlation was performed with hierarchical clustering based on correlation distance (1 − r). Note that the correlation scale is confined to a narrow range near 1.0 highlighting minor differences between cell cycle stages. (D) Heat map and clustering analysis of variable genes during the cell cycle. Genes are represented by row-wise z-scored log2(TPM) values and clustered by k-means (k = 2). Only genes expressed in ≥2 cell cycle stages and with ≥4× Δ log2TPM were considered. (E) Reactome ontology analysis of the major cluster (blue) is shown. (F) Exemplary chromosome regions harboring variable genes that are induced during cell cycle progression (CDK1, TOP2A, and CCNB1).
PARTAGE also enables the profiling of cell cycle phase-resolved transcriptome profiling, providing a direct measurement of dynamic regulation of gene expression patterns across the cell cycle. We first determined the degree of similarity between the transcriptomes obtained from the distinct stages of the cell cycle. After removing nonexpressed genes, applying batch correction, and performing pairwise correlation analysis, we observed strong correlations (r ≥ 0.95) within a narrow, high range between all cell cycle stages (Fig. 5C), indicating technical reproducibility of our methods and global stability of the HepG2 transcriptome across the cell cycle in unperturbed conditions. To identify transcriptional changes during the cell cycle, we identified genes expressed in a minimum of two samples and filtered those with ≥4× change across cell cycle stages. Under these criteria, we detected 83 protein-coding genes changing expression patterns during the cell cycle. Clustering analysis revealed a group of coregulated genes that increase expression during cell cycle progression, containing cell cycle regulators (Fig. 5D). As expected, ontology analysis confirmed that these variant genes are associated with processes regulating the cell cycle progression (Fig. 5E; Supplemental Fig. S7). Notably, the number of cell cycle regulated genes is substantially smaller than previous reports based on synchronized cells. Exemplary PARTAGE tracks of cell cycle-regulated genes are shown in Figure 5F. Overall, these analyses illustrate how PARTAGE provides an integrated framework with the potential to detect CNV, RT, and gene expression regulation during the cell cycle from the same experimental samples.
Discussion
Analyses of genome organization and function have unveiled the coregulation between RT and gene expression (Rivera-Mulia et al. 2015, 2019a; Dileep et al. 2019; Sima et al. 2019). Recent studies of early mammalian embryogenesis have begun to elucidate when RT emerges during development and how this relates to chromatin states and transcriptional activity (Halliwell et al. 2024; Nakatani et al. 2024, 2025; Takahashi et al. 2024; Xu et al. 2024), but they reach differing conclusions regarding the presence and consolidation of RT programs in the zygote and earliest cell divisions. These discrepancies highlight the complexity of RT regulation and the need for technological advances to jointly interrogate RT and gene expression. Notably, previous studies rely on separate profiling of these genomic properties, which might obscure the complex coregulation between the epigenome and transcriptome. Multiomic approaches can help to address these questions; however, rigorous testing is necessary to avoid artifacts in RT mapping (Rivera-Mulia 2025). In this work, we present PARTAGE, a multiomics approach that enables the parallel analysis of RT and gene expression, as well as CNV, from the same samples.
The major limitation for the joint mapping of transcriptomes and RT was that methods for cell cycle analysis by FACS relied on methods that do not preserve the RNA. Standard cell cycle analysis by flow cytometry depends on the precise DNA content measurement using propidium iodide (PI) or diamidino-2-phenylindole (DAPI) staining. PI staining requires RNase treatment, whereas DAPI staining requires permeabilization that leads to RNA loss. To overcome this challenge, we exploited nontoxic, cell-permeant dyes with narrow emission spectra (DyeCycle dyes, Invitrogen). These dyes enable the stoichiometric DNA staining for accurate cell cycle analysis based in DNA content (Bradford et al. 2006) and can be used for Repli-seq sample preparation (Meyer-Nava et al. 2023). Because these dyes are not RNA sensitive, they allow precise DNA content measurement and S-phase gating of nuclei without the need for RNase treatment, thereby preserving nuclear RNA for downstream sequencing. Here, we processed isolated nuclei in order to enrich for nuclear RNA, which is closer to ongoing transcriptional activity and less influenced by cytoplasmic mRNA stability and export, as supported by analyses of intronic (nuclear) versus exonic reads in RNA-seq (Zaghlool et al. 2013; Gaidatzis et al. 2015; Bakken et al. 2018; Oh et al. 2022; Engström et al. 2025). In principle, our PARTAGE workflow can be adapted to whole-cell preparations to obtain steady-state RNA-seq from the same sorted populations, although specific library preparations would need validation. We demonstrated that PARTAGE sample preparation allows the copurification of DNA and RNA of high quality and in sufficient amounts for parallel processing for Repli-seq and RNA-seq. In fact, we found increasing yields of DNA that reach 2× in G2/M as compared to G1, highlighting the accuracy of this method to collect cell populations throughout the cell cycle (Fig. 2).
Conventional methods for CNV detection rely on deep sequencing (≥30× coverage) to accurately identify variations by ensemble methods. Here, we demonstrated that PARTAGE can accurately detect major amplifications in the HepG2 genome with only ≤0.5× coverage. Despite the shallow sequencing (∼8 M single-end 100-bp reads), PARTAGE reproduced the large CNV detected by the ENCODE mapping (Fig. 4). This was achieved by the focused analysis of purified nuclei in G1-phase, which ensures the analysis of uniform diploid populations by removing S-phase and G2/M-phase nuclei, and reducing cell cycle-associated artifacts in CNV detection. Thus, PARTAGE enables the accurate detection of CNV, providing additional tools to better understand genome instability in development, disease, and cancer states. Importantly, PARTAGE's capacity to detect CNVs with this sequencing depth makes this approach cost-effective and scalable for clonal tracking, genome integrity screenings, and cancer alterations studies, with the addition of joint profiling of RT and gene regulation.
For Repli-seq analysis in PARTAGE, we took advantage of our optimized Repli-seq workflow, which reduced DNA loss and technical noise by minimizing transfer/handling steps, and increasing yields by 3× (Rivera-Mulia et al. 2022). We also applied the multifraction Repli-seq approach and exploited the data from the G1-purified nuclei to normalize signals and generate accurate heat maps of RT. Although standard E/L Repli-seq is also compatible with our PARTAGE workflow, it is sufficient to accurately detect RT and is more cost-effective; we selected multifraction Repli-seq to enable a more accurate detection of cell cycle regulation when integrating the data with the matching transcriptomes. The combined implementation of multifraction analyses and optimized library preparation strategies allowed us to generate highly reproducible RT data across replicates that recapitulate the RT data obtained from the standard method.
We also evaluated the robustness of the RNA-seq data generated in PARTAGE and found high reproducibility across replicates. Moreover, although PARTAGE RNA-seq was performed on low-input samples (20 K nuclei), it faithfully recapitulated the transcriptome obtained with standard RNA-seq from 0.5 M cells (Fig. 4). Moreover, PARTAGE sample preparation allows us to collect nuclei at distinct phases of the cell cycle. Thus, we asked whether PARTAGE could detect transcriptional changes across the cell cycle. Notably, PARTAGE profiles RNA in native conditions without the need of forced cell synchronization. Conventional studies of cell cycle-regulated transcriptional activity depend on synchronization strategies through serum starvation, mitotic shake-off, or using drugs that activate checkpoint mechanisms. However, these approaches trigger cellular stress responses, DNA damage pathways, and/or metabolic reprogramming that alter normal transcriptional regulation (Cooper 2003; Karlsson et al. 2017; Mahdessian et al. 2021). Thus, PARTAGE is a valuable alternative that does not require any external perturbation and enables a more accurate assessment of the cell cycle regulation of the transcriptome. Indeed, the simultaneous profiling of RT and transcriptomes in PARTAGE facilitated the integration of multimodal signals under physiological conditions and confirmed the strong enrichment of actively transcribed genes in early replicating regions (Fig. 5). Moreover, PARTAGE identified a subset of genes that increase transcriptional levels 4× as the cells progress in the cell cycle and confirmed that these genes are indeed involved in cell cycle regulation. The relatively small number of cell cycle-regulated genes detected in PARTAGE can be due to different factors: (1) the reduced synchronization-induced artifacts by processing cells in native conditions; (2) the processing of nuclei instead of whole cells to reduce confounding effects from cytoplasmic post-transcriptional regulation (e.g., mRNA stability and export); and (3) the use of stringent thresholds to identify variable genes.
Important open questions, such as whether RT changes precede or follow chromatin and transcriptome programs, can be addressed by jointly profiling these genomic properties across defined developmental trajectories. In its current implementation, PARTAGE provides a robust multiomic method that is directly applicable to differentiation systems of pluripotent stem cells. A limitation of PARTAGE is that it is an ensemble method, and further optimization and adaptations to low-input and single-cell formats will be necessary to better evaluate the temporal emergence of RT relative to chromatin and the transcriptome, especially during early embryogenesis.
Overall, PARTAGE is a robust method that provides accurate mapping of CNV, RT, and transcriptomes. We validated PARTAGE as a multiomic approach capable of generating highly accurate data consistent with those produced using the gold-standard separate methods (Repli-seq, RNA-seq, and CNV mapping). This high consistency was quantitatively demonstrated by the near complete overlap pf CNV calls, by strong genome-wide correlation (r ≥ 0.95) between PARTAGE and standard Repli-seq RT profiles, and by the high concordance (r ≥ 0.945) found between PARTAGE and standard transcriptome profiling, despite using significantly lower input material. This approach reduces the required sample input as compared to the corresponding separate approaches, minimizes heterogeneity and technical noise between batches of samples, and enables a direct assessment and integration of multimodal signals. Moreover, PARTAGE enables the characterization of cell cycle regulation of genome function in unperturbed conditions. PARTAGE is well suited for investigating coordinated changes in RT, genome stability, and transcriptional activity in dynamic system such as differentiation trajectories of pluripotent stem cells. In disease contexts, such as cancer and other conditions characterized by genome instability, PARTAGE provides a practical approach to concurrently monitor RT alterations, CNV burden, and transcriptional dysregulation, thereby enabling more comprehensive assessments of genome integrity and its functional implications.
Methods
Cell culture
The HepG2 cell line was obtained from ATCC and expanded according to the supplier's protocol. Briefly, cells were maintained in Eagle's Minimum Essential Medium (ATCC, #30-2003) supplemented with 10% FBS (Corning. #35011CV). Cell cultures up to 60% confluency were treated with BrdU (Sigma-Aldrich, #B5002) at a final concentration of 100 µM at 37°C for 2 h and processed for nuclei preparation for PARTAGE analyses.
Intact nuclei preparation for PARTAGE
BrdU-labeled HepG2 cells were dissociated into single-cell suspensions by treatment with trypsin-EDTA (Life Technologies, #A011105-01), and intact nuclei were isolated as previously described (Meyer-Nava et al. 2023). Briefly, cell pellets were resuspended in 1 mL of DMEM/F-12 50/50 (1×), counted, and aliquoted into batches of 2.0 × 106 cells. Then, cells were triturated in a lysis buffer: 0.025% IGEPAL (Millipore Sigma, #542334) in 10 mM TrisHCl, 10 mM NaCl, 3 mM MgCl2, and 1× PBS (Thermo Fisher Scientific, #20012027). Lysed cells were washed three times and resuspended in 1 mL PBS/1% FBS. Finally, nuclei were stained with Vybrant DyeCycle Violet Stain (Thermo Fisher Scientific, #V35003) at 1.5 µL per 1 × 10−6 cells for 30 min at 37°C and filtered with a 36-µm nylon mesh (Fisher Scientific, #NC05156770).
Cell cycle analysis and cell sorting by flow cytometry
Stained nuclei were processed in a SONY SH800 cell sorter. Single nuclei were identified and cell cycle stages determined based on DNA content (Vybrant DyeCycle Violet intensity). Twenty thousand cells were collected per fraction of G1, S1, S2, S3, S4, and G2M cell cycle stages and sorted directly into wells of a 96-well plate (Axygen, #14-223-345) containing 400 µL of DNA/RNA Shield Buffer (Zymo, #R1100). If not processed immediately, the plates were sealed with an adhesive film and stored at −80°C.
Nucleic acid extraction
For DNA and RNA copurification, the samples were thawed at room temperature and nucleic acids isolated using the Quick DNA/RNA MicroPrep Plus kit (Zymo, #D7005) according to the manufacturer's instructions. Purified DNA and RNA were quantified using Qubit kits (Invitrogen, #Q32852 and #Q32851).
CNV library preparation and whole-genome sequencing
For CNV analysis, we used the genomic DNA purified from G1 cells and performed WGS. Briefly, purified DNA from G1 sorted cells was directly processed using the KAPA HyperPlus library preparation kit (Roche, #07958978001) according to the manufacturer's instructions. Enzymatic fragmentation was performed at 37°C for 30 min, followed by end-repair and ligation of KAPA dual-indexed adapters (Roche, #08278555702). Libraries were purified using the Zymo DNA Clean & Concentrator-5 kit, amplified using the KAPA HiFi HotStart from the KAPA HyperPlus kit according to the manufacturer's instructions, and purified using AMPure XP beads. Low-coverage whole-genome sequencing libraries were sequenced at the University of Minnesota Genomics Center (UMGC), on the Illumina NextSeq P2 Illumina platform with 100 bp of single-end sequencing and ∼0.5× genome coverage.
Repli-seq
DNA samples were processed using the optimized Repli-seq protocol as previously described (Rivera-Mulia et al. 2022). Briefly, purified DNA from sorted cells was directly processed using the KAPA HyperPlus library preparation kit (Roche, #07958978001) according to the manufacturer's instructions. Enzymatic fragmentation was performed at 37°C for 30 min, followed by end-repair and ligation of KAPA dual-indexed adapters (Roche, #08278555702). Libraries were purified using the Zymo DNA Clean & Concentrator-5 kit and processed for immunoprecipitation using antibodies against BrdU. Immunoprecipitated libraries were amplified using the KAPA HiFi HotStart from the KAPA HyperPlus kit according to the manufacturer's instructions and purified using AMPure XP beads. DNA concentration and size distribution were evaluated by Qubit quantification (Invitrogen, #Q32851), and size estimation was performed on TapeStation High Sensitivity DNA ScreenTape (Agilent, #5067–5592). Repli-seq libraries were pooled at a final concentration of 10 NM and sequenced at the University of Minnesota Genomics Center (UMGC), on the Illumina NextSeq P2 Illumina platform at ∼8 M reads per library and 100 bp of single-end sequencing.
Standard RNA-seq
RNA samples were isolated from 0.5 million HepG2 cells using the RNeasy Plus kit (Qiagen, #74004), and libraries were prepared with the TruSeq Stranded mRNA Library Prep (Illumina) according to the manufacturer's instructions. Standard RNA-seq libraries were sequenced on the Illumina NovaSeq platform at ∼20 M reads per library and 150 bp of pair-end sequencing.
PARTAGE RNA-seq
RNA samples were processed using the SMARTer Stranded Total RNA-seq Pico for ultralow inputs (Takara, #634411). RNA concentration and size distribution were evaluated by Qubit quantification (Invitrogen, #Q32852) and size measured using the Bioanalyzer RNA 6000 Nano kit (Agilent, #5067–1511). RNA-seq libraries were pooled at a final concentration of 10 nM and sequenced at the University of Minnesota Genomics Center (UMGC), on the Illumina NovaSeqX platform at ∼40 M reads per library and 150 bp of pair-end sequencing.
Data analyses and visualization
Data analyses were performed with resources at the Minnesota Supercomputing Institute (MSI).
PARTAGE CNV data
Sequencing reads obtained from G1 fractions were trimmed for adapter sequences using cutadapt 4.9 (Martin 2011) and aligned to the reference genome with Burrows-Wheeler Aligner 0.7.18 (Li and Durbin 2010). Sequence Alignment Map (SAM) files were converted into Binary Alignment Maps (BAM) with SAMtools 1.18 (Danecek et al. 2021). Quality filter and removal of PCR duplicates were performed with SAMtools using the view and markdup functions, respectively. Mapped, filtered, and deduplicated reads were counted into genomic windows of 1 Mb using the intersect function from BEDTools 2.31.1 (Quinlan and Hall 2010). CNV tracks were visualized using the Integrative Genomics Viewer (IGV) browser (Robinson et al. 2023).
CNV calling
CNVs were called using CNVpytor (Suvakov et al. 2021). Briefly, aligned whole-genome sequencing reads from ENCODE (60× coverage) and from PARTAGE libraries (∼0.5× coverage) were processed with CNVpytor following the developers’ recommendations. Read-depth histograms were generated, normalized, and used to infer copy-number states, and CNV segments were defined using CNVpytor's built-in segmentation and significance thresholds. CNV calling for PARTAGE and ENCODE data sets was performed with the same pipeline to enable direct comparison.
CNV overlap analysis
To assess the concordance between CNVs detected in PARTAGE and those detected in the high-coverage ENCODE reference, we used ProOvErlap (Gualandi et al. 2025) to test the statistical significance of overlap between CNV segments. CNVs identified by CNVpytor in ENCODE were used as the reference set, and CNVs from each PARTAGE replicate were used as query sets. ProOvErlap was used to calculate the expected overlap under a random model and to obtain P-values for the observed overlap between reference and query CNVs.
Standard Repli-seq data
Sequencing reads were processed as for CNV (trimmed for adapter sequences, aligned with Burrows-Wheeler Aligner 0.7.18, and PCR duplicates removed). Mapped, filtered, and deduplicated reads were counted into genomic windows of distinct size (from 5- to 100-kb) using the intersect function from BEDTools 2.31.1 (Quinlan and Hall 2010). Downstream processing, including quantile normalization and LOESS smoothing, and data visualization, was performed in R (R Core Team 2017) as previously described (Marchal et al. 2018; Rivera-Mulia et al. 2022) and IGV browser (Robinson et al. 2023).
PARTAGE Repli-seq data
Binned RPKM data for each sample was obtained as above and further processed into 50-kb bins. For each set of samples, the log2([sample/G1_sample] + 1) was calculated. Data were scaled linearly from 0 to 100 for visualization. Bedgraphs were generated and displayed using the IGV browser (Robinson et al. 2023).
For collapsed Repli-seq from PARTAGE data, FASTQ files were concatenated for S1 and S2 as the early sample and S3 and S4 as the late sample. Data were then processed as above, including loess smoothing and quantile normalization for comparison to standard Repli-seq data.
RNA-seq data
Raw read counts were trimmed using Trimmomatic 0.39 (Bolger et al. 2014), aligned using HISAT2 2.1.0 (Kim et al. 2015), and alignment filtering and counts using SAMtools 1.18. Raw counts were converted to TPM counts by calculating reads per kilobase (RPK) and scaling by the sum of RPK/1,000,000. The log2(TPM + 1) was used for downstream analyses. For analysis of expression patterns, samples were median-centered, low-expressed genes (TPM < 1) and all noncoding genes were removed, and batch correction was calculated using ComBat 2.0 (Johnson et al. 2007). Mean values were calculated across samples for each time point. Only genes expressed in at least two samples and with a fourfold TPM change were considered. k-means clustering was performed on the remaining genes with a cluster count of two. Gene set enrichment was performed using gseapy (Fang et al. 2023) and Enrichr (Chen et al. 2013) on the following terms: “ChEA_2022”, “ENCODE_and_ChEA_Consensus_TFs_from_ChIP-X”, “GO_Biological_Process_2025”, “GO_Cellular_Component_2025”, “GO_Molecular_Function_2025”, “Reactome_Pathways_2024”. Significantly enriched terms were filtered by gene count (>1) and adjusted Benjamini–Hochberg P-value <0.05. For each category, the top 10 most significant gene categories were plotted using seaborn.
For comparisons of RNA and RT data, plots were created comparing the number of bins with expressed genes compared to their corresponding replication timing values. For each sample, genome wide RT data were sorted by value and fit into 100 bins. For each bin, gene expression values were binarized into 0 sec and 1 sec based on log2(TPM) expression > 1. A linear regression model was fit to the data using statsmodels (Seabold and Perktold 2010).
Data access
All raw sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1312882. Processed data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE307022. Open-source code used in the analysis of PARTAGE data and generation of the figures is available at GitHub (https://github.com/Rivera-Mulia-Lab/PARTAGE) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by National Institutes of Health/National Institute of General Medical Sciences grants R35GM137950, R35GM137950-02S1, and R35GM137950-04S1 to J.C.R-M.; and by Regenerative Medicine Minnesota RMM-091621-DS-006 to J.C.R-M.; and institutional support from the University of Minnesota Medical School.
Author contributions: Conceptualization: J.C.R-M. Methodology: J.C.R-M. Investigation: L.S.S.M., Q.D., S.M-N., and J.C.R.-M. Formal analysis: Q.D. and J.C.R-M. Visualization: Q.D. and J.C.R-M. Data curation: Q.D. and J.C.R.-M. Writing–original draft: L.S.S.M., Q.D., and J.C.R-M. Writing–review and editing: L.S.S.M., Q.D., and J.C.R-M. Funding acquisition: J.C.R-M. Resources: J.C.R-M. Supervision: J.C.R-M. Project administration: J.C.R-M.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.281532.125.
-
Freely available online through the Genome Research Open Access option.
- Received October 6, 2025.
- Accepted March 2, 2026.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








