
Diverse evolutionary trajectories of mitocoding DNA in mammalian and avian nuclear genomes
- Yu-Chi Chen1,2,
- David L.J. Vendrami3,4,5,6,
- Maximilian L. Huber2,
- Luisa E.Y. Handel2,
- Christopher R. Cooney7,
- Joseph I. Hoffman3,4,5,6,8 and
- Toni I. Gossmann1,2,3,5
- 1Computational Systems Biology, Faculty of Biochemical and Chemical Engineering, TU Dortmund University, 44227 Dortmund, Germany;
- 2Department of Evolutionary Genetics, Faculty of Biology, Bielefeld University, 33615 Bielefeld, Germany;
- 3Department of Animal Behavior, Faculty of Biology, Bielefeld University, 33615 Bielefeld, Germany;
- 4Department of Evolutionary Population Genetics, Faculty of Biology, Bielefeld University, 33501 Bielefeld, Germany;
- 5Joint Institute for Individualization in a Changing Environment (JICE), Bielefeld University and University of Münster, 48149 Münster, Germany;
- 6Center for Biotechnology (CeBiTec), Faculty of Biology, Bielefeld University, 33615 Bielefeld, Germany;
- 7Ecology and Evolutionary Biology, School of Biosciences, University of Sheffield, Sheffield S10 2TN, United Kingdom;
- 8British Antarctic Survey, High Cross, Cambridge CB3 OET, United Kingdom
Abstract
Sporadically, genetic material that originates from an organelle genome integrates into the nuclear genome. However, it is unclear what processes maintain such integrations over evolutionary time. Recently, it was shown that nuclear DNA of mitochondrial origin (NUMT) may harbor genes with intact mitochondrial reading frames despite the fact that they are highly divergent from the host's mitochondrial genome. Two major hypotheses have been put forward to explain the existence of such mitocoding nuclear genes: (1) recent introgression from another species and (2) long-term selection. To investigate whether these intriguing possibilities play a role, we scanned the genomes of more than 1000 avian and mammalian species for NUMTs. We show that a subclass of divergent NUMTs harboring mitogenes with intact reading frames is widespread across mammals and birds. We also show that some of these NUMTs appear to be similar across species. In addition, we demonstrate that many mitochondrial-coding NUMTs exhibit signs of long-term selection. In a subset of these NUMT genes, we detected evolutionary signals consistent with adaptive evolution, including one human NUMT shared among seven ape species. These findings suggest that NUMT insertions may occasionally be functional.
A key question in evolutionary biology is how genetic novelties arise and contribute to adaptation (Sangster and Luksenburg 2021; Bomblies and Peichel 2022). Mutations are the ultimate source of genetic novelties that are subsequently subject to evolutionary forces such as selection and drift (Huang et al. 2016). There is compelling evidence that most mutations do not show signatures of positive selection on the molecular level but instead are subject to random genetic drift and purifying selection (Bank et al. 2014). As novel mutations may be insufficient in many cases to generate adaptive variation, alternative mechanisms may explain the generation and maintenance of novel beneficial genetic elements.
Mutations that have already been exposed to evolutionary forces may provide a means of generating adaptive variation. For example, the acquisition of novel, preadapted genetic elements can occur via the lateral transfer of DNA. There is considerable evidence for such exchanges of genetic material among bacteria and even in eukaryotes (Danchin 2016; Dunning et al. 2019). For example, the substantial exchange of genetic material between eukaryotic species occurs through hybridization and introgression (Gabaldón 2020; Setter et al. 2020; Popadin et al. 2022). Although it is known that an organism's genomic architecture is fundamental to whether hybridization/introgression occurs successfully, we have limited knowledge of the extent to which lateral gene transfer, hybridization, and introgression contribute to genomic diversification driven by the acquisition of adaptive elements across species.
An intriguing possibility for the acquisition of novel, genetically adapted elements is the transfer of organelle DNA (Popadin et al. 2022; Butenko et al. 2024). In many eukaryotic species, organelles such as mitochondria or plastids possess their own genomes. Mitochondrial DNA integration into the nuclear genome (NUMT) is a continuous and ongoing mutagenic process that occurs across different species and tissues (Ricchetti et al. 2004). After NUMTs were first observed in the domestic cat (Felis catus) (Lopez et al. 1994) and in humans (Hu and Thilly 1994; Lopez et al. 1997), they were then found in many different eukaryotic species from yeast (Ricchetti et al. 1999) to many other mammals (Tsuji et al. 2012; Calabrese et al. 2017; Gossmann et al. 2019; Biró et al. 2022; Uvizl et al. 2023) and birds (Nacer and do Amaral 2017; Liang et al. 2018; Lucas et al. 2022; Baltazar-Soares et al. 2023). NUMT insertion number, size, and sequence composition vary not only across species (Richly and Leister 2004; Hazkani-Covo 2022) but also within populations (Soto-Calderón et al. 2014; Lucas et al. 2022; Wei et al. 2022). In humans, ∼14% of individuals possess NUMTs that are present in <0.1% of the broader population, and some of these NUMTs have been shown to be passed on from parents to their offspring (Wei et al. 2022). This suggests that NUMT integration is a frequent mutagenic event that produces inheritable changes, potentially relevant to future evolution. The germline NUMT mutation rate in humans is estimated at 2.44 × 10−4 mutations per generation (Wei et al. 2022), illustrating that novel NUMT genetic variation is frequently and constantly being generated.
Although NUMTs are widespread in the animal kingdom, there is little evidence of functional integrations of mitochondrial DNA (Noutsos et al. 2007; Pozzi and Dowling 2019; Wei and Chinnery 2020). Instead, it is generally assumed they are exposed to genetic drift and will quickly accumulate novel disruptive mutations. In line with this, it is frequently observed that mitocoding genes integrated into the nuclear genome accumulate missense mutations that ultimately lead to a broken reading frame. However, if the integration is very recent, if the local mutation rate is very low, or if selection actively maintains the coding properties of the integration, the former mitochondrial coding genes may retain intact reading frames over longer periods of evolutionary time. Such NUMTs containing genes with intact mitochondrial reading frames can be considered coding NUMTs (cNUMTs).
Recently, in the genome of the Antarctic fur seal (Arctocephalus gazella), several cNUMTs were identified (Vendrami et al. 2022). The coding genes of three of the cNUMTs contained numerous silent mutations (i.e., showed substantial divergence at neutral sites) but very few amino acid–changing mutations relative to fur seal mitochondrial genes. The authors concluded that the signature of cNUMTs that are divergent (dcNUMTs) at synonymous sites was consistent with purifying selection and hinted at functionality and introgression. As the dcNUMTs observed in the Antarctic fur seal genome cannot be explained by a recent nuclear integration from its own mitochondrial genome, alternative scenarios of noncanonical integrations might explain the existence of dcNUMTs (Fig. 1). Currently, it is unclear how common and frequent dcNUMTs are across larger taxonomic groups and whether they harbor signatures of functionality and introgression. Hence, their potential role as adaptive elements of the genome has yet to be thoroughly investigated. In this study, we aimed to unravel the role of organelle DNA in creating novel and potentially adaptive variation in nuclear genomes. For this, we investigate (1) whether mitocoding DNA may stem from lateral gene transfer/introgression and (2) whether we find any evidence of selection on nuclear mitocoding DNA.
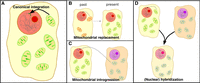
Potential sources of NUMTs. (A) Canonical integration through host species’ mitochondrial genome. (B) Integration of the host species’ mitochondrial genome stemming from mitochondrial replacement such as a consequence of hybridization (Javaheri Tehrani et al. 2021). (C) Integration of the mitochondrial DNA of another species, for example, through a retrovirus (Tsuji et al. 2012). (D) Integration of nuclear DNA from another species, for example, through nuclear hybrid genome formation (Popadin et al. 2022), which happens to carry a NUMT.
Results
Class-wide NUMT identification
To examine the evolutionary impact of NUMT integration, we identified mitochondrial insertions in the nuclear genomes of more than 1000 mammalian and avian species. Specifically, we analyzed 680 mammalian and 458 avian genomes with both nuclear and mitochondrial assemblies available from public databases. Using a previously published bioinformatic pipeline (Vendrami et al. 2022), we detected 85,100 NUMTs across 435 mammalian species (66.62%) and 28,947 NUMTs across 458 avian species (100%). These results align with earlier studies showing that NUMT integration is a frequent, ongoing process in vertebrates (Hazkani-Covo 2022). On average, mammals had more NUMTs than birds (about 125 vs. about 63 per species), with NUMT count positively correlated with genome size in mammals (P < 2.2 × 10−16) but not in birds (P = 0.79, phylogenetic generalized least-squares regression).
Phylogenetic patterns of NUMTs in mammals and birds
Given the contrasting patterns in NUMT abundance between mammals and birds, we investigated the phylogenetic distribution of NUMTs. Established species phylogenies for mammals and birds (Kumar et al. 2017; https://timetree.org) were used to reconstruct NUMT counts as a phylogenetic trait through a Bayesian approach implemented in BayesTraits v4.0.1 (Pagel and Meade 2022). Several clear phylogenetic patterns were observed.
First, NUMT counts showed substantial variation across clades (Fig. 2). Among mammals, most marsupials exhibited either an absence or low numbers of NUMTs, with the exception of the Tasmanian devil (Sarcophilus harrisii) (Hazkani-Covo 2022). In contrast, clades such as Old World monkeys and cetaceans displayed elevated NUMT counts. In birds, NUMT enrichment was particularly pronounced in three clades within the order Passeriformes: (1) warblers, cowbirds, and blackbirds; (2) sparrows (e.g., Zonotrichia albicollis), juncos, seedeaters, tanagers, and finches (e.g., Geospiza fortis); and (3) cardinals (Cardinalis cardinalis) (Sin et al. 2020), buntings, grosbeaks, longspurs, and grackles.
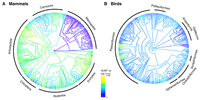
Phylogenetic distribution of NUMT occurrences in mammals and birds. Shown are the species cladograms from TimeTree, with the colors representing the reconstructed number of NUMTs for mammals (A) and birds (B). Note that the colorized representation refers to the log-transformed (i.e., log(1 + x)) NUMT number.
Second, NUMT counts varied significantly among closely related species. For example, the highly endangered spoon-billed sandpiper (Calidris pygmaea) exhibited extensive NUMT integration, suggesting a high level of genome degeneration (Chowdhury et al. 2022). Similarly, among mammals, the Tasmanian devil (S. harrisii, 1546 NUMTs), gray short-tailed opossum (Monodelphis domestica, 811 NUMTs), and yellow-footed antechinus (Antechinus flavipes, 792 NUMTs) showed elevated NUMT counts. In birds, in addition to C. pygmaea (13,170 NUMTs), the island finch (Nesospiza acunhae, 403 NUMTs) and northern cardinal (C. cardinalis, 373 NUMTs) also exhibited significant NUMT accumulation.
Distinct distributions of cNUMTs
Although mitochondrial integration into the nuclear genome is frequent and ongoing across many species, the origins and evolutionary fate of NUMTs, particularly those retaining intact mitochondrial coding frames over long timescales, remain unclear. To address this, we focused on a subset of NUMTs with at least one intact coding gene (cNUMTs) identified using the vertebrate mitochondrial genetic code. Of the 114,047 NUMTs identified, 6429 (5229 in 379 mammalian species and 1200 in 319 avian species) retained at least one intact mitochondrial gene. In mammals, the number of cNUMTs is positively correlated with noncoding NUMTs (P < 2.2 × 10−16), whereas no such correlation exists in birds (P = 0.9), as shown by phylogenetic generalized least-squares regression. This suggests that cNUMTs may arise as a byproduct of NUMT integration in mammals but not in birds. To estimate the evolutionary age of cNUMTs, we calculated pairwise synonymous divergence (dS) between cNUMT genes and their mitochondrial counterparts (Wang et al. 2010). The distribution of dS values provides insight into integration times across species (Fig. 3). We observed a decline in cNUMT abundance with increasing dS values in mammals, whereas birds exhibited a pronounced peak around dS ≈ 1.
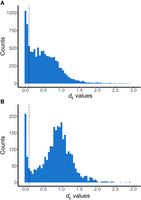
Distribution of dS values of genes in cNUMTs. Shown are the distribution of dS values between genes in the NUMTs and the host species mitochondrial DNA. NUMTs that possess at least one gene with dS > 0.1 are considered dcNUMTs; dS = 0.1 is indicated by the dashed lines. Distributions are shown for mammals (A) and birds (B). Note that count data for dS values greater than three are not shown.
Noncanonically integrated NUMTs
As recently integrated NUMTs will show little to no sequence divergence from their mitochondrial counterparts, they cannot tell us very much about the evolutionary consequences of NUMT integration. We hence focus on those cNUMTs that show substantial synonymous divergence from the respective gene in the host mitochondrial genome (dcNUMTs with at least one gene with dS > 0.1 relative to the respective host mitochondrial gene), because these are likely the consequence of noncanonical NUMT integrations (Fig. 1). We identify 4182 and 917 NUMTs that are dcNUMTs with 8406 and 2599 coding genes with dS > 0.1 in mammals and birds, respectively, meaning that some dcNUMTs contain multiple divergent mitocoding genes.
We identify 148 dcNUMTs (3.54%) in mammals and 249 dcNUMTs (27.15%) in birds that we can assign to a nonhost mitochondrial genome with high confidence (Table 1, Fig. 4; Supplemental Fig. S1). We find that 77.19% and 40.46% of the dcNUMTs in mammals and birds, respectively, have no hit with any of the available mitochondrial genomes at the 95% identity level. To test whether these NUMTs perhaps originate from species outside of the two taxonomic clades, we conducted further BLAST analyses but did not identify any further hits. The phylogenetic origins of these dcNUMTs are therefore located in their respective taxonomic groups, but their exact origins remain obscure.

Phylogenetic pattern of NUMT integrations of nonhost mitochondrial DNA for mammals (A) and birds (B). Shown are species that either show evidence of NUMTs of nonhost origin or that appear be the donor species of the NUMT. Note that some species can be either a donor and/or a recipient and that not all species included in our analysis are resolved phylogenetically in TimeTree, so not all pairs can be assigned. A complete list can be found in Supplemental Tables S2 and S3.
Number of BLAST hits of dcNUMTs against available mitochondrial genomes for birds and mammals
dcNUMT gene analysis
Because dcNUMTs contain at least one gene with an intact reading frame, it is possible to investigate the evolutionary pressures that prevailed since the split from the last common ancestor using DNA codon models. For example, if genetic drift was the predominating force since the split from the last common ancestor of the NUMT and the host’s mitochondrial genome, one would expect the ratio of nonsynonymous to synonymous substitutions (dN/dS) to be approximately one. It is important to note that in our analysis, we do not assume that NUMTs are transcribed or that their gene products are imported into mitochondria. The evolutionary patterns we observe do not depend on whether NUMTs are expressed or functional in a traditional sense. Instead, the signatures of selection may reflect broader genomic processes, such as linkage to functional genomic regions under selection or historical constraints on these sequences prior to their nuclear integration.
To test selection on dcNUMTs, we divided our data set into (1) single NUMT genes (Fig. 5A), which are highly divergent from the other NUMT genes and therefore seem to be isolated (“single”), and (2) NUMT gene clusters (Fig. 5B) that are highly similar, for instance, owing to shared speciation events (Vendrami et al. 2022). We then aligned the single NUMTs and NUMT gene clusters together with available mitochondrial genes of related species to calculate dcNUMT gene (Fig. 5A) and gene cluster–specific dN/dS ratios (Fig. 5B) using codeml (Yang 2007). codeml does not incorporate heterogeneity in codon composition or transition-to-transversion ratio (κ), which may differ between the nuclear and mitochondrial genome (Belle et al. 2005). However, we performed a series of simulations (Supplemental Fig. S2; Supplemental Table S1) that incorporate such heterogeneity (Fletcher and Yang 2009) that show little evidence for selection under a neutral model. We therefore consider our approach suitable to infer selection for nuclear integrated mitochondrial sequences.

Schematic illustration of the single gene and clustered genes codeml analysis. (A) Foreground branch in red (dN/dS = ω1) for a single NUMT gene; background branches (black, dN/dS = ω0) are for the respective mitochondrial genes of related species. (B) Foreground branch in red for NUMT gene clusters (dN/dS = ω2, here illustrated with a cluster size of two); background branches (black, dN/dS = ω0; gray, dN/dS = ω1) are for the respective mitochondrial genes of related species. Note that the gray branch is a separate background branch because it denotes the branch when the potential nuclear genomic integration took place.
Selection analysis on isolated dcNUMT genes imply noncanonical NUMT integrations and “missing” mitochondrial genomes
We first conducted a codon analysis of the “single” dcNUMT genes. We identified 3549 single dcNUMT genes in mammals (42.2% of total dcNUMT genes) and 2246 in birds (86.4% of total dcNUMT genes). The differences in these proportions between the two taxonomic groups is likely because dcNUMTs genes in birds have greater synonymous divergence and are therefore generally more ancient (Fig. 3). We conducted branch-specific analyses to test for positive and negative selection, as well as drift (Table 2). For the majority of single dcNUMT genes (Table 2), we identify sequence evolutionary patterns consistent with neutral evolution (62.4% for mammals and 83% for birds). We also observe a smaller number of cases (37.6% for mammals and 17% for birds) that are consistent with purifying selection, as well as five cases of positive selection.
Evolutionary pressure analysis on single dcNUMT genes for mammals and birds
Selection analysis on NUMT gene clusters suggests positive and negative selection on NUMTs
A proper evolutionary analysis of a single phylogenetically isolated NUMT gene is limited by the availability of the last mitochondrial lineage from which the mitochondrial DNA was transferred into a nuclear genome. To circumvent this limitation, we clustered the NUMT genes according to their similarity to each other independent of the host species. We then aligned these groups of gene sequences together with the most closely related mitochondrial genes available in our database. Based on these alignments, we then created gene trees and conducted clade-specific tests of selection (Table 3). We excluded gene trees that did not form monophyletic NUMT clusters. We then evaluated the selection pressures acting on the clade representing the diversification of the NUMTs (i.e., a node-based last common ancestor approach [Lee 1998] in contrast to branch-based last common ancestor, e.g., excluding the gray branch in Fig. 5B). It is therefore likely that the considered clade reflects an evolutionary time when the NUMT was truly integrated into the nuclear genome, for example, a pattern one would observe owing to speciation events (Vendrami et al. 2022). Consequently, the estimated selective pressure analysis should reflect selection on the NUMT, not the mitochondrial DNA.
Evolutionary pressure analysis on dcNUMT gene clusters for mammals and birds
We identified approximately 10 times more of these NUMT gene clusters for mammalian species than for birds (996 vs. 98). The average number of species with NUMT genes per cluster was three for both taxonomic groups (2.98 and 3.01 for mammals and birds, respectively). Although mammalian dcNUMTs are generally less divergent from the host's mitogenome than are avian dcNUMTs (Fig. 3), they are as frequently shared between species. Based on the evolutionary rate analysis, we identify more than 250 gene clusters that show evidence of purifying selection, most of them specific to mammals. Moreover, we identify an additional 24 gene clusters, 23 for mammals and one for birds, that show evolutionary signatures consistent with positive selection.
Evidence of positive selection in a human NUMT
We identified one NUMT on Chromosome 2 in humans that is part of a MT-ND4L gene cluster with evidence for positive selection (one out of 23 NUMT clusters for mammals) (Table 4). This particular NUMT is shared among seven ape species (Nomascus leucogenys, Pan troglodytes, Gorilla gorilla gorilla, Pan paniscus, Homo sapiens, Hylobates moloch, Pongo abelii), but the NUMT itself clusters with mitochondrial sequences of species of the family Callitrichidae (Fig. 6). In the human genome, this NUMT is flanked by the genes ARHGAP15, GTDC1, ZEB2, KYNU, and LRP1B. We also conducted an AlphaFold structural prediction (Mirdita et al. 2022) using the NUMT's gene sequence translated into a protein with the vertebrate mitochondrial code (Supplemental Fig. S3) and find very little 3D structural variation between the human MT-ND4L protein and the 3D protein prediction of the NUMT gene.
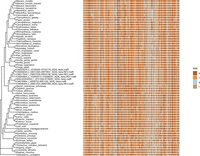
Phylogenetic tree and sequences used for codeml analysis of a NUMT cluster that contains dcNUMTs of seven ape species along with mitochondrial sequences of 59 (sub)species. The NUMT gene with an intact reading frame is MT-ND4L. Evolutionary pressures were estimated with codeml using the mitochondrial genetic code. The coding sequence is shown in color using the four-letter-DNA notation illustrating no pinpointed divergence of the NUMT sequences.
Codeml analysis results of a mitocoding gene located on a NUMT that is shared among seven ape species
Discussion
Here, we present a comprehensive analysis of mitochondrial DNA that has been integrated into the nuclear genomes (NUMTs) of more than 1000 mammalian and avian species and identify more than 80,000 NUMTs in mammals and more than 29,000 NUMTs in birds (Table 5). Generally, there are more NUMTs in mammalian genomes, which is not unexpected given that mammalian genomes tend to be larger (Kapusta et al. 2017). We also find a positive relationship between genome size and the number of mitochondrial integrations in mammals but not birds. A possible explanation for this is that mammals contain NUMT copies resulting from intragenomic duplications. Birds are known to exhibit very little variation in their genome sizes in general, which might also explain the lack of intragenomic copies, such as segmental duplications. For many of the identified NUMTs, it is unclear on which chromosome in the genome they are located. Instead, they are mainly located on small scaffolds that are often not much bigger than a mitochondrial genome (e.g., <20 kb) (Supplemental Fig. S4). However, large-scale application of long-range haplotype-based genomic sequencing (Rhoads and Au 2015; Lu et al. 2016) will overcome the current limitations of identifying the precise genomic locations of nuclear DNA of organelle origin in the near future (Wilcox et al. 2022).
Number of NUMTS, coding NUMTs (cNUMTs), and divergent coding NUMTs (dcNUMTS) in birds and mammals
Some of the identified NUMTs are very distantly related to the host's mitochondrial DNA. As our approach of identifying NUMTs tends to detect NUMTs that show a reasonable degree of similarity between the NUMT and the host's mitochondrial genome, it is likely that we did not identify all distantly related NUMTs. Therefore, our estimates of the number of NUMTs per species are potentially underestimates. For example, we do not find any NUMTs that are highly similar to a species outside of the respective animal class. Because we do not attempt to identify NUMT integration events as such, some of the NUMTs may result from structural mutations, such as segmental genome copies (Wilcox et al. 2022). Hence, our NUMT numbers are likely an overestimate of the number of NUMT integration events from nonnuclear DNA. However, because we focus on dcNUMTs for our evolutionary analysis, it is irrelevant whether a NUMT has undergone intragenomic copying or is the result of a “original” genomic integration.
Our method also depends on an accurate database assignment of the mitochondrial genome to the respective species. However, because of biological mitochondrial diversity (Clark et al. 2023) or simply erroneous entries in databases (Caswell et al. 2019; Laine et al. 2019; Sangster and Luksenburg 2021), mitochondrial DNA and the NUMT DNA could appear more divergent. To limit the impact of this in our dcNUMT analysis, we focused on cNUMTs with at least 10% synonymous divergence to the respective mitochondrial gene. To investigate the possibility of erroneous database entries, we checked the mitochondrial genomes of 22 bird species that show a large number of highly dissimilar NUMTs. For this, we assembled the mitochondrial genomes of these species from raw sequencing data using an established pipeline (Jin et al. 2020). Although we observed some genetic variation between the assembled and the canonical mitochondrial genomes in the database (Supplemental Table S4), we did not find any evidence for an incorrect assignment of mitochondrial genomes. We conclude that falsely assigned mitochondrial genomes play a very limited role in our analysis.
dcNUMTs
We find more than 5000 NUMTs in mammals and birds (Table 5) that contain genes with an intact mitochondrial reading frame and that are highly divergent from the respective host's mitochondrial encoding genes (dcNUMTs with genes with dS > 0.1, i.e., a synonymous per site divergence of >10%). In mammals, we find that the number of dcNUMTs becomes lower for increasing dS values, but in birds, there is a clear peak at dS ≈ 1. This might reflect a historic episode of high transposable element activity common to all birds. However, as the nuclear and mitochondrial mutation rates may vary substantially across species, we speculate that the dS estimates should be considered only as a rough proxy of the integration time of the NUMT.
It is possible that dcNUMTs are transcribed (Cohen et al. 2016) and may have a function on the RNA level (Plazzi et al. 2024) or that the DNA itself has some regulatory function as observed for mitochondrial DNA (Noutsos et al. 2007; Van Acker et al. 2023). For example, the transcription of noncoding genomic DNA is very common (Gao et al. 2020), and active retroviruses (Bolisetty et al. 2012) may lead to the NUMTs being expressed as well. However, this remains speculative and would need to be supported by genomic signatures consistent with functionality. Further experimental validation, such as RNA-seq analysis of these genomic regions, is needed to confirm whether NUMTs in bird species are indeed transcribed.
Noncanonical integration
To identify potential origins of noncanonically integrated NUMTs, we compared dcNUMTs against available mitogenomes that also contain mitochondrial genomes that were not in our initial analysis (e.g., because there was no nuclear genome available for the species). The excess of synonymous mutations observed in these NUMTs likely reflects purifying selection in the original mtDNA. Therefore, these patterns likely represent evolutionary signatures acquired prior to integration, rather than resulting from ongoing selection in the nuclear genome. These NUMT insertions may appear relatively intact because they have had less time to accumulate mutations. We observe a greater number of distant hits in birds than in mammals. This is possibly because the bird genomes in our data set diverged earlier than the mammalian genomes, which means that their mitogenomes are more diverged, allowing for better discrimination of noncanonical NUMT origins. In mammals, the majority of the noncanonically integrated NUMTs occur within the ungulates (Fig. 4A; Supplemental Table S2), a taxonomic group in which hybridization frequently occurs (Iacolina et al. 2019).
In birds, we find several examples of noncanonical integration events in which NUMTs are similar to the mitochondria of distantly related species (Fig. 4B; Supplemental Table S3). For example, we identified a NUMT in the gweela (Alectura lathami), a mound-building bird from the family Megapodiidae found in eastern Australia that shows high similarity to the tawny frogmouth mitogenome (Podargus strigoides), a species native to the Australian mainland and Tasmania. A possible explanation might be that some NUMTs colocalize with retrotransposon elements (Bolisetty et al. 2012; Tsuji et al. 2012; Dayama et al. 2014; Schiavo et al. 2017), and the transmission of retroviruses between species has been documented in mammals and invertebrates (Flavell 1999; Diehl et al. 2016). We also note that the W Chromosome is a refugium for retroviruses in birds (Peona et al. 2021), which might imply that we may have missed NUMTs that are located on the W if the respective genome of a species was obtained from a male individual, which has the homogametic genome in birds. We cannot pinpoint the exact genomic locations of many of the identified NUMTs, because they tend to be located on very short scaffolds (Supplemental Fig. S4), and this appears to particularly true for avian dcNUMTs. It has been noted previously that some avian chromosomes have not been assembled in many bird genomes, such as Chromosome 16 (Laine et al. 2016), smaller microchromosomes (Kapusta et al. 2017), and the germline-restricted chromosome (Kinsella et al. 2019). This reduced ability to locate the precise genomic locations hampers the possibility to pinpoint integration events that happened as a consequence of past genomic hybridization. In the future, this can be overcome by the application of long-range sequencing approaches, which will subsequently allow us to use dcNUMTs as markers for genomic hybridization and could facilitate the unraveling of their potential role as adaptive elements.
We also identify 3179 and 384 dcNUMTs in mammals and birds, respectively, for which we cannot identify a homologous sequence with a sequence identity of >95% (Table 1). Although this could mean that these are simply older NUMTs that are highly diverged but that have remained in the genome for a long time, it could also mean that some mitogenomes are lacking from our database. This could be either because the respective species has not been yet sampled or because it has already gone extinct. The number of “orphan” NUMTs is almost 10-fold higher in mammals than in birds. Sequencing more mammalian genomes and the retrieval of ancient mammalian (mito)genomes might enable us to reveal the evolutionary origins and fates of further dcNUMTs in the future.
Choice of genetic code
Here, we use of the mitochondrial genetic code for dN/dS calculations. In our analysis, we opted for the mitochondrial genetic code because our primary focus is to investigate the (ancient) integration of mitochondrial sequences into the nuclear genome and how these sequences have evolved over time relative to the mitochondrial genome of the host species. The maintenance of mitochondrial reading frames and the accumulation of mutations that disrupt these frames in the nuclear context can provide important insights into the evolutionary history of these elements. However, it is crucial to recognize that if some of these NUMTs are functional in a nuclear environment (e.g., through transcriptional or regulatory roles), then a reassessment using the standard nuclear code might be warranted for a more nuanced understanding of their evolutionary trajectories.
Tests of selection
For the majority of single NUMT genes, we observe a signature consistent with drift (e.g., dN/dS = 1) but we also identify more than 2000 genes showing signatures of purifying selection (Table 2). We also identify five NUMT genes showing signatures of positive selection. Although it is intriguing that many of these genes have been evolving under selection pressure, this analysis is hampered by the fact that, for the tested branches, the single NUMT genes may have existed as mitochondrial genome sequences. Unfortunately, we cannot incorporate the precise date of the nuclear integration, as we potentially lack a substantial amount of mitodiversity (Table 1) and therefore cannot be sure that we include a sequence that did not diverge before the NUMT integration took place. To address this limitation, we conducted clade-specific tests of selection on groups of NUMT genes. Therefore, the considered branches in the clade with the NUMTs may reflect an evolutionary time when the NUMT was truly integrated into the nuclear genome. However, it is also possible that this approach groups sequences that have evolved the same sequence independently (Hazkani-Covo et al. 2010; Song et al. 2013).
Positive selection in a human NUMT gene
Finally, we identified one gene within a human NUMT that is part of a cluster of seven ape NUMTs. For humans, the genes located in the genomic region where the NUMT is located are known to have evolved under positive selection in the ancestral lineage of modern humans (Green et al. 2010; Prüfer et al. 2013; Racimo 2016). Moreover, structural mutations, such as deletions and duplications, in the respective region are known to cause Mowat–Wilson syndrome in humans (Baxter et al. 2017; Goyal et al. 2022), a rare but severe genetic disorder. Because the NUMT is linked to functionally important genes, it is a possible explanation of the signal of positive selection; the NUMT might be hitchhiking with a gene under positive selection (Barton 2000). The fact that one mitochondrial gene on the NUMT shows a signature of positive selection at the (mitochondrial) protein-coding level that is shared between many ape species fuels speculation to a potential functional role of this NUMT or one of its genes. Our hypothetical 3D prediction (Supplemental Fig. S3) does not suggest that the accumulated mutations in the NUMT strongly alter the three-dimensional structure. It therefore appears unlikely that the signal of positive selection of the mitocoding nuclear DNA is the consequence of a false positive, for example, through misalignment or rapid accumulation of random amino-acid-altering mutations (Mallick et al. 2009).
Haplotype-resolved sequencing
Here we have shown that NUMTs may originate from noncanonical integration and that signatures of purifying and positive selection are common and widespread across two main vertebrate clades. Currently, in many genome assemblies, NUMTs cannot be placed onto chromosomes because they are usually flanked by highly repetitive sequences owing to retrovirus integration. Haplotype-resolved sequencing will likely be able to place more NUMTs on chromosomal locations and thereby reveal the evolutionary signatures of NUMTs as a consequence of linkage (Wilcox et al. 2022). Our example of a human NUMT has shown that it is crucial to know the genomic context to deduce the likely functional roles of these intriguing genetic elements. Moreover, recent and ancient integration may be understood better when placing NUMTs into phylogenetic context. Even if NUMTs are not inherently functional, they serve as valuable markers of selection and may arise as byproducts of genomic introgression or hybridization events. Additionally, their integration near functional elements suggests a potential role for background selection in maintaining coding-like NUMTs within nuclear regions, particularly in areas of low recombination (Gossmann et al. 2014). This selective pressure could further suppress the rate of mutation accumulation in NUMTs.
Methods
Data collection and selection
Genome assemblies for all mammalian species (available before November 11, 2021) and all avian species (available before November 2, 2021) were downloaded from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) (Sayers et al. 2021) using NCBI-genome-download v0.3.1 (https://github.com/kblin/ncbi-genome-download). For species with multiple assemblies, the representative genome or the assembly with higher coverage (if no representative was specified) was selected.
Mitogenomes containing at least one partial coding gene for all mammalian species (available before November 22, 2021) and all avian species (available before December 16, 2021) were obtained from GenBank to build BLAST databases. Mitogenomes from the same species or closely related subspecies with genome assemblies were prioritized for NUMT identification. When multiple mitogenomes existed for a species, the reference assembly was used. Unverified mitochondrial genomes were excluded. Protein-coding gene annotations were obtained from GenBank, and mitogenomes lacking annotations were annotated via the MITOS2 webserver (Donath et al. 2019). Supplemental Table S5 lists the genomes, mitochondrial genomes, and their accession numbers.
NUMT identification
NUMTs were identified using local BLASTN v2.6.0+ (Camacho et al. 2009), with a word size of 20. Mitochondrial genomes were used as queries against genome assemblies from the same species, following a modified approach from previous studies (Lammers et al. 2017; Vendrami et al. 2022). Hits <200 bp were discarded, and NUMT sequences were extracted using BEDTools v2.26.0 (Quinlan and Hall 2010).
To identify cNUMTs, NUMTs and their reverse complements (generated with the EMBOSS v6.6.0.0 revseq algorithm) (Rice et al. 2000) were aligned to the protein-coding genes of the corresponding mitochondrial genome using MAFFT v7.310 (Katoh and Standley 2013).
NUMTs were mapped to species phylogenies from TimeTree (Kumar et al. 2017) to assess evolutionary trends. Evolutionary rates were estimated using an independent contrast model with reversible jump Markov chain Monte Carlo (RJMCMC) in BayesTraits v4.0.1 (Pagel and Meade 2022). Each data set was run for 12,000,000 iterations, with sampling every 2000 iterations following a 2,000,000-iteration burn-in. Ancestral reconstructions of NUMT counts, adjusted for branch length rate changes, were performed using the ace function in the phytools package (Revell 2012) in R v4.2.3 (R Core Team 2023).
Substitution rate analysis of NUMTs with coding genes
We first assessed whether cNUMTs were in-frame by checking pairwise alignments with mitochondrial protein-coding genes for the absence of internal stop codons. In-frame cNUMTs and their corresponding mitochondrial genes were used to calculate synonymous substitution rates (dS) using the KaKs_Calculator 2.0 (Wang et al. 2010) under the NG substitution model (Nei and Gojobori 1986). NUMTs with at least one coding gene and dS > 0.1 were classified as potentially dcNUMTs.
These dcNUMTs were used as queries in BLAST searches (Camacho et al. 2009) against mitogenomes of all mammalian and avian species (Supplemental Table S5). The best BLAST hits were defined as those with the highest bit score, whereas high-confidence hits were defined by at least 95% sequence identity to the query. dcNUMTs with high-confidence hits to mitogenomes from other species, in which the percentage of identical matches differed by at least 5% from hits to their host species, were considered potential cases of lateral transfer between species pairs.
Clustering NUMTs and selection analysis
To investigate functional constraints on coding sequences in cNUMTs, we first identified NUMTs derived from the same source and then analyzed the ratio of nonsynonymous mutations to synonymous mutations (dN/dS) for each NUMT cluster. We used MAFFT v7.310 (Katoh and Standley 2013) to align protein-coding sequences identified in cNUMTs and reconstructed the coding sequence trees with IQ-TREE v2.0.3 (Minh et al. 2020). Then, we used codeml implemented in PAML (Yang 2007) to calculate pairwise dN/dS comparisons and substitutions per codon on all the protein-coding genes from cNUMTs that correspond to the same mitochondrial protein-coding genes. The coding sequence pairs with substitutions per codon less than 0.1 were examined for their best hits against the mitogenomes. The pairs in which both coding sequences had a percentage of identical matches <98% were examined if any of the coding sequences matched with coding sequences in other pairs. The coding sequences matched into groups were grouped into clusters.
We then translated the mitochondrial protein-coding genes from the species we used to identify NUMTs with Biopython (Cock et al. 2009) and aligned each mitochondrial protein-coding gene with MAFFT v7.310 (Katoh and Standley 2013). The translated sequences with lengths 20% shorter than the longest sequences in the alignment were removed to exclude incomplete mitochondrial protein sequences. The alignments of corresponding mitochondrial protein-coding genes were used to align with each dcNUMT clusters generated in the previous step with prank v.170427 (Löytynoja 2014). The alignments were used to reconstruct coding sequence trees with FastTree v2.1.11 (Price et al. 2010). To increase computational efficiency, we extracted the subtrees with the clades containing cNUMT genes and three hierarchical levels above the focal clades using Newick utilities v1.7.0 (Junier and Zdobnov 2010). The codon alignments of the corresponding subtrees were converted from mitochondrial gene sequences and their translated protein sequences using pal2nal v14 (Suyama et al. 2006). Then, we used codeml implemented in PAML (Yang 2007) for the selection analysis. We implemented two different branch models: a three-ratio model (model = 2, NSsites = 0) and a three-ratio model with the omega of NUMT clades fixed to one (model = 2, NSsites = 0, fix_omega = 1, omega = 1) for each cNUMT gene cluster. The likelihood differences among the models were used to derive statistical significance. Benjamin–Hochberg adjustments (Benjamini and Hochberg 1995) were applied for multiple testing corrections.
Heterogenous substitution models
We also conducted a set of analysis to test for the effect of heterogeneous versus homogeneous substitution models. Potential differences in mitochondrial and nuclear DNA might be reflected in the transition-to-transversion ratio (ts/tv; κ), which is much higher in mitochondrial DNA relative to nuclear DNA in humans, although there is lots of variation across animals (Belle et al. 2005). Although codeml does not incorporate heterogeneity in κ or codon composition, it is possible to conduct sequence simulation using branch-specific models of sequence evolution (Fletcher and Yang 2009). For this, we performed simulations with INDELible and used estimated κ and codon frequencies from our data (i.e., cluster depicted in Fig. 6, with NUMT and mitochondrial specific codon composition and κ estimated separately) as input parameters. We also simulated a homogenous model in which we assumed codon composition and κ from the mitochondrial sequences only. We then tested these simulated sequences using codeml assuming homogeneous codon composition and a single κ across branches similar to our test setup (Supplemental Fig. S2; Supplemental Table S1). The INDELible input file control.txt is provided as Supplemental Code and is available at GitHub (see Software availability).
Software availability
Scripts are available at GitHub (https://github.com/chnyuch/numt_vertebrate [NUMT detection] and https://github.com/tgossmann/numt_vertebrate [INDELible simulations]) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We sincerely thank Justin Wilcox for his valuable input and insightful contributions to this project. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme grant agreement no. 947636. This work was supported by the BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI; 031A532B, 031A533A, 031A533B, 031A534A, 031A535A, 031A537A, 031A537B, 031A537C, 031A537D, 031A538A) and the CeBiTec computer cluster at Bielefeld University. This research was funded by the German Research Foundation (DFG) as part of the SFB TRR 212 (NC3)—Project Numbers 316099922 and 396774617. C.R.C. was supported by a Natural Environment Research Council Independent Research Fellowship (NE/T01105X/1).
Author contributions: T.I.G. designed the study with input from J.I.H. and D.L.J.V. Y-C.C. conducted the majority of the analyses. M.L.H., L.E.Y.H., C.R.C., D.L.J.V., and T.I.G. contributed to analyses. T.I.G. and Y-C.C. drafted the manuscript with input from all of the authors. All authors contributed to finalizing the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279428.124.
- Received April 3, 2024.
- Accepted March 20, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








