
Analysis of canine gene constraint identifies new variants for orofacial clefts and stature
- Reuben M. Buckley1,
- Nüket Bilgen2,
- Alexander C. Harris1,
- Peter Savolainen3,
- Cafer Tepeli4,
- Metin Erdoğan5,
- Aitor Serres Armero1,
- Dayna L. Dreger1,
- Frank G. van Steenbeek6,
- Marjo K. Hytönen7,8,9,
- Heidi G. Parker1,
- Jessica Hale1,
- Hannes Lohi7,8,9,
- Bengi Çınar Kul2,
- Adam R. Boyko10,11 and
- Elaine A. Ostrander1
- 1National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA;
- 2Department of Animal Genetics, Faculty of Veterinary Medicine, University of Ankara, Ankara 06110, Türkiye;
- 3KTH Royal Institute of Technology, School of Chemistry, Biotechnology and Health, Science for Life Laboratory, SE-100 44 Stockholm, Sweden;
- 4Department of Animal Science, Faculty of Veterinary Medicine, University of Selcuk, Konya 42100, Türkiye;
- 5Department of Veterinary Biology and Genetics, Faculty of Veterinary Medicine, Afyon Kocatepe University, Afyonkarahisar 03200, Türkiye;
- 6Department of Clinical Sciences, Faculty of Veterinary Medicine, Utrecht University, 3584 CM Utrecht, The Netherlands;
- 7Department of Medical and Clinical Genetics, University of Helsinki, 00014 Helsinki, Finland;
- 8Department of Veterinary Biosciences, University of Helsinki, 00014 Helsinki, Finland;
- 9Folkhälsan Research Center, 00290 Helsinki, Finland;
- 10Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, Ithaca, New York 14853, USA;
- 11Embark Veterinary, Inc., Boston, Massachusetts 02210, USA
Abstract
Dog breeding promotes within-group homogeneity through conformation to strict breed standards, while simultaneously driving between-group heterogeneity. There are over 350 recognized dog breeds that provide the foundation for investigating the genetic basis of phenotypic diversity. Typically, breed standard phenotypes such as stature, pelage, and craniofacial structure are analyzed through genetic association studies. However, such analyses are limited to assayed phenotypes only, leaving difficult-to-measure phenotypic subtleties easily overlooked. We investigated coding variation from over 2000 dogs, leading to discoveries of variants related to craniofacial morphology and stature. Breed-enriched variants were prioritized according to gene constraint, which was calculated using a mutation model derived from trinucleotide substitution probabilities. Among the newly found variants is a splice-acceptor variant in PDGFRA associated with bifid nose, a characteristic trait of Çatalburun dogs, implicating the gene's role in midline closure. Two additional LCORL variants, both associated with canine body size are also discovered: a frameshift that causes a premature stop in large breeds (>25 kg) and an intronic substitution found in small breeds (<10 kg), thus highlighting the importance of allelic heterogeneity in selection for breed traits. Most variants prioritized in this analysis are not associated with genomic signatures for breed differentiation, as these regions are enriched for constrained genes intolerant to nonsynonymous variation. This indicates trait selection in dogs is likely a balancing act between preserving essential gene functions and maximizing regulatory variation to drive phenotypic extremes.
The genomes of domestic species hold immense promise for the discovery of novel genotype–phenotype associations (Andersson and Purugganan 2022; Buckley and Ostrander 2024). The domestic dog comprises over 350 breeds (Rogers and Brace 1995; Fogle 2000), many of which exhibit extreme levels of phenotypic diversity, including those associated with morphology, behavior, and disease susceptibility (Ostrander et al. 2017; Leeb et al. 2023). Identifying genetic variants that drive canine phenotypic variation will further define gene functions.
Genetic association studies typically require an initial and robust characterization of a phenotype, a practice that ultimately limits the scope of potential findings to only those that can be recognized and sufficiently defined. In this study, we explore how gene constraint can be used to identify variants with potential to impact phenotypes as trait-associated candidates, later determining association by investigating breed traits and characteristics of dogs carrying these alleles. Because variants are first prioritized according to functional impact, we can identify trait associations for variants often overlooked by traditional genome-wide association studies (GWASs).
The rigid population structure and extreme phenotypic diversity across breeds is an essential component of our analysis. By definition, dog breeds are closed populations, such that every registered dog is descended from a small number of historic founders and shares extensive genetic homogeneity (Mellanby et al. 2013; Lampi et al. 2020). Breed members have multigeneration pedigrees and are judged against morphologic standards that further enforce genetic homogeneity, resulting in a reduction in the locus heterogeneity that typifies human traits, where dozens or more genes may be foundational for a given trait. The ease of association studies in dogs is further enhanced by the fact that most breeds were developed within the last 200 years and, as such, there are minimal numbers of generations for meiotic recombination to occur (Worboys et al. 2018). This unique population structure means that breed membership can be used as a surrogate when mapping breed-associated morphologic traits, thus avoiding the time and expense of collecting individual metrics (Gough et al. 2018; Plassais et al. 2019; Dutrow et al. 2022; Morrill et al. 2022).
In humans, candidate trait and disease variants are generally identified using a combination of strategies, including multiple species alignment, protein structure predictions, gene mutation tolerance, and machine learning (Ng and Henikoff 2001; Adzhubei et al. 2010; Havrilla et al. 2019; Rentzsch et al. 2019; Karczewski et al. 2020; Cheng et al. 2023; Gao et al. 2023; Sullivan et al. 2023). Most such tools benefit from extensive catalogs of human variation featuring large amounts of clinical and personal data (Landrum et al. 2014; Lek et al. 2016; Bycroft et al. 2018; All of Us Research Program Genomics 2024). Although efforts to catalog trait-associated genetic variation exist for domestic mammals (Plassais et al. 2019; Nicholas 2021; Meadows et al. 2023; Nicholas et al. 2024), the resources for doing so are limiting. While mapping functional gene/genomic annotations over from humans can theoretically be used for improving variant annotation, it is estimated that ∼13% of human pathogenic ClinVar variants are common in dogs, and thus likely benign (Gao et al. 2023). Systems such as the dog are therefore rife with lost opportunities for identifying genetic variants that play fundamental roles in mammalian biology. Nevertheless, the recent Dog10K release of whole-genome sequence (WGS) data from over 2000 dogs (Ostrander et al. 2019; Meadows et al. 2023), inclusive of 321 breeds and populations, means the time is ripe to use measures of functional and evolutionary constraint to identify new variants with a high probability of impacting gene function.
This study focuses, specifically, on identifying genotypes that are rare overall, but common within a single breed or groups of related breeds that share a particular trait. We characterize coding variation across 2291 canine genomes from over 200 phylogenetically informed breeds/subpopulations and develop a canine-specific mutation model to determine whether gene/site constraint metrics are predictive of variant fitness consequences. From this, we identify breed-enriched variants predicted to cause loss of function (LoF) in genes under evolutionary constraint, identifying trait-causing alleles requiring additional study. Finally, we investigate the contribution of coding variation to breed differentiation, identifying the central role constrained genes play in shaping phenotypic differences between breeds. Collectively, our work provides a road map for performing constraint-based analysis of genetic variation in other domesticated species with similar genomic resources, such as cattle, swine, and horses.
Results
A framework for evaluating gene sequence constraint metrics
Genomic sequence constraint metrics are often used to predict whether specific mutations are likely to be deleterious. Characteristic breed traits are often phenotypic extremes that can be linked to deleterious health outcomes, such as in brachycephalic breeds (Marchant et al. 2019; O'Neill et al. 2020). Sequence constraint can therefore be used to prioritize likely trait-causing variants. To explore the utility of gene sequence constraint for this purpose, we assembled a data set of over 2291 high-quality genomes and used phylogenetic clustering to group them into 200 breeds/groups of five or more (Supplemental Figs. S1, S2; Supplemental Tables S1–S3). Throughout the coding sequence (CDS) and splice sites, we captured 611,644 single-nucleotide variants (SNVs), 28,970 small deletions, and 28,123 small insertions, including multiallelic variants (Methods; Supplemental Fig. S3; Supplemental Table S4).
Three separate metrics for constraint are available in the dog: constrained CDS, human ortholog LOEUF (loss of function observed over expected upper fraction), and variant phyloP score. Constrained CDS and variant phyloP score both represent evolutionary conservation of canine genomic sequence derived from the Zoonomia consortium's alignment of 240 mammals. Variant phyloP score is the level of conservation of an individual nucleotide position. Constrained CDS is the fraction of nucleotides within a gene's CDS that have a phyloP score >2.27 (genome-wide FDR > 0.05) (Christmas et al. 2023; Sullivan et al. 2023). Human ortholog LOEUF indicates whether a human–dog ortholog is depleted of predicted LoF variation in humans (Karczewski et al. 2020), where LoF here specifically represents nonsense, start loss, splice disruptor, and frameshift variant annotations (MacArthur et al. 2012). Importantly, these metrics do not measure canine coding constraint directly. Instead, evolutionary-based metrics, such as constrained CDS and variant phyloP score, identify sites and genes conserved across many species and are blind to lineage-specific effects. Similarly, because LOEUF values were calculated in human genes, they are blind to lineage-specific selection in canids.
We evaluated the capacity of each constraint measure to predict gene tolerance to mutations within dogs by measuring their concordance with the ratio of observed to expected mutations in canine genes. Calculation of the observed mutation rates must only capture negative selection effects and not canine demographic history, which has been largely shaped by recurrent population bottlenecks and artificial selection. One approach for achieving this is to analyze recent spontaneous mutations whose presence in the genome is not reflective of population history. Singleton variants in dogs with highly represented ancestry represent the best candidates for this variant class, as variant scarcity alone is insufficient for determining how recently the variant arose and the likely demographic forces to which it was subjected.
Canine ancestry representation was determined using individual singleton counts, where excess singletons per individual indicated either underrepresented ancestry or poor sequencing quality. Seventy percent of dogs carried <40 singletons each within their CDSs, allowing this value to serve as a cutoff (Supplemental Fig. S4A). These dogs belonged to breeds with the greatest number of individual members in the data set (greater Wilcoxon rank sum test; P-value < 10−15), and larger fractions of their genomes were within runs of homozygosity (FROH) (greater Wilcoxon rank sum test; P-value < 10−15), than was observed for individuals with >40 singletons (Supplemental Fig. S4B). Only 11.2% of all singletons were inferred to be recent spontaneous mutations by this method, and most low-frequency variants were contributed by a minority of individuals with poorly represented ancestry (Supplemental Fig. S4C), mostly consisting of wolves, village dogs, and breeds of eastern origin, which have greater diversity than western dogs (Supplemental Fig. S2; Supplemental Table S3).
To evaluate the fit of the mutation model, we used the Pearson correlation coefficient between observed frequency of synonymous mutations and synonymous mutation probability per gene. From a total of 8660 observations, the model achieved a modest correlation of 0.53, which was lower than anticipated and likely due to an insufficient number of observations. Sparsity in observations was addressed first by relaxing criteria for defining recent spontaneous mutations and, second, by grouping genes based on mutation probabilities. The 95% confidence intervals for expected fit were also calculated, allowing the mutation model to be evaluated as a departure from expected fit values. Although relaxing criteria led to increased correlations, the departure from expected model fit increased (Fig. 1A), as the inclusion of additional variants to the analysis diluted the fraction of true recent spontaneous mutations. The alternative strategy of placing genes into groups based on mutation probability was more successful. For 1000 groups or less, correlations greater than 0.9 were reached and the model fit remained within expected ranges, unlike a competing model based on CDS length (Fig. 1B). These analyses indicate that accumulation of synonymous mutations follows expected mutation probabilities, demonstrating that this framework can be used to determine if genes have significantly fewer nonsynonymous mutations than expected, thus detecting negative selection effects.
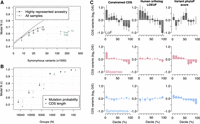
Rare canine variants are used to estimate mutational tolerance in constrained genes. (A) Pearson correlation coefficient between observed rare variants and gene mutation probabilities. Variants are organized by the group of samples they were observed in and the maximum allele count threshold for defining rare variants, which is displayed near each data point. The gray band represents the 95% expected interval of the model fit based on simulated accumulation of synonymous mutations. (B) Pearson correlation coefficient of rare variant frequency and gene mutation probability across gene groups. Genes were grouped according to their mutation probabilities. The x-axis shows the number of groups used to calculate correlation coefficients. The number of groups decreases as group sizes get larger. Two models are tested; one is the mutation probabilities calculated using the mutation model and the other is mutation probabilities according to gene CDS length. The 95% confidence intervals for the expected model fit are also displayed. (C) Gene mutation frequency according to three different constraint metrics. The expected number of mutations was calculated following the mutation model. The observed number of mutations are from singletons in dogs with highly represented ancestry. Error bars indicate whether the observed mutation frequency is outside the 95% expected bounds.
The functional constraint of dog genes
Using 907 LoF and 14,947 missense singleton mutations, we tested whether constraint metrics were associated with negative selection effects (Methods). Gene-based constraint metrics, i.e., constrained CDS and human ortholog LOEUF, were significantly associated with intolerance to LoF mutations (Fig. 1C), indicating that a significant portion of the expected LoF mutations in the most constrained genes were deleterious. The strongest effects were observed in the top human ortholog LOEUF decile, where only half the expected number of LoF mutations were observed (Fig. 1C). Conversely, no significant negative selection effects were observed for LoF mutations among the top phyloP constraint deciles, indicating the importance of sequence context when evaluating the fitness impacts of evolutionarily conserved sites. For example, most reference genomes used in multispecies alignments do not contain LoF variants, whereas population analyses show they are often tolerated (Lek et al. 2016).
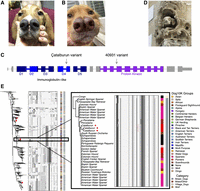
PDGFRA LoF mutations are associated with bifid nose. (A) Çatalburun dog with the distinctive bifid nose phenotype. (B) A mixed-breed dog (Dog ID: 40931) with a bifid nose phenotype. The dog is heterozygous for an additional PDGFRA splice-acceptor variant. (C) Canine PDGFRA transcript showing the locations of each splice-acceptor variant found within dogs with orofacial clefts. The first variant is the Çatalburun variant and the second variant belongs to Dog 40931. (D) Photograph of a statue depicting a dog with a bifid nose phenotype. The statue is located at the Palace of Bertemati in Andalusia, Spain and was constructed in the eighteenth century. (E) Haplotype analysis of the PDGFRA locus. A neighbor-joining tree is shown alongside a matrix of phased haplotypes. Red diamonds on the tree show the locations of Çatalburun haplotypes. The inset shows the Çatalburun haplotypes containing the PDGFRA splice-acceptor mutation. The position of the mutation within the haplotype is indicated in red.
All three constraint metrics were associated with intolerance to missense mutations; however, selection effects for gene-based metrics were less pronounced than those observed for LoF mutations (Fig. 1C). By comparison, synonymous mutations demonstrated no significant negative selection effects among the top gene-based constraint deciles, as expected. Conversely, there was significant intolerance to synonymous substitutions for the top phyloP score deciles. Because such scores are based on genomic positions, this is likely due to synonymous mutations causing cryptic-splice sites, impacting mRNA stability, or inducing change in functionally relevant GC content, e.g., methylation sites (Sarkar et al. 2022; Gudkov et al. 2024).
We next tested whether constraint metrics were associated with known canine trait variants from the Online Mendelian Inheritance in Animals (OMIA) database (Supplemental Table S5), a catalog of variants and genes associated with inherited disorders and traits in nonmodel organism species (Nicholas 2021; Nicholas et al. 2024) (https://www.omia.org/home/). We incrementally increased the minimum threshold for defining constraint and measured the enrichment of trait associations for the remaining variants. To ensure these variants were segregating in dogs and did not represent sporadic cases, we only focused on OMIA variants with an allele count >2 within the combined cohort. Altogether, 31 out of 305 LoF and 50 out of 154 missense OMIA variants met these criteria (Supplemental Table S5). Importantly, supporting evidence for trait associations varies widely throughout OMIA, ranging from candidate alleles found in a single individual, to mutations shown to be causative in functional studies. Although we detected instances of LoF and missense OMIA variant enrichment for evolutionary-based constraint metrics (Supplemental Fig. S5), increases to the minimum constraint threshold often led to a loss of OMIA enrichment (Supplemental Table S6). Similarly, no enrichment of LoF OMIA variants was detected for human ortholog LOEUF constraint, indicating most OMIA LoF variants segregating within the combined cohort are likely not severely deleterious.
Detection of new genotype–phenotype associations using gene constraint
We used gene sequence constraint to investigate genotypes for their potential to impact canine breed phenotypes, focusing specifically on breed-enriched genotypes. Analysis of variant breed specificity showed that single-breed enrichment of OMIA variants was highest at a minimum breed allele frequency of 20% (Supplemental Fig. S6). Therefore, we only considered variants with breed allele frequencies above this threshold in at least one breed. To counteract the overrepresentation of breeds with small population sizes and common variants, we also ensured that variants had a minimum allele count of three, a minimum breed allele number of six, and a maximum nonbreed allele frequency of 5%.
For each constraint metric, potential LoF and missense candidates for canine traits were identified and ranked according to the number of breeds the variant was found in and the proportion of individuals in those breeds carrying the variant (Supplemental Fig. S7; Supplemental Tables S7–S12). Four variants with the potential to impact breed-characteristic phenotypes were selected for further discussion (Table 1). These included a splice-acceptor mutation in PDGFRA in Çatalburun dogs as a candidate for bifid nose and orofacial clefts, a stop-gained mutation in CSF1 fixed in Chow Chows (Chow) as a candidate for their distinctive skull shape, a frameshift in LCORL segregating in large breeds (>25 kg), and a frameshift in ITGA11 fixed in small brachycephalic breeds.
Breed genotypes and allele frequencies of select candidate variants
The PDGFRA splice acceptor was heterozygous in all three WGS Çatalburun dogs, a breed characterized by a split/bifid nose phenotype (Fig 2A; Yilmaz et al. 2012; Özarslan et al. 2021). Mouse knockout models demonstrate that Pdgfra is essential for medial nasal process development and palatogenesis (He and Soriano 2013; Qian et al. 2017), aligning with the hypothesis that the PDGFRA splice-acceptor variant causes the Çatalburun's bifid nose phenotype. The variant was genotyped in 14 additional Çatalburun dogs. Although phenotypes were not available, 13 dogs were heterozygotes and one was homozygous for the reference allele (Supplemental Table S13).
Eight additional bifid nose dogs, all lacking significant Çatalburun ancestry were recruited and underwent WGS and candidate variant filtering (Supplemental Figs. S8, S9; Supplemental Methods). Six of the dogs were unrelated to one another (Supplemental Table S14), while two were littermates and were analyzed in combination with an unaffected parent (Supplemental Table S15). Twelve heterozygous candidate variants were identified across six of the non-Çatalburun bifid nose dogs, either in evolutionarily constrained or orofacial cleft-associated genes (Supplemental Table S16). One sample, 40931 (Fig. 2B), carried a heterozygous insertion that also disrupted a PDGFRA splice acceptor and was the only dog other than the Çatalburun's to have a LoF variant in this gene (Fig. 2C), further supporting the gene as a candidate for orofacial clefts.
An additional breed, the Pachón Navarro, which originates in Spain, is also known for its bifid nose phenotype. Unfortunately, no samples from this breed were available for study. However, masonry on the outside of the eighteenth century Palace of Bertemati (Andalusia, Spain) shows a gargoyle-like dog with a bifid nose (Cobo 2017), attesting to the continuous selection for this trait in the Pachón Navarro breed (Fig. 2D).
Phylogenetic clustering of the Çatalburun haplotypes surrounding PDGFRA indicates a high degree of heterozygosity at this locus. Despite this, the haplotypes carrying the mutant allele clustered together among Spaniel and Setter haplotypes, appearing almost identical except for the splice-acceptor mutation (Fig. 2E). The depiction of a bifid nose dog in an eighteenth century Spanish statue, combined with observed haplotype similarity with Spaniels and Setters, indicates a Western European/Iberian origin for the mutation.
The identified frameshift in LCORL (c.1978_1981delACAG) causes a premature stop in the long isoform of the gene, much like the previously identified c.3768_3769insA variant (Fig. 3A; Plassais et al. 2019). Importantly, the newly identified c.1978_1981delACAG variant is not detectable in GWASs for either breed standard height or weight, indicating that constraint-based approaches can identify otherwise overlooked trait-associated candidate variants (Fig 3B; Supplemental Table S17). Association analysis of the LCORL loci using GEMMA also identified a third previously undescribed intronic variant common in small breeds (<40 cm in height and weighing <10 kg). This variant, c.525-14A > T, had a more significant association with breed standard height than c.3768_3769insA and was also associated with breed standard weight (Supplemental Fig. S10; Zhou and Stephens 2012). All three variants were in close proximity but were not in linkage disequilibrium (LD) (Fig. 3C; Supplemental Fig. S11). Conversely, since c.1978_1981delACAG has a similar impact on protein structure as c.3768_3769insA, it is found primarily in large breeds (>60 cm and >25 kg in height and weight, respectively) (Fig. 3D; Supplemental Fig. S11), such as the Spanish mastiff, where the variant is fixed (Table 1). Haplotype analysis of the region showed an ancestral haplotype shared between dogs and wolves and a modern haplotype that is exclusive to dogs (Fig. 4). The large body size alleles are embedded within each of these haplotypes separately, with the c.1978_1981delACAG variant belonging to the ancestral haplotype and the c.3768_3769insA variant to the modern haplotype. Despite this, different combinations of the three variants segregate within multiple breeds, indicating that selection for these size alleles likely predates breed development (Supplemental Table S18). There are also instances of compound heterozygotes, further supporting this hypothesis (Supplemental Fig. S11).
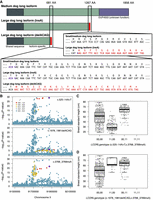
Identification of new LCORL variants associated with canine body size. (A) Mutations found in the long isoform of LCORL. Early termination of translation and loss of the DUF4553 domain caused by frameshift mutations is associated with larger dog size (>55 cm/>25 kg). (B) Manhattan plots at the LCORL locus showing association with breed standard height for three different variants in separate linkage groups. (C,D) Boxplots showing associations between homozygous LCORL size-associated genotypes and breed standard height.
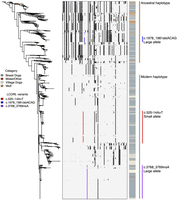
Haplotype analysis of the LCORL locus. A neighbor-joining tree is displayed alongside a matrix of phased haplotypes. Each of the LCORL size variants form distinct haplotypes with few recombinants. Depending on whether haplotypes are shared between wolves and dogs or are exclusive to dogs, they are classified as either ancestral or modern, respectively.
Selection for breed traits is associated with constrained genes and not CDS variation
Artificial selection for characteristic breed traits in dogs is associated with extended regions of homozygosity and strong signatures of population differentiation between breeds (Akey et al. 2010; Boyko et al. 2010; Vaysse et al. 2011; Parker et al. 2017a; Kim et al. 2018; Plassais et al. 2019; Morrill et al. 2022). However, due to the large size of these regions, it is difficult to identify specific variants that contribute to breed characteristic traits. To enhance detection of trait associations, potentially functional variants in constrained genes were analyzed for their association with signatures of breed differentiation.
Breed-differentiated regions were defined as 50 kb genomic windows of extreme FST that overlapped 1 Mb regions of breed homozygosity (Methods; Fig. 5A). A total of 1743 were identified, representing 2085 genes (Supplemental Table S19). Importantly, 92.3% and 55.2% of breed-differentiated regions were within 1 Mb and 100 kb of each other, respectively, indicating that most breed-differentiated regions are likely part of larger selective sweeps. In addition, 17 of 25 autosomal trait-associated loci described by Plassais et al. (2019) are within 100 kb of a breed-differentiated region, indicating genomic signatures for breed differentiation are associated with trait selection (Fig. 5B).
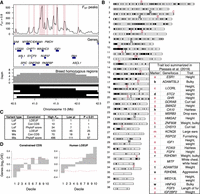
Noncoding variation at constrained genes drives breed differentiation. (A) Identification of breed-differentiated regions at Chromosome 15 IGF1 locus. Genomic windows with FST > 0.8 between breed pairs were plotted across the genome. Peak FST regions were then classified as regions of breed differentiation. Top FST peaks are shown in the first panel and highlighted in red. The second panel shows genes found in the region. The third panel shows the density of regions of breed homozygosity that were identified as 1 Mb genomic regions with breed nucleotide diversity <0.02. The final panel shows black bars as individual breed homozygosity stretches. (B) Genome-wide distribution of breed-differentiated regions. Trait loci summarized in Plassais et al. (2019) are displayed as lower-case characters. Trait loci in red are within 100 kb of a breed-differentiated region. (C) Number of genes in highly differentiated regions that contain potentially functional variants that pass minimum constraint thresholds. The “High FST” column is the number of genes that contain variants of interest. The “Low pi” column is the number of genes that contain variants in breeds with low nucleotide diversity across breed-differentiated regions. The final column is the number of genes that contain variants associated with breed differentiation (Fisher's exact test, P-value < 0.01). (D) Observed over expected frequency of constrained genes within breed-differentiated regions. The x-axis indicates constraint decile with 10 being the most constrained genes.
There were between 10 and 34 genes across all breed-differentiated regions that contained LoF mutations with potential functional consequences (Fig. 5C). Of these, only two were found in breeds that exhibited signatures of differentiation at these loci, specifically the CSF1 premature stop codon found in Chows (Table 1), and the SGK3 mutation causing hairlessness in American Hairless Terriers (Parker et al. 2017b). Although not significantly associated with breed differentiation, there were 115 variants within these regions that segregated in breeds with long stretches of homozygosity (Supplemental Table S20), indicating these potentially functional variants may be in LD with traits under selection.
Altogether, 70 genes contained potentially impactful missense mutations significantly associated with breed differentiation (Fig. 5C). The strongest result was for a missense variant in RYR1 (Supplemental Table S20), a calcium ion channel primarily expressed in the sarcoplasmic reticulum of skeletal muscle and involved with muscle contraction (Hernández-Ochoa et al. 2016). The variant was almost exclusively found within Bull Terriers and American Staffordshire Terriers, two breeds with shared ancestry known for their musculature and agility.
Most breed-differentiated regions were not associated with nonsynonymous variants, indicating that breed selection may instead be acting upon noncoding variants, which is consistent with most mapping studies for breed-defining traits and breed/lineage differentiation (Dutrow et al. 2022; Morrill et al. 2022). Human ortholog LOEUF constraint was significantly overrepresented among genes within breed-differentiated regions (Fig 5D), suggesting that phenotypic breed differentiation is primarily driven by noncoding variation surrounding and likely regulating highly constrained genes. Further investigation is required to identify trait-associated noncoding variants and candidate gene regulatory regions.
Discussion
Artificial selection for desired traits has made the domestic dog one of the most phenotypically diverse mammals in the world (Wayne and vonHoldt 2012). Rigid population structure, enforced by breed standards, has promoted high levels of homozygosity within breeds while also maintaining isolation between breeds (Parker et al. 2004; Karlsson and Lindblad-Toh 2008). Together, these factors make the domestic dog an invaluable system for identifying genes related to breed characteristics, which are often morphological extremes (Ostrander et al. 2017; Buckley and Ostrander 2024). We therefore investigated the genetic basis of canine traits by using genomic constraint to identify variants with potential phenotypic impacts. This work provides significant advances to the field by: (1) defining a variant set whose presence in the genome is unaffected by canine population dynamics, thus making it possible to determine genes under negative selection; (2) evaluating three constraint metrics for prioritizing variants for potential phenotypic impacts; (3) identifying new variants with strong impacts on craniofacial morphology and body size; and (4) applying genomic constraint to support a model of breed differentiation based on noncoding variation.
Using gene constraint, we identified strong candidates for phenotypic impacts as previously unreported LoF variants in PDGFRA and LCORL. Two PDGFRA variants were identified in dogs with bifid noses. This gene may therefore be one of a few for which functional mutations affecting midline closure occur, but are tolerated, as long as they are in the heterozygous state. In humans, a missense variant in this gene within a single family is associated with left nonsyndromic cleft lip and palate but is not fully penetrant (Yu et al. 2022). PDGFRA is a type III receptor tyrosine kinase that consists of an extracellular region with five immunoglobulin-like domains for ligand binding, a transmembrane helix, and an intracellular tyrosine kinase domain (Williams 1989; Chen et al. 2013). The Çatalburun variant occurs in the seventh intron of the gene within the fourth immunoglobulin-like domain, while the other variant in Dog 40931 is further downstream within the protein kinase domain (Fig 4C). Both mutations are splice acceptors, making it difficult to determine their impact on transcription. The affected exons may be skipped entirely, or an alternative splice site may be used, resulting in either partial exon retention or retention of nearby intronic sequence. Dogs carrying the PDGFRA mutations had no reported health concerns, which is surprising as PDGFRA is a broadly expressed developmental gene and strongly associated with gastrointestinal stromal tumor-plus syndrome in humans, with onset most likely to occur between 40 and 70 years of age (Manley et al. 2018; Huang et al. 2022).
Two other breeds share a similar phenotype as the Çatalburun; one is the Andean tiger hound, about which little is known, and the other is the Spanish Pachón Navarro. The Pachón Navarro, like the Çatalburun, is a pointing breed with only some members displaying the characteristic bifid nose phenotype. However, because Pachón Navarro dogs are susceptible to cleft palate, they have been used as surgical animal models for palate repair (Paradas-Lara et al. 2014). Evidence for bifid nose dogs in Spain dates back at least four hundred years, as the trait is depicted in a 1623 painting of a hunting party titled “Diane et ses nymphes s'apprêtant à partir pour la chasse” (Diana and her nymphs getting ready to go hunting) by Pierre Paul Rubens and Jan Brueghel l'Ancien. There are also descriptions of bifid nose pointing dogs in the south of France that date back to the turn of the twentieth century (Arkwright 1906). Although it is unknown whether this phenotype in the Çatalburun and Pachón Navarro breeds shares a common origin, Türkiye and Spain were both under the rule of the Umayyad Caliphate in the eighth century CE, providing a possible route for cultural exchange between the two regions. In addition to the recurrent literary and artistic depictions of bifid nose dogs throughout Spain and France, the haplotype similarity between Çatalburun dogs and Setters and Spaniels, whom have Spanish origins, indicates the phenotype likely arose in Western Europe or the Iberian Peninsula.
The newly discovered LCORL c.1978_1981delACAG allele was identified because of its high frequency in Spanish Mastiffs, Berger Picards, and Briards (Table 1). Like the previously described LCORL c.3768_3769insA variant, the c.1978_1981delACAG allele causes a frameshift and premature stop and loss of the DUF4553 DNA-binding domain from the long isoform of the protein, contributing to increased stature and weight (Plassais et al. 2019). We also identified a third variant in the gene that corresponded to smaller body size, indicating recurrent selection at this locus. Importantly, the LCORL locus is associated with stature across many domesticated mammals including goats (Saif et al. 2020; Graber et al. 2022), cattle (Bouwman et al. 2018; Chen et al. 2020), pigs (Rubin et al. 2012), and horses (Metzger et al. 2013; Tetens et al. 2013). As with dogs, large body size in most of these species is caused by truncation of the long isoform of the LCORL protein (Bai et al. 2025). Moreover, two additional candidate size variants upstream of LCORL have also been described based on size differences between Bull Terriers and Miniature Bull Terriers (Wade 2025), further indicating the complexity of the LCORL locus for controlling body size in dogs.
An additional variant revealed by this study that may correspond to a breed trait is the stop-gained at position p.Glu391* in CSF1 in Chows. The variant was homozygous in all six unrelated Chows, heterozygous in one of five Elos, and absent from two Eurasiers, the latter two breeds being the product of Chow admixture (Table 1), thus indicating a high degree of breed specificity. The Chow is a medium-sized spitz-type dog that traces its origins to China, and is characterized by its thick double coat, black pigmented tongue, straight hind legs, broad square skull and muzzle, and supernumerary third incisors (Yang et al. 2017). The genomic region containing CSF1 is under strong positive selection in Chows (Yang et al. 2017), suggesting genes in this region contribute to the breed's characteristic phenotypes. The p.Glu391* mutation is located in exon six of the secreted isoform of the CSF1 protein (Cosman et al. 1988). Rodent knockout models implicate Csf1 in skeletal and craniofacial development, specifically a domed skull, missing incisors, short thickened limb bones, fertility, and macrophage differentiation (Marks and Lane 1976; Yoshida et al. 1990; Dai et al. 2002; Van Wesenbeeck et al. 2002; Sehgal et al. 2021). While the precise function of the p.Glu391* mutation in dogs is unknown, the rodent phenotypes of a domed skull and altered number of incisors, suggest p.Glu391* may contribute to the Chow's broad square skull and two additional incisors, which contrast sharply with the Eurasier and Elo breed requirements. Similarly, the identified frameshift within ITGA11 may also correspond to craniofacial structure (Table 1). The variant is fixed in small brachycephalic breeds, and mouse knockouts of the gene demonstrate its association with small size and craniofacial abnormalities, specifically the eruption of incisors (Popova et al. 2007). Consistent with this, small brachycephalic breeds (<5 kg) have delayed tooth eruption times compared to other breed sizes and skull types (Van den Broeck et al. 2022). Also, associations between the locus and brachycephaly have been described previously but were sensitive to corrections for canine size (Schoenebeck et al. 2012; Marchant et al. 2017). Thus, the variant could contribute to a range of phenotypes, including size, craniofacial structure, and development; however, functional analyses are needed.
One of the defining features of the canine system is that often few loci contribute to large phenotypic variation, particularly for morphological traits like breed standard weight and height or ear shape and position (Rimbault et al. 2013; Plassais et al. 2019), while in the human genome, there are often large numbers of loci contributing to complex phenotypes (Yengo et al. 2022). Loci contributing to extreme phenotypic differences between breeds exhibit strong signatures of positive selection because of artificial selection for desired traits (Plassais et al. 2019; Morrill et al. 2022; Plassais et al. 2022). Our analysis shows that highly constrained genes are overrepresented in such regions and are therefore the likely targets of trait selection, driving phenotypic variation. This suggests an explanation for why CDS variation is rarely associated with morphological differences between breeds and more often associated with disease. Constrained genes are often essential for development and survival and are frequently implicated in highly penetrant genetic disorders and syndromes (Lek et al. 2016; Karczewski et al. 2020; Sun et al. 2024; Zeng et al. 2024). Trait selection in dogs is therefore a balancing act between preserving essential gene functions and maximizing regulatory variation of essential genes to drive phenotypic extremes. Future work should thus focus on assigning noncoding variants under positive selection to their target genes.
Central to the analysis is the identification of variants with phenotypic impacts, which was assessed according to evolutionary/functional constraint acting on genes/sites containing the variant in question. However, specific factors impact the interpretation of these results. For instance, the measured selection effects only capture negative selection acting on heterozygous variants with highly deleterious impacts on fitness (Fuller et al. 2019). Our analysis is therefore blind to recessive deleterious alleles and genes associated with late age of onset conditions such as breed-prevalent diseases like cancer. Since singletons are the input to the mutation model, depletion of variants with little to no deleterious impact in a heterozygous state remains unobservable, as do variants that produce no phenotype until after a dog's reproductive years have elapsed. This is relevant to canines, where a high degree of inbreeding makes dogs especially susceptible to genetic load and recessive disease allele burden (Marsden et al. 2016; Mooney et al. 2021; Donner et al. 2023). Moreover, OMIA variants segregating in canine populations likely persist because they do not have strong effects on fitness in a heterozygous state. In fact, many of the OMIA variants underlie traits like coat color or recessive and late-onset diseases, such as the many forms of progressive retinal atrophy (Miyadera et al. 2012; Downs et al. 2014; Bunel et al. 2019). Therefore, strong human ortholog LOEUF constraint lacks an association with OMIA variants because OMIA LoF variants have a low impact on fitness, and human ortholog LOEUF constraint indicates genes where heterozygous LoF mutations have a strong deleterious impact on fitness.
Another factor impacting interpretation of results is whether the unit for measuring constraint is gene or base pair. The increased resolution provided by site-specific phyloP scores is only beneficial for analyzing missense mutations, which tend to have localized effects and primarily negligible impacts on biology unless interrupting important functional domains (Gerasimavicius et al. 2022). Conversely, LoF mutations, defined throughout as premature stop codons, frameshifts, start-losses, and splice-site disruptions, impact the entire downstream transcript. Therefore, in dogs, human ortholog LOEUF is the best of the three constraint metrics for identifying LoF mutations associated with phenotypes with severe impacts on fitness while variant phyloP scores are best for prioritizing missense mutations as having potential phenotypic impacts.
The dog genome project began with two goals and both remain a high priority today: improve the health of canine companions through informed genetics and translate those findings to advance human health. While many breeds feature largely healthy, long-lived animals, others are characterized by an excess of disease alleles and associated health problems (Axelsson et al. 2021). These breeds offer a unique opportunity to make use of the approaches and data provided here to identify underlying genetic flaws, particularly for diseases of relevance to human health. Of equal priority are studies of morphologic and behavioral genetics, as identification of alleles that control extreme phenotypes associated with these traits can improve our understanding of human growth and development.
Methods
Defining a canine mutation model and callable gene set
We built a mutation model in dogs following the methods described in Samocha et al. (2014). Briefly, trinucleotide substitution probabilities, normalized according to the generation mutation rate and trinucleotide frequencies, were calculated for each possible substitution for each site within gene models. For each gene model, substitution probabilities were summed according to their consequences on CDS, such as synonymous, missense, stop-gained, stop-lost, start-lost, etc. The result was per gene probabilities for each mutation type per generation (Supplemental Fig. S12). Intronic and intergenic Dog10K variants at least 20 bp from an exon boundary and within callable loci were used to calculate trinucleotide substation rates (Meadows et al. 2023). A canine generation mutation rate of 9.02 × 10−9 was used for normalization (Supplemental Fig. S13; Bergeron et al. 2023). We calculated transcript-specific mutation probabilities using NCBI gene annotations from Dog10K, only considering sites that were callable loci, obviating the need to normalize probabilities by mean exon coverage (Supplemental Table S21). There was insufficient variant data to adjust mutation probabilities by evolutionary conservation as was performed in Samocha et al. (2014). We classified genes according to intact start and stop codons, low-quality annotation, and partial annotation. Also, for each gene, a representative transcript was chosen based on reciprocal best hit BLAST results with protein sequences of human representative transcripts. For genes with no reciprocal best hit, we used the isoform annotated as “transcript variant X1” as the representative transcript. BLAST hits were also defined as dog–human orthologs and used to assign human LOEUF constraint metrics to dog genes. For phyloP constraint, we used the Zoonomia values mapped to UU_Cfam_GSD_1.0 as part of Dog10K, which were further summarized according to gene models (Supplemental Table S22). Expected mutations per gene were calculated by sampling n gene names with replacement weighted by gene/gene group mutation probabilities, where n is the number of observations to which the expected rate is being compared. Sampled gene names were tabulated to determine the expected per gene mutation count. This process was replicated 10,000 times to generate 95% expected ranges.
Preparing genotypes for analysis
Genotyping and filtering procedures are described in full in the Supplemental Methods. Briefly, a VCF comprised of Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) genomes was constructed using GATK to complement the publicly available Dog10K VCF (Poplin et al. 2017). All WGS data were mapped to the UU_Cfam_GSD_1.0 reference assembly (Wang et al. 2021). For each VCF, samples were filtered according to multiple genotype metrics (Supplemental Table S23), which included coverage statistics (Supplemental Fig. S14) and heterozygous allele balance (Supplemental Fig. S15) in addition to others (Supplemental Fig. S16). A total of 1146 SRA and 1778 Dog10K samples remained (Supplemental Table S24). Duplicates and first-degree relatives were removed from both data sets (Supplemental Fig. S17), leaving a total of 2,291 samples in the final cohort (Supplemental Table S25). Sites were filtered according to variant quality score recalibration (VQSR) using a common training set (Supplemental Fig. S18). Autosome and Chromosome X PAR sites (Supplemental Table S26) were analyzed separately from Chromosome X non-PAR sites (Supplemental Table S27). Sites that passed VQSR filtering in both data sets were used to create a merged VCF for population-scale analyses. Sites that passed filtering in either data set and were within the CDS underwent further filtering (Supplemental Fig. S19; Supplemental Table S28). Criteria for filtering CDS genotypes depended on chromosome, sex, and genotype (Supplemental Table S29). CDS indels and SNVs were reclassified and atomized in both data sets to improve harmonization (Supplemental Fig. S20; Supplemental Table S30). Over 95% of SNVs and 80% of indels passed filtering (Supplemental Table S31). Genotype quality was the criteria that failed most often (Supplemental Table S32). Sample genotype pass rates were higher for SNVs than indels (Supplemental Table S33). CDS filtering removed genotyping biases between each data set for SNVs (Supplemental Fig. S21) and indels (Supplemental Fig. S22). Genotypes were merged and annotated with variant effect predictor (VEP) (McLaren et al. 2016). All VCF-based operations were performed with BCFtools (Danecek et al. 2021), PLINK (Purcell et al. 2007), VCFtools (Danecek et al. 2011), and vcflib (Garrison et al. 2022). LOEUF constraint values were obtained from gnomAD v4.1.0 and are based on expected rates of LoF variation from a combined set of over 800,000 human exomes and genomes (Chen et al. 2024). Zoonomia canine phyloP scores were obtained from Dog10K (Meadows et al. 2023). Details regarding sequencing of additional bifid nose dogs are presented in the Supplemental Methods.
Defining breeds and groups
Illumina CanineHD array genotypes from 2156 samples were lifted over to the UU_Cfam_GSD_1.0 reference genome using LiftoverVCF (Supplemental Table S34). WGS sites were downsampled to match array positions, and data sets were merged. After filtering at MAF > 0.01, genotyping rate > 0.9, and LD < 0.5, a total of 76,570 sites remained (Purcell et al. 2007). A consensus neighbor-joining tree was built using PHYLIP with a golden jackal as the outgroup (Felsenstein 1993). Branch support was calculated from 10% jackknifing 10,000 times. Branches needed at least 50% support. Breed/group clusters were determined by optimizing for shared breed membership on the same branch. Samples that did not match the majority breed on a branch were assigned new breed membership, matching the majority breed. Branches that consisted of overlapping breeds were merged. Breeds that could not form a branch of at least three members were placed in the unassigned category.
Canine population statistics, breed-differentiated loci, and haplotype structure
Population analyses were only performed on the 200 breeds/groups that had five or more members. Genome-wide population statistics were calculated across 200,000 randomly sampled sites. Nucleotide diversity within breeds was measured as pi, the average distance between two randomly sampled sequences in a breed (Nei and Li 1979). Breed differentiation was measured as FST, using the Weir and Cockerham estimator on a per-site basis (Weir and Cockerham 1984). FROH was calculated using the same procedures as specified in Meadows et al. (2023). We detected breed-differentiated regions by calculating FST between all 200 pairs of breeds with five or more members across 50 kb windows using a 10 kb step size. Windows with FST values >0.8, which corresponded to ∼0.1% of all pairwise comparisons, were extracted for further analysis. The density of high FST windows was then measured across the genome and the “peaks” function of IDPmisc, a peak-calling software R package, was used to define breed-differentiated regions (https://CRAN.R-project.org/package=IDPmisc). For each region we extracted breeds with pairwise FST values > 0.8 within the region's peak window. Extracted breed pairs with FST < 0.05 were defined as sharing many of the same alleles and were assigned the same cluster ID. We also defined genomic segments of extended breed homozygosity if half of the 50 kb windows along a megabase segment had π values <0.02. Breed-differentiated regions were defined as selection loci in individuals where there was overlap with extended homozygosity regions. All haplotype analyses were performed using the phased reference panel from Dog10K (Meadows et al. 2023). Only variants within 20 kb upstream of and downstream from the target variant and have an allele frequency of >1% were included. Trees were built using the “ape” v5.8 R package (Paradis and Schliep 2019).
Software availability
Computer codes generated in this analysis are available at GitHub (https://github.com/ReubenBuck/constraint_dogs) and as Supplemental Code.
Data access
All whole-genome sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under the accession numbers PRJNA726547 and PRJNA1185472. All genotyping array data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE278600. Individual accessions for all samples used in this study are listed in Supplemental Tables S1 and S34. Both tables indicate all newly released data sets associated with this study.
Competing interest statement
A.R.B. is a co-founder and board member of Embark Veterinary, Inc., a private canine DNA testing company. All other authors declare no competing interests.
Acknowledgments
We thank Tatiana Feuerborn for her helpful advice and insight throughout the project and Gabby Spatola for assistance in recruiting samples. We also thank Zachary Lounsberry and Embark Veterinary for assistance in recruiting samples and Sruthi Hundi for bioinformatic assistance. For careful reading of the manuscript and providing helpful feedback, we also thank Christopher B. Kaelin and Wesley C. Warren. Most importantly, we thank all citizen scientists who donated samples for analysis, without whom this work would be impossible. E.A.O., R.M.B., A.C.H., D.L.D., A.S.A., and H.G.P. are funded by the Intramural Program of the National Human Genome Research Institute at the National Institutes of Health (HG200377). H.L. is funded by the Jane and Aatos Erkko Foundation.
Author contributions: The project was conceptualized by R.M.B. and E.A.O. R.M.B. and A.S.A. were responsible for methodology. Software was written by R.M.B. and A.S.A., and validation experiments were performed by N.B., F.G.v.S., and M.K.H. R.M.B. led the formal analysis and R.M.B., N.B., M.K.H., H.G.P., and D.L.D. performed the investigation. Resources were provided by N.B., P.S., C.T., M.E., F.G.v.S., M.K.H., J.H., H.L., B.Ç.K., A.R.B., D.L.D., R.M.B., and E.A.O. R.M.B. and A.C.H. were responsible for data curation. Writing of the original draft was completed by R.M.B. and E.A.O., and all authors contributed to review and editing. R.M.B. created all visualizations, and E.A.O. was the primary supervisor. Project administration was carried out by R.M.B. and E.A.O., while E.A.O. was primarily responsible for funding acquisition. All photos of individual pet dogs in Figure 2B and Supplemental Figure S9 are published with permission from their owners. N.B. is credited for the photograph in Figure 2A. We also thank Antonio Virgil Guzmán, who is credited with providing the photograph used in Figure 2D.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280092.124.
-
Freely available online through the Genome Research Open Access option.
- Received October 4, 2024.
- Accepted March 10, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








