
Genetic regulation of nascent RNA maturation revealed by direct RNA nanopore sequencing
Abstract
Quantitative trait loci analyses have revealed an important role for genetic variants in regulating alternative splicing (AS) and alternative cleavage and polyadenylation (APA) in humans. Yet, these studies are generally performed with mature mRNA, so they report on the outcome rather than the processes of RNA maturation and thus may overlook how variants directly modulate pre-mRNA processing. The order in which the many introns of a human gene are removed can substantially influence AS, while nascent RNA polyadenylation can affect RNA stability and decay. However, how splicing order and poly(A) tail length are regulated by genetic variation has never been explored. Here, we used direct RNA nanopore sequencing to investigate allele-specific pre-mRNA maturation in 12 human lymphoblastoid cell lines. We find frequent splicing order differences between alleles and uncover significant single-nucleotide polymorphism (SNP)-splicing order associations in 17 genes. This includes SNPs located in or near splice sites as well as more distal intronic and exonic SNPs. Moreover, several genes showed allele-specific poly(A) tail lengths, many of which also have a skewed allelic abundance ratio. HLA class I transcripts, which encode proteins that play an essential role in antigen presentation, show the most allele-specific splicing orders, which frequently co-occur with allele-specific AS, APA, or poly(A) tail length differences. Together, our results expose new layers of genetic regulation of pre-mRNA maturation and highlight the power of long-read RNA sequencing for allele-specific analyses.
Premature messenger RNAs (pre-mRNAs) undergo several maturation steps before their release from chromatin. Most human pre-mRNAs have many introns that are removed through splicing, which takes place either cotranscriptionally or soon after transcription (Pandya-Jones and Black 2009; Tilgner et al. 2012; Yeom et al. 2021). Cleavage of the 3′-end occurs cotranscriptionally, after RNA polymerase II has transcribed through the poly(A) cleavage site, and is rapidly followed by polyadenylation, which consists of the addition of 200–250 adenines to the 3′-end of pre-mRNAs (Nicholson and Pasquinelli 2019). Alternative splicing (AS) and alternative cleavage and polyadenylation (APA), which are observed in 95% and 70% of human genes, respectively, allow extensive diversification and tailoring of cell proteomes (Pan et al. 2008; Wang et al. 2008; Derti et al. 2012).
Population-wide and quantitative trait loci (QTL) analyses have revealed an important role for common genetic variants in regulating AS and APA (Pickrell et al. 2010; Lappalainen et al. 2013; Ferreira et al. 2016; Li et al. 2016; Mariella et al. 2019; Mittleman et al. 2020; Garrido-Martín et al. 2021). Importantly, these variants harbor as much disease risk as expression QTLs, for pathologies ranging from immune disorders to schizophrenia (Li et al. 2016; Raj et al. 2018; Walker et al. 2019; Garrido-Martín et al. 2021). Yet, because these studies are generally performed with mature mRNA, they report on the outcome rather than the process, and may overlook ways in which variants can modulate pre-mRNA maturation. Indeed, mapping of alternative polyadenylation QTLs (apaQTL) in nuclear RNA revealed apaQTLs that were absent in whole cell RNA, while it also allowed to distinguish between co- and posttranscriptional regulatory mechanisms for whole cell apaQTLs (Mittleman et al. 2020). This highlights the necessity to understand how genetic variants impact RNA processing during the production and maturation of pre-mRNAs, which could in turn help explain their potential role in disease susceptibility.
Introns play a crucial role in AS control, as they include numerous sequence elements called splicing enhancers or silencers, which are bound by RNA-binding proteins (RBPs) that regulate AS (Fairbrother and Chasin 2000; Zhang and Chasin 2004; Barash et al. 2010; Blencowe 2012). As such, the order in which introns are removed may determine how long splicing regulatory elements remain within a transcript to exert their influence on AS. Early gene-specific studies (Kessler et al. 1993; Schwarze et al. 1999; Takahara et al. 2002) and recent transcriptome-wide short- or long-read sequencing studies (Kim et al. 2017; Drexler et al. 2020; Sousa-Luís et al. 2021; Zeng et al. 2022; Gohr et al. 2023) demonstrated that introns are not always removed in the order in which they appear in the nascent transcript. Our recent work extended these results by studying posttranscriptional splicing order for three to six consecutive introns (Choquet et al. 2023) using long-read direct RNA nanopore sequencing (Garalde et al. 2018). We revealed that multi-intron splicing order is predetermined, meaning that only a small subset of the possible splicing orders is used, which contributes to maintaining splicing fidelity. We found that multi-intron splicing order is largely conserved between several human cell types, consistent with the conclusions from another independent study (Gohr et al. 2023). These findings suggest that the determinants of splicing order are hardcoded in the genome.
Direct nanopore RNA sequencing (dnRNA-seq) (Garalde et al. 2018) yields long reads that allow for the simultaneous detection of several introns in the same molecule, making it ideal for investigating splicing order (Choquet et al. 2023). In addition, splicing and 3′-end processing are reciprocally regulated through interactions between RBPs over the last exon (for review, see Kaida 2016), emphasizing the importance of simultaneously interrogating distinct pre-mRNA maturation steps. Indeed, long-read sequencing studies have shown direct coupling between AS and APA in Drosophila and humans (Hardwick et al. 2022; Alfonso-Gonzalez et al. 2023; Zhang et al. 2023). Furthermore, long reads are particularly advantageous for allele-specific transcriptome analyses (Tilgner et al. 2014; Workman et al. 2019; Glinos et al. 2022), enabling the detection of several heterozygous single-nucleotide polymorphisms (SNPs) in the same read and the assignment of reads to each parental allele to uncover splicing patterns that are specific to each allele. In addition, as pre-mRNAs from both alleles share the same cellular environment and potential technical variations during sample preparation (Demirdjian et al. 2020), any RNA processing differences between the two parental alleles should result from genetic variation rather than sample-to-sample variation. Lastly, long reads capture the maturation of pre-mRNAs that were previously difficult to characterize with short-read sequencing, such as transcripts from the highly polymorphic HLA genes (Tilgner et al. 2014; Cole et al. 2020).
In this study, we used dnRNA-seq of chromatin-associated, polyadenylated RNA to investigate allele-specific posttranscriptional pre-mRNA maturation in 12 human lymphoblastoid cell lines (LCLs). We aimed to determine whether splicing order and poly(A) tail length are modulated by genetic variation, and how these key features of pre-mRNA maturation relate to one another and to APA.
Results
Subcellular dnRNA-seq across 12 lymphoblastoid cell lines
To explore the impact of genetic variants on nascent RNA processing, we leveraged naturally occurring heterozygous variants in LCLs from different individuals. We selected 12 LCLs derived from Yoruba (YRI) individuals that were part of the 1000 Genomes Project, and for which gene regulatory variations have already been extensively studied (Pickrell et al. 2010; Lappalainen et al. 2013; Li et al. 2016; Mittleman et al. 2020). We performed cellular fractionation to collect cytoplasmic and chromatin-associated RNA from each LCL, followed by dnRNA-seq of poly(A)-selected RNA (Supplemental Table S1). Overall, we obtained medians of 6.07 and 1.70 million reads per LCL for the chromatin and cytoplasm fractions, respectively, for a total of 73.5 and 20.6 million reads per fraction. Moreover, our chromatin and cytoplasmic RNA data sets were characterized by median read lengths of 1255 and 854 nt, respectively (Supplemental Table S1), consistent with the presence of longer pre-mRNAs in the chromatin fraction (Supplemental Fig. S1A). As demonstrated previously (Choquet et al. 2023), chromatin-associated RNA was enriched for partially spliced reads compared to cytoplasmic RNA across samples (Supplemental Fig. S1B).
To enable allele-specific analyses, we used the software LORALS (Glinos et al. 2022) to map nanopore sequencing reads to two personalized reference genomes per LCL and we determined their allele of origin using HapCUT2 (Supplemental Fig. S1C; Edge et al. 2017). We identified known allele-specific AS events (Supplemental Fig. S1D,E; The GTEx Consortium 2020), such as in the gene DPP7 (Fig. 1A), confirming the validity of our approach. Thus, dnRNA-seq enables allele-specific analyses of nascent and mature mRNA.
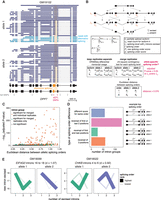
Subcellular dnRNA-seq reveals allele-specific splicing orders. (A) Example of allele-specific splicing in DPP7 in chromatin-associated RNA from LCL GM19102. Ten percent of reads mapping to each allele were randomly sampled. The gene structure is shown at the bottom, with exons as rectangles and introns as horizontal lines. The arrow represents the transcription start site. Each dark or light blue arrow represents one read, with light blue reads highlighting AS of the orange exon. The alternative intron is shaded in orange and reads are sorted based on the excision status of this intron. Heterozygous SNPs in LCL GM19102 are shown as vertical bars below the gene, with dark pink bars representing previously identified sQTL SNPs. (B) Schematic of splicing order computation for groups of three introns. Each intermediate isoform k at splicing levels 1 (one intron excised, L1) and 2 (two introns excised, L2) is depicted. The associated read counts ck are used to calculate splicing order scores for each of the six possible orders. The DRIMSeq differential transcript usage is used to test for differential splicing order using individual replicates (Ra, Rb), while allelic splicing orders from merged replicates are compared using a χ2 contingency test and the Euclidean distance between orders. (C) Volcano plot showing the results of allele-specific splicing order analysis from merged replicates. Each dot represents one intron group. Intron groups that showed a significant difference in the analyses with individual replicates and merged replicates are shown in orange. (D) Type of splicing order difference as a function of the Euclidean distance between allelic splicing orders. The different categories are schematized on the right, with the number representing the order in which each intron is removed, and are defined in more detail in the Methods. The transcript in gray has a lower top splicing order score than the ones in black. (E) Splicing order plots showing allele-specific splicing orders for two example intron groups. The thickness and opacity of the lines are proportional to the frequency at which each splicing order is used, with the top-ranked order per intron group set to the maximum thickness and opacity. d indicates the Euclidean distance between alleles.
Splicing order variation between alleles
Previous work from our group and others suggest that splicing order is mostly determined by features that do not vary across cell types (Choquet et al. 2023; Gohr et al. 2023). We hypothesized that nucleotide sequence is the primary determinant of splicing order, in which case we would expect that some genetic variants lead to changes in splicing order. Thus, we sought to determine the extent of splicing order variation across individuals. We previously found good agreement between co- and posttranscriptional splicing order (Choquet et al. 2023). We focused on posttranscriptional splicing order because cotranscriptional splicing order is challenging to study with current dnRNA-seq read lengths, as most reads corresponding to elongating transcripts are completely unspliced (Drexler et al. 2020; Choquet et al. 2023). In addition, splicing quantitative trait locus (sQTL) SNPs were found to be moderately enriched in introns that are removed posttranscriptionally (Garrido-Martín et al. 2021), making this intron population of particular interest. As in our previous study (Choquet et al. 2023), our analysis was limited to groups of three introns in highly expressed genes due to the current read length and coverage of dnRNA-seq (Supplemental Fig. S1F). For each group of three proximal introns that met our coverage thresholds (see Methods) on both alleles, we extracted read counts for each isoform containing a combination of introns that had already been excised and introns that were still present (intermediate isoforms). We calculated their frequency relative to other intermediate isoforms with the same number of excised introns (splicing level). For each possible splicing order, the splicing order score was obtained by multiplying the frequency of the corresponding two intermediate isoforms (one per splicing level), as we recently described (Fig. 1B; Methods; Choquet et al. 2023). We applied our strategy to each allele separately, yielding “allelic splicing orders” (Supplemental Table S2). Similar to what we observed without allele specificity (Choquet et al. 2023), most intron groups displayed only 1–2 predominant splicing orders with scores >0.25 on each allele, out of the six possible splicing orders (Supplemental Fig. S2A; Supplemental Table S2). We were able to compute allelic splicing orders for 58–1947 intron groups in each LCL (median of 638), depending on the sequencing depth and the number of heterozygous loci in highly expressed genes (Supplemental Fig. S1F). We also assembled splicing orders without allele specificity (“allele-agnostic” splicing orders) for each LCL, which were highly correlated with allelic splicing orders (Supplemental Fig. S2B–D).
Analysis of biological or technical replicates from the same cell line (Supplemental Table S1) showed strong reproducibility of allelic splicing orders (Supplemental Fig. S3A). Conversely, comparing splicing orders between alleles within each LCL revealed numerous intron groups with strong differences in splicing order scores (Supplemental Fig. S3B). Of note, due to sequencing coverage constraints, allelic splicing order analysis was possible in only one LCL for almost half of the intron groups (Supplemental Fig. S4A). HLA class I genes were a notable exception, with all intron groups meeting our allelic analysis thresholds in five or more LCLs (Supplemental Fig. S4B), likely due to their high density of polymorphisms (Voorter et al. 2016), thus enabling deeper insight into these genes.
To systematically identify splicing order differences between alleles across our data set, for each intron group and LCL, we computed the Euclidean distance between vectors consisting of the splicing order scores for each allele (Fig. 1B). Intron groups that displayed the same splicing orders between alleles were characterized by a small distance, while differences in splicing order scores led to an increased distance (Fig. 1C; Supplemental Fig. S4C). Analysis of the Euclidean distance distribution between replicates of the same allele was used to define the threshold (interquartile range × 1.5 = 0.379) for identifying differences between alleles (Supplemental Fig. S4C; Supplemental Methods). As an orthogonal approach, we compared the distribution of intermediate isoform counts between alleles at each splicing level using a χ2 contingency test (Supplemental Fig. S4D,E). Hereafter we refer to intron groups with a Euclidean distance >0.379 and χ2 test false discovery rate (FDR) < 0.05 for at least one splicing level as displaying “allele-specific splicing order.”
More than half of intron groups displaying allele-specific splicing orders were detected in a single LCL (Supplemental Fig. S4F), with those in HLA class I genes representing outliers that were detected in many LCLs (Supplemental Fig. S4G). Of the 3564 unique intron groups that were analyzed, 159 (4.5%) showed differences in splicing order between alleles in at least one LCL (Fig. 1C). When counting all instances of intron groups with allele-specific orders even when they were observed in multiple LCLs, we detected 272 intron groups with allele-specific splicing order (Supplemental Table S2). Intron groups that showed differences most frequently (57%) displayed changes in the splicing order of 2 introns (e.g., EIF4G2 and CHKB) (Fig. 1E), while 22% showed changes in the relative order of all three introns (Fig. 1D). To assess the reproducibility of allele-specific splicing orders, we analyzed differential intermediate isoform usage between alleles without merging replicates using DRIMSeq (Nowicka and Robinson 2016), a package developed for differential transcript usage (DTU) analysis in small sample sizes (Fig. 1B). This revealed 62 unique intron groups with reproducible differences in differential intermediate isoform usage in at least one cell line. All of these overlapped with significant intron groups in the χ2 contingency test and 50 (81%) had an Euclidean distance above the threshold with merged replicates. Thus, although the sequencing depth does not allow the study of allelic splicing order across multiple replicates for all intron groups, this analysis demonstrates the reproducibility of allele-specific splicing orders. Lastly, we investigated splicing order within 8292 unique intron pairs that did not meet our filters to be considered as part of groups of three introns, of which 114 showed significant allele-specific splicing order (Supplemental Table S3). These findings indicate that splicing order is frequently modulated by allelic nucleotide sequence, solidifying our hypothesis that genetic sequence is the primary determinant of splicing order.
Specific genetic variants are associated with allele-specific splicing orders
Considering our frequent observations of allele-specific splicing orders, we next wondered whether specific genetic variants were associated with splicing order variation. We tested the association between each SNP and intermediate isoform counts across LCLs using the DRIMSeq transcript usage QTL (tuQTL) tool (Nowicka and Robinson 2016). SNPs within one gene that shared the same genotypes across samples were grouped into “haplotype blocks.” This revealed 472 unique SNPs in 127 haplotype blocks that were significantly associated with allele-specific splicing order of 29 intron groups in 17 genes (Supplemental Table S4). Previous work has revealed moderate associations between splicing order and several sequence features, including splice site strength (Drexler et al. 2020; Zeng et al. 2022; Choquet et al. 2023; Gohr et al. 2023). Consistently, a SNP at the last nucleotide of exon 8 in CCM2, which reduced the 5′SS strength of intron 8 from 8.81 to 2.69 (Yeo and Burge 2004), was associated with an excision order change of introns 6–8 (Fig. 2A). Moreover, SNPs near the branchpoint of intron 4 in CHKB and near the 5′SS of intron 3 in CYBA were associated with excision order changes of the corresponding intron and the second-next intron (Fig. 2A).

Splicing order changes are associated with specific genetic variants. (A, Left) Schematic representation of each gene, with exons as rectangles and introns as horizontal lines. SNPs represented in the right plots and other SNPs in the same haplotype blocks are depicted in red, while SNPs that are significantly associated with splicing order of the same intron group but in a different haplotype block are shown in light orange. The distance between the SNPs represented in the right plots and the closest splice site are depicted in blue. (Right) Examples of splicing orders in five genes that are significantly associated with the genotypes of the indicated SNPs. Each point represents one allele and the identity of the splicing order is shown at the top of each plot, with intron numbers separated by arrows. Each LCL is displayed in a different shape. (B) Splicing order plot showing allele-specific splicing orders for introns 3–5 of RPS2 in five LCLs. The thickness and opacity of the lines are proportional to the frequency at which each splicing order is used, with the top-ranked order per intron group set to the maximum thickness and opacity. The gene structure is shown at the top, with exons as rectangles, introns as lines, and associated SNPs as in (A).
We also identified intron groups for which associated SNPs were outside of splice sites. In OXA1L introns 4–6, the only associated SNP within the intron group was 46 nt from the 5′SS (Fig. 2A). Moreover, RPS2 intron 3 was removed before introns 4 and 5 on one allele, while this order was reversed on the second allele in five different LCLs (Fig. 2B). Multiple SNPs in linkage disequilibrium were associated with these RPS2 allele-specific splicing orders (Fig. 2B). The majority were located within intron 3 but at least 59 nt from the splice sites, suggesting that some of these more distal intronic variants influence the timing of removal for this intron relative to its neighbors (Fig. 2B). Lastly, in EEF1A1 introns 3–5, the closest associated SNP was located two exons and 504 nt downstream from the considered intron group (Fig. 2A). Thus, our analysis suggests that splicing order may be regulated by changes to the consensus splice site sequences as well as more distal features, including outside of the affected exon.
We next explored potential genetic coregulation of splicing order and AS. We found little overlap between SNPs associated with splicing order and previously published sQTL SNPs (Supplemental Table S4; Li et al. 2016; The GTEx Consortium 2020). Accordingly, there were few allele-specific AS events in intron groups with allele-specific splicing orders (Supplemental Table S2). Although the small number of intron groups studied here prevents us from drawing global conclusions, these findings hint that genetic regulation of splicing order is frequently distinct from that of AS in the analyzed genes. Nevertheless, we note that RPS2 intron 3 displayed an alternative 5′SS on the same alleles that show delayed removal of this intron in three LCLs (Fig. 2B; Supplemental Table S2), suggesting an interplay between AS and splicing order might occur in some genes.
Most splicing orders lead to productive splicing and nuclear export
We next inquired whether all the observed splicing orders represent productive splicing paths, as some could lead to long-term intron retention and subsequent nuclear retention and decay. To determine whether allele-specific splicing orders impact the allelic balance of cytoplasmic isoforms, we compared allelic transcript ratios between chromatin and cytoplasm for all genes that showed allele-specific splicing order differences and for which alleles could be distinguished in the cytoplasm (n = 92 gene/LCL pairs, “splicing order genes”). Globally, we found a significant correlation (R = 0.58) between chromatin and cytoplasmic allelic ratios for these splicing order genes (Fig. 3A), which was higher than the correlation observed when all genes detected in both compartments were considered (R = 0.37) (Supplemental Fig. S5A). The majority of gene/LCL pairs had an allelic ratio between 0.4 and 0.6 in both compartments (e.g., GAPDH) (Fig. 3B). Overall, only 7% of gene/LCL pairs showed a substantial difference in allelic ratios between compartments. The most notable outlier was RPS2, for which we had detected strong splicing order differences (Fig. 2B). We observed that the allele associated with delayed removal of intron 3 accumulated on chromatin, while the allelic ratio was closer to 0.5 in the cytoplasm (Fig. 3C). Observation of the expected allelic ratio in the cytoplasm suggests that while removal of intron 3 is delayed on one allele, all transcripts are eventually spliced and exported to the cytoplasm. Moreover, the correlation between compartments for the splicing order genes increased (R = 0.72) when RPS2 was removed. Thus, adopting different splicing orders does not appear to substantially influence relative mRNA abundance between alleles in the cytoplasm.
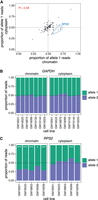
Splicing order changes do not alter cytoplasmic mRNA abundance. (A) Correlation in allele-specific mRNA abundance between chromatin and cytoplasm for intron groups that showed significant allele-specific splicing orders. The proportion of allele 1 reads divided by the total number of reads for alleles 1 and 2 is shown for each subcellular compartment. Each dot represents one gene in one LCL. RPS2 is highlighted in blue dotted circles for the five LCLs that showed splicing order changes in this gene and that are displayed in Figure 2B. (B) and (C) Proportion of (B) GAPDH (B) or (C) RPS2 reads mapping to each allele in chromatin and cytoplasm. The number of reads mapping to each allele on chromatin-associated or cytoplasmic RNA was compared using Qllelic (Mendelevich et al. 2021) or a two-sided binomial test, respectively. (***) P-value < 0.001 and proportion of allele 1 reads <0.4 or >0.6; (*) P-value < 0.01 and proportion of allele 1 reads <0.45 or >0.55.
Genetic regulation of nascent poly(A) tail length
Poly(A) tails are added to newly synthesized pre-mRNAs on chromatin immediately after 3′-end cleavage. Following nuclear export, poly(A) tails are progressively shortened throughout the mRNA lifetime (Nicholson and Pasquinelli 2019). Nascent poly(A) tail length was long thought to be relatively constant across all nuclear pre-mRNAs (200–250 nt) (Nicholson and Pasquinelli 2019), but we recently found substantial intergene variability of poly(A) tail length on chromatin-associated RNA (Ietswaart et al. 2024), which was closely associated with splicing progression (Choquet et al. 2023). However, how nascent poly(A) tail length is regulated or connected to splicing remains unknown. Therefore, we sought to establish whether poly(A) tail length is influenced by genetic variation. Poly(A) tail lengths correlated well between replicates (Supplemental Fig. S5B,C). As expected, poly(A) tails were longer and less variable on chromatin than in the cytoplasm, with respective medians of 176 and 97 nt (Supplemental Fig. S6A,B). We uncovered 37 genes (65 gene/LCL pairs) with statistically significant differences (Wilcoxon rank-sum test) in poly(A) tail length between alleles on chromatin-associated RNA, with only one gene, HLA-DQA1, identified as having a tail length difference in the cytoplasm (Supplemental Table S5; Supplemental Fig. S6C). This number is likely an underestimation, as poly(A) tail length measurements with dnRNA-seq are inherently noisy and require substantial coverage to detect differences. Indeed, 87% of genes with significant differences had at least 200 reads across both alleles, but this level of coverage was achieved by only 23% of assessed genes (Supplemental Table S5). A correlation between allelic chromatin and cytoplasmic poly(A) tail lengths was not observed (Supplemental Fig. S6D), consistent with the progressive deadenylation occurring in the cytoplasm (Nicholson and Pasquinelli 2019) and the positive correlation between the extent of deadenylation after nuclear export and cytoplasmic mRNA half-lives (Ietswaart et al. 2024). Among genes with allele-specific nascent poly(A) tail lengths, some showed robust differences across several LCLs. For example, in six LCLs, ERAP2 displayed an average difference of 89 nt between the two alleles, while in five LCLs, RPS2 exhibited an average difference of 39 nt (Fig. 4A,B). Thus, our results suggest that nascent poly(A) tail length can be influenced by genetic variants, while such an association is rarer for cytoplasmic mRNA.
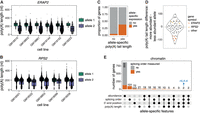
Genetic regulation of poly(A) tail length. (A,B) Examples of allele-specific poly(A) tail length in (A) ERAP2 and (B) RPS2. Each dot represents one read. A two-sided Wilcoxon rank-sum test was used to compare tail length distributions between alleles for each LCL. All tests were statistically significant (adjusted P-value < 0.001). (C) Proportion of genes that showed a skewed chromatin-associated RNA abundance ratio (allele 1 reads/total reads) out of genes with significantly different poly(A) tail lengths or not. The two groups were compared with a two-sided Fisher's exact test (P-value = 9.232 × 10−16). (D) Violin plot of the distribution of the difference in poly(A) tail length between the more abundant and the less abundant allele for genes with a skewed chromatin-associated RNA abundance ratio. Each dot represents one gene in one LCL. (E) UpSet plot showing the number of genes with skewed chromatin-associated RNA allelic abundance ratio (<0.4 or >0.6), allele-specific splicing order, significant differences in 3′-end position and/or significant differences in poly(A) tail length between alleles.
Hyperadenylation of nascent RNA has been associated with targeting for nuclear decay (Bresson and Conrad 2013; Bresson et al. 2015). Thus, we asked whether poly(A) tail length differences were accompanied by abundance imbalances between alleles. Genes with allele-specific poly(A) tail length were significantly enriched among genes with skewed nascent RNA abundance ratios (P-value = 9.232 × 10−16, Fisher's exact test) (Fig. 4C). However, we found that the allele with the longest poly(A) tails could either be the least abundant (e.g., ERAP2) or the most abundant (e.g., RPS2) (Fig. 4A,B,D), suggesting that long poly(A) tails are not solely due to nuclear decay targeting and indicating a complex relationship between these two variables. Indeed, allele-specific abundance could also be the result of different RNA synthesis levels that may in turn impact poly(A) tail length.
Given the connections between 3′-end cleavage, polyadenylation, and splicing (Kaida 2016; Choquet et al. 2023; Zhang et al. 2023), we investigated whether allele-specific poly(A) tail lengths, splicing order and/or APA co-occurred in some transcripts. We first asked whether tail length differences on chromatin were primarily driven by partially spliced or fully spliced reads. The majority of genes showed significant differences in tail length for each read class (i.e., all reads, fully spliced reads only, partially spliced reads only) or only when all reads were considered, indicating that splicing progression is not the main reason for differences in poly(A) tail length, with some exceptions (Supplemental Fig. S6E,F). Overall, 21 gene/LCL pairs showed both allele-specific splicing order and poly(A) tail length differences (Fig. 4E), including RPS2 (Fig. 4B). To analyze APA, we extracted the genomic location of the 3′-end of each read and compared the distribution of 3′-ends between alleles, recapitulating known allele-specific APA events, such as in IRF5 (Supplemental Fig. S6G; Graham et al. 2007). Most genes with allele-specific APA did not show differences in splicing order or poly(A) tail length (n = 88 gene/LCL pairs), while 10 and 25 gene/LCL pairs also displayed poly(A) tail length or splicing order differences, respectively (Fig. 4E; Supplemental Table S6). Together, these results indicate that poly(A) tail length can be genetically regulated, and that this sometimes, but not necessarily, overlaps with other features such as pre-mRNA abundance, splicing progression, and APA.
Multilayered genetic regulation of HLA transcripts on chromatin
While many transcripts were heterozygous in a small number of LCLs, we reasoned that focusing on genes with higher level of variation across LCLs would provide greater power to dissect the influence of genetic variants on the different steps of pre-mRNA maturation and the interrelation between them. HLA genes are the most polymorphic loci in humans (Voorter et al. 2016) and are highly expressed in LCLs (Supplemental Fig. S7A). Accordingly, we detected HLA class I transcripts (HLA-A, -B, -C) in most cell lines in our data set (Supplemental Fig. S4B,G). Given that alternative RNA processing can alter the identity or the levels of HLA proteins and affect their ability to present peptides to the immune system (Voorter et al. 2016), it is critical to understand how polymorphisms impact HLA pre-mRNA maturation. However, due to the technical challenges associated with analyzing HLA transcripts using short-read RNA-seq reads, HLA genetic variation has been primarily studied at the DNA level (Voorter et al. 2016; Cole et al. 2020), with little information as to whether and how this variation impacts pre-mRNA maturation. While the majority of intron groups with splicing order differences displayed only one or two different top splicing orders across LCLs (Supplemental Fig. S4F), HLA class I genes frequently exhibited three or more orders (Fig. 5A), consistent with their higher degree of polymorphism. Furthermore, the majority of intron groups in these genes showed splicing order differences between alleles in most LCLs (Fig. 5A; Supplemental Fig. S7B–D). For example, six HLA-B alleles (blue box in Fig. 5B) had strong usage of splicing orders 3 → 5 → 4 and 6 → 5 → 4, which were never observed on other alleles. Splicing orders for both intron groups were associated with numerous SNPs, including some in introns 4 and 5 (Supplemental Table S4). The other alleles displayed removal of intron 4 before intron 5, suggesting that some of these SNPs may be responsible for this strong splicing order reversal.
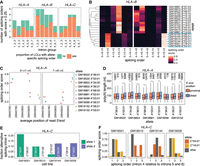
Multilayered regulation of nascent HLA class I transcripts. (A) Number of splicing orders observed across LCLs for each intron group in HLA class I genes. Each bar is colored based on the proportion of LCLs showing allele-specific splicing order for that intron group. (B) Heatmap representing splicing orders for introns 3–5 and 4–6 in HLA-B across LCLs. Each line shows one allele and each column shows one possible splicing order. The squares are colored based on the splicing order score. Alleles were clustered based on their genotypes across the HLA-B gene, so genetically similar alleles are located close to one another. The blue box highlights alleles that have delayed removal of intron 4 relative to other alleles. (C) Splicing order scores as a function of average genomic read 3′-end position in HLA-A. Each dot represents one allele. The splicing order is shown at the top of each plot, with intron numbers separated by arrows. (D) Poly(A) tail length distribution of HLA-A reads separated by allele, based on whether the read ends near the proximal or the distal 3′-end site. Poly(A) tail lengths were compared using a two-sided Wilcoxon rank-sum test. (***) P-value < 0.001, (**) P-value < 0.01. (E) Fraction of reads showing alternative excision of intron 4 in HLA-C, corresponding to exon 5 skipping. (F) Splicing order score as a function of the position of intron 4 in each possible splicing order (removed first, second, or third). The alleles for which there is AS of intron 4 in (E) (HLA-C*04:01 and *16:01) show delayed removal of this intron relative to the other allele.
HLA-A was one of only two genes that displayed allele-specific splicing order, poly(A) tail length, 3′-end position, and abundance (Fig. 4E). These features appear to be coregulated at the genetic level. Indeed, predominant usage of the proximal (e.g., HLA-A*36:01) or distal (e.g., HLA-A*03:01) 3′-end cleavage sites were, respectively, associated with strong usage of the splicing orders 6 → 7 → 5 or 7 → 6 → 5 (Fig. 5C). In addition, 3′-end cleavage at the distal site was associated with slightly longer poly(A) tails across most alleles (Fig. 5D), consistent with a previously reported positive correlation between 3′ UTR and poly(A) tail lengths (Alles et al. 2023). These observations suggest that 3′-end choice, poly(A) tail length, and excision order of the last two introns are coupled, although we cannot exclude that genetic variants independently act on each feature.
Additionally, we observed AS of exon 5 on HLA-C*04:01 and *16:01 alleles in four LCLs (Fig. 5E), as previously reported (Ehlers et al. 2022). While the splicing orders varied between HLA-C*04:01 and *16:01 alleles, they were all associated with delayed removal of flanking intron 4 (Fig. 5F), suggesting that AS and splicing order may be intimately linked in some genes. Despite allelic differences in pre-mRNA maturation, the allelic ratio was close to 0.5 for HLA class I genes across most LCLs, both on chromatin and in the cytoplasm (Supplemental Fig. S7E), indicating that allele-specific splicing orders do not substantially impact mRNA abundance in these LCLs. Nevertheless, AS, APA, and poly(A) tail length could impact translation, while splicing order may influence these aforementioned features, emphasizing the importance of understanding how the HLA transcript life cycle is regulated. Our analyses highlight that HLA class I genes can be regulated at these distinct levels, with potential downstream functional consequences.
Discussion
Here, we performed dnRNA-seq of cytoplasmic and chromatin-associated, poly(A)-selected RNA in LCLs from 12 individuals for which multiple other functional genomics data sets already exist (Pickrell et al. 2010; Lappalainen et al. 2013; Li et al. 2016; Mittleman et al. 2020). To our knowledge, this is the first long-read subcellular RNA sequencing data set across multiple individuals. In this study, we focused on the allele-specific analysis of two key aspects of pre-mRNA maturation, revealing that both proximal and distal variants are associated with splicing order changes and that nascent poly(A) tail lengths can differ by tens of nucleotides between alleles. Our findings highlight how analyzing specific RNA subpopulations can uncover new layers of genetic regulation during gene expression.
We observed that several groups of three introns displayed allele-specific splicing orders. As both alleles were subjected to the same cellular environment and experimental procedures (Demirdjian et al. 2020), our findings demonstrate the crucial role of nucleotide sequences in splicing order determination and expand on previous studies showing that splice site and branchpoint sequences are moderately associated with splicing order (Drexler et al. 2020; Zeng et al. 2022; Choquet et al. 2023; Gohr et al. 2023). Consistently, several of the SNPs that we identified were located in splice sites and were associated with robust splicing order changes. Nevertheless, other SNPs were located further away from splice sites or from the intron groups themselves, though we note that short indels, which were not considered in this study, could also influence splicing order. Although future studies will be needed to confirm the causality of these genetic variants in modulating splicing order, our findings suggest that splicing order determination can involve regulatory elements located distally from intron–exon boundaries through mechanisms yet to be identified. Genetic variants could modulate splicing order by modifying RBP binding sites or altering the secondary structure of pre-mRNA, which was shown to impact splicing efficiency and AS (McManus and Graveley 2011; Taliaferro et al. 2016; Saha et al. 2020; Saldi et al. 2021) through local or long-range intramolecular RNA–RNA interactions that are enriched in posttranscriptionally removed introns (Lovci et al. 2013; Kalmykova et al. 2021; Vorobeva et al. 2023; Margasyuk et al. 2023a,b). Genetic variants, either individually or in combinations, could disrupt these structures to favor earlier or delayed removal of an intron relative to its neighbors, leading to the allele-specific splicing orders observed herein, especially when the associated SNPs are located away from splice sites. Future studies aimed at studying short- and long-range intramolecular RNA interactions in LCLs would help to shed light on such mechanisms.
Similar to our previous study (Choquet et al. 2023), we focused on posttranscriptional splicing order, through which 30%–40% of mammalian introns are removed (Khodor et al. 2012; Tilgner et al. 2012; Yeom et al. 2021; Choquet et al. 2023). While our previous study suggested a high concordance between post and cotranscriptional splicing orders (Choquet et al. 2023), posttranscriptional splicing may provide additional time for genetic variants to influence splicing and may result in a higher occurrence of allele-specific splicing orders. As dnRNA-seq read lengths continue to improve, similar analyses of cotranscriptional splicing will help unravel the link between splicing timing and the effect of genetic variants. Furthermore, we were limited to analyzing 3564 groups of three introns in highly expressed genes due to the current read length and coverage restrictions of dnRNA-seq (relative to the 119,936 intron groups in genes expressed in LCLs). Thus, the number of genes with allele-specific splicing orders or poly(A) tail lengths may not be reflective of the entire transcriptome and could be an under- or overestimation. In many intron groups, we did not find any genetic variant whose genotype was significantly associated with allele-specific splicing orders. While this is likely partly due to our small sample size, it is also possible that some allele-specific splicing orders result from combinatorial effects of several SNPs, as was shown for variants in DNA that physically interact to regulate gene expression (Corradin et al. 2016). This could be especially relevant if RNA structure is an important determinant of splicing order, and several genetic variants in a pre-mRNA favor the formation of alternative local and/or long-range interactions. Studies with larger cohorts will provide more power to identify instances consistent with such a mechanism.
We observed that splicing order changes did not affect cytoplasmic mRNA abundance, raising the question of the functional role of splicing order. We previously found that perturbing splicing order by blocking or mutating splice sites or depleting U2 snRNA severely impaired splicing fidelity (Choquet et al. 2023). However, most allele-specific splicing orders were not accompanied by increased AS or aberrant splicing. We propose that SNPs that impair splicing fidelity by modifying splicing order would be deleterious and have been negatively selected through evolution. Thus, common genetic variants such as the ones studied here are likely enriched for genes in which different splicing orders can be used without detrimental consequences or have advantages that led to their evolutionary selection. For example, in HLA-C, we found an association between exon 5 skipping and delayed intron 4 removal (Fig. 5E,F), consistent with our previous observations that introns flanking alternative exons tend to be removed later in motor neurons (Choquet et al. 2023). Future splicing order analyses of sQTL genes that did not have sufficient coverage to be investigated in this study could help to shed light on the relationship between AS and splicing order. Moreover, several recent studies have shown a connection between posttranscriptional splicing timing and response to stimuli (e.g., cell differentiation, neuronal stimulation, heat shock, etc.) (Shalgi et al. 2014; Mauger et al. 2016; Yeom et al. 2021; Mazille et al. 2022), where delayed splicing and nuclear transcript retention allow to form a pool of almost mature pre-mRNAs that can be spliced and exported upon specific signals. Some allele-specific splicing orders may provide a functional advantage for the response to stress or environmental signals that would only become apparent when performing a similar study in combination with different perturbations.
The use of dnRNA-seq enabled us to query allelic differences in poly(A) tail lengths of chromatin-associated RNA. Some genes, where the least abundant allele had the longest tail (e.g., ERAP2) (Fig. 4A), were consistent with hyperadenylation of nuclear mRNAs to target them for decay through the nuclear exosome (Bresson and Conrad 2013; Bresson et al. 2015). On the other hand, genes for which the most abundant allele had the longest tail (e.g., RPS2) (Fig. 4B) are more in line with our previous observations of a positive correlation between poly(A) tail length and the time that transcripts spend on chromatin (Ietswaart et al. 2024), suggesting that polyadenylation continues as long as the transcript is on chromatin. How polyadenylated mRNAs remain tethered to the chromatin is still unknown, but one hypothesis is that splicing completion acts as a licensing step for transcript release (Brody et al. 2011; Hochberg-Laufer et al. 2019; Yeom et al. 2021; Ietswaart et al. 2024). Although the individual contribution of these mechanisms to nascent poly(A) tail length control remains to be elucidated, our findings highlight previously underappreciated regulation at this level of gene expression.
HLA class I genes were not subjected to the sample size and heterozygosity limitations noted above, which allowed us to investigate the interplay between different pre-mRNA maturation steps in an allele-specific manner. Notably, we found an association between the removal order of the last three introns and 3′-end APA in HLA-A, suggesting that the order or timing in which terminal introns are removed relative to their neighbors may be important for 3′-end choice (Kaida 2016). Further experiments are required to establish whether the co-occurrence of splicing order changes and APA is a special case in HLA-A or is widespread. Nonetheless, our results further emphasize the power of long-read sequencing to uncover connections between distinct pre-mRNA maturation steps and to decipher how genetic variants modulate these steps, which will help to unravel the mechanisms linking variants to human traits and diseases.
Methods
Cell lines
Human LCLs (Supplemental Table S1) were purchased from the Coriell Institute for Medical Research. LCLs were maintained at 37°C and 5% CO2 in RPMI 1640 medium (Gibco 11875119) containing 10% fetal bovine serum (Gibco 10437036), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco 15140122). Cells were split every 3–5 days when they reached a density of ∼1 million cells/mL.
Direct RNA sequencing of chromatin-associated and cytoplasmic RNA
Cellular fractionation and RNA extraction steps were performed as previously described (Drexler et al. 2021) and are outlined in the Supplemental Methods. Poly(A)+ RNA was purified using the Dynabeads mRNA purification kit (Invitrogen 61006) according to the manufacturer's instructions, starting with up to 40 μg of chromatin-associated RNA and 75 μg of cytoplasmic RNA. Direct RNA library preparation was performed using the SQK-RNA002 or SQK-RNA004 kits (Oxford Nanopore Technologies) with 500–700 ng of poly(A)+ RNA according to the manufacturer's instructions with the following exceptions: the RNA Calibration Strand was omitted and replaced with 0.5 μL water and the ligation of the reverse transcription adapter was performed for 15 min. For some initial samples (see Supplemental Table S1), a yeast spike-in control was added to the libraries, as described in Choquet et al. (2023). Sequencing was performed for up to 72 h with FLO-MIN106D, FLO-PRO002, or FLO-PRO004RA flow cells on a MinION or a PromethION 2 Solo device (Oxford Nanopore Technologies) (Supplemental Table S1). Biological replicates are defined as samples from the same cell line for which the RNA was collected and sequenced independently, while technical replicates correspond to different sequencing libraries from the same RNA sample (Supplemental Table S1). For cell lines sequenced on a MinION, 3–6 flow cells were used on biological or technical replicates, while a single flow cell was used for each replicate sequenced on the PromethION 2 Solo.
Nanopore data processing
Nanopore sequencing was performed with MinKNOW (release 22.03.5 or later). High accuracy basecalling was performed with Dorado v0.7.0 using default parameters and optional parameter ‐‐estimate-poly-a. Basecalled BAM files from Dorado were converted to FASTQ using the SAMtools fastq command (Li et al. 2009). Reads were aligned to the reference human genome (Ensembl GRCh38 [release-86]) using minimap2 (Li 2018) with parameters -ax splice -uf -k14, followed by haplotype-aware alignment using LORALS (Supplemental Methods; Glinos et al. 2022).
Computation of splicing order
Splicing order for groups of three introns was computed as described in Choquet et al. (2023). Briefly, intron groups were analyzed when they met the following criteria: (1) each intron was retained in at least 10 reads
in each allele; (2) each splicing level, defined as the number of excised introns within each read for the considered intron
group, was represented by at least 10 reads that spanned all introns in the intron group of interest for each allele; and
(3) the number of reads per allele at each splicing level was greater than twice the number of reads for which the allele
could not be determined. For duplicated intron groups with the same genomic coordinates within different transcripts, only
one instance was kept. For overlapping intron groups in distinct transcripts that differ by an AS event (e.g., exon inclusion
or exclusion), splicing order was measured separately for the two possible intron groups using different sets of reads that
match each junction. For each splicing level L and allele A, the frequency fk of each possible intermediate isoform k was recorded by dividing the number of reads matching this intermediate isoform by the total number of reads at that splicing
level. Next, we iterated through each level L, where for each observed intermediate isoform k, we identified the intermediate isoform(s) at the previous splicing level L − 1 from which the isoform under consideration could originate. Those intermediate isoforms were connected within a possible
splicing order path and their frequencies fk were recorded. After iterating through each level, the frequencies of patterns supporting each possible splicing order i were multiplied to yield the raw splicing order score Pi, where N is the total number of intermediate isoforms supporting a given splicing order (4 for groups of 3 introns):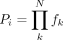
These raw scores Pi were further divided by the sum of all raw scores for the considered intron group, where n is the total number of observed splicing orders for the intron group. This yielded the final splicing order score pi such that the sum of all scores pi was equal to 1: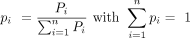
Only introns with “excised” or “not excised/present” statuses were considered for this analysis. Additional splicing order analyses are described in the Supplemental Methods.
Identification of splicing order differences between alleles
To identify statistically significant differences in allelic splicing order for groups of three introns, we compared the distribution of intermediate isoform counts between alleles for each splicing level (one intron excised or two introns excised) using a χ2 contingency test, followed by multiple testing correction with the Benjamini–Hochberg method. To measure the extent of the difference between alleles, we calculated the Euclidean distance between splicing order scores by defining a vector with the six possible splicing order scores per intron group and allele, with the same order of splicing orders for the two alleles. Intron groups were considered to display allele-specific splicing when the χ2 contingency test FDR < 0.05 for one or both splicing levels and Euclidean distance between allelic splicing order vectors >0.379 (see Supplemental Methods for details on threshold selection). As an orthogonal approach, we used the DRIMSeq (Nowicka and Robinson 2016) v1.30.0 DTU workflow to identify allele-specific splicing orders without merging replicates. We extracted intermediate isoform counts from intron groups for which allelic splicing orders could be computed according to the filtering thresholds outlined in the “Computation of splicing order” section. Intermediate isoform counts were separated by splicing level (e.g., one intron excised or two introns excised), yielding three possible intermediate isoforms per splicing level. We used the script detect_differential_isoforms.py from rMATS-long v1.0.0 (https://github.com/Xinglab/rMATS-long) to execute the DRIMSeq DTU workflow, with each unique intron group at one splicing level representing a “gene” and each intermediate isoform representing a “transcript.” For each LCL, the two alleles represented the two groups being compared and the replicates were used as individual samples. Intron groups with adjusted P-value < 0.05 for at least one splicing level were considered to display allele-specific splicing order. Additional analyses to characterize allele-specific splicing orders are described in the Supplemental Methods.
Testing for association between splicing orders and genetic variants
To identify SNPs associated with allele-specific splicing order, we used the tuQTL analysis workflow from DRIMSeq (Nowicka and Robinson 2016) with intermediate isoform counts from merged replicates. We extracted intron groups for which at least two LCLs displayed allele-specific splicing orders (χ2 contingency test adjusted P-value < 0.05 for one or both splicing levels and Euclidean distance >0.379). We retrieved the genotypes for all SNPs in the corresponding genes, including 500 nt upstream and downstream, using the phased and corrected VCF files generated with LORALS (see above) and the window argument in the dmSQTLdata function. Each allele was considered to be a separate sample, with the genotype 0 for the reference allele and 2 for the alternative allele. Each unique intron group at one splicing level represented a “gene” and each intermediate isoform represented a “transcript.” Intron groups were filtered using the command dmFilter, requiring a minimum of ten alleles with at least 10 reads each and a minor allele frequency of at least two alleles. Within the tuQTL framework, SNPs within a gene that shared the same genotypes were grouped into a haplotype block (Nowicka and Robinson 2016). SNPs or haplotype blocks were considered to be significantly associated with splicing order if adjusted P-value < 0.05. Analyses for characterizing SNPs associated with splicing orders are included in the Supplemental Methods.
Estimation and comparison of poly(A) tail lengths
Poly(A) tail lengths were estimated during basecalling with Dorado using the parameter ‐‐estimate-poly-a. We found that samples sequenced with SQK-RNA004 tended to have longer poly(A) tails than the corresponding replicates sequenced with SQK-RNA002, though poly(A) tail lengths remained highly correlated (Supplemental Figs. S5B,C, S6C). Therefore, for cell lines sequenced with the two different chemistries, replicates were analyzed separately. Cell lines for which replicates were all sequenced with SQK-RNA002 on the MinION device had lower coverage and were therefore merged for further analyses. To compare poly(A) tail length between alleles, we filtered for genes that (1) had more than 20 reads assigned to each allele and (2) the number of reads assigned to each allele was at least two times higher than the number of undetermined reads. Poly(A) tail length distributions were compared using a two-sided Wilcoxon rank-sum test. Multiple testing correction was performed with the Benjamini–Hochberg method. Genes with adjusted P-value < 0.05 were considered to have statistically significant differences in poly(A) tail length. For cell lines for which the replicates were not merged due to different chemistries being used, we required adjusted P-value < 0.05 in both replicates in order for the gene to have allele-specific poly(A) tail length. For comparing poly(A) tail lengths of partially spliced or fully spliced reads (as defined above), the same approach and thresholds were used. Quantification of allele-specific transcript abundance and APA is described in the Supplemental Methods.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE256190. All the code is provided as Supplemental Code and is available at GitHub (https://github.com/churchmanlab/allele_specific_RNA_maturation).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank members of the Churchman lab for helpful discussions, advice, and assistance; Chantal Guegler, R. Stefan Isaac, Nicholas Kramer, Inés Patop, and Ana-Maria Raicu for critical reading of the manuscript; and the Biopolymers facility at Harvard Medical School for sequencing services. This work was supported by the NIH (R01-GM136794, R21-HG011682, and R01-HG010538 to L.S.C.), the Fonds de Recherche du Québec—Santé, the Canadian Institutes of Health Research (postdoctoral fellowship awards to K.C.), Université de Sherbrooke, and the Research Centre on Aging (start-up funds to K.C.).
Author contributions: K.C. and L.S.C.: conceptualization; K.C. (lead) and L.S.C.: methodology; K.C. (lead), L.-P.C., S.B., and A.R.B.-K.: investigation; K.C., L.-P.C., and S.B.: software/formal analysis; K.C. and L.S.C.: writing—original draft; K.C., L.-P.C., S.B., A.R.B.-K., and L.S.C.: writing—review and editing; K.C. and L.S.C.: funding acquisition; K.C. and L.S.C.: supervision.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279203.124.
- Received February 28, 2024.
- Accepted October 31, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








