
The genomic consequences and persistence of sociality in spiders
- Jilong Ma1,2,
- Jesper Bechsgaard2,
- Anne Aagaard2,
- Palle Villesen1,
- Trine Bilde2,3,4 and
- Mikkel Heide Schierup1,4
- 1Bioinformatics Research Center, Aarhus University, Aarhus C, DK-8000, Denmark;
- 2Department of Biology, Aarhus University, Aarhus C, DK-8000, Denmark;
- 3Center for Ecology and Conservation, University of Exeter, Penryn, TR10 9FE, United Kingdom
-
↵4 These authors contributed equally to this work.
Abstract
In cooperatively breeding social animals, a few individuals account for all reproduction. In some taxa, sociality is accompanied by a transition from outcrossing to inbreeding. In concert, these traits reduce effective population size, potentially rendering transitions to sociality “evolutionarily dead-ends.” We addressed this hypothesis in a comparative genomic study in spiders, in which sociality has evolved independently at least 23 times, but social branches are recent and short. We present genomic evidence for the evolutionary dead-end hypothesis in a spider genus with three independent transitions to sociality. We assembled and annotated high-quality, chromosome-level reference genomes from three pairs of closely related social and subsocial Stegodyphus species. We timed the divergence between the social and subsocial species pairs to be from 1.3 million to 1.8 million years. Social evolution in spiders involves a shift from outcrossing to inbreeding and from an equal to a female-biased sex ratio, causing severe reductions in effective population size and decreased efficacy of selection. We show that transitions to sociality only had full effect on purifying selection at 119, 260, and 279 kya, respectively, and follow similar convergent trajectories of progressive loss of diversity and shifts to an increasingly female-biased sex ratio. This almost deterministic genomic response to sociality may explain why social spider lineages do not persist. What causes species extinction is not clear, but either could be selfish meiotic drive eliminating the production of males or could be an inability to retain genome integrity in the face of extremely reduced efficacy of selection.
The evolution of cooperative breeding, in which some individuals forego their own reproduction to become helpers that assist other individuals in raising the offspring, is widespread in the animal kingdom (Alexander 1974; Rubenstein and Abbot 2017). In the most extreme cases, as in eusocial insects, strict division of labor results in highly complex societies with one or a few reproducing queens and countless nonreproducing helpers that are further organized into specialized sterile worker castes that each have specific tasks to perform (Crespi and Yanega 1995). In other cooperatively breeding taxa, individuals can switch between roles of reproducer and helper depending on age, hierarchy, and ecological conditions (Alexander 1974; Koenig et al. 1992; Ross and Keller 1995; Faulkes et al. 1997; Lubin and Bilde 2007). Cooperative breeding has evolved independently in divergent taxa, including insects, arachnids, birds, mammals, and fish. Common to all social cooperative species are that the evolution of cooperative breeding occurs in family groups and that helpers gain inclusive fitness benefits by assisting related individuals in reproducing, highlighting the importance of relatedness and kin selection in the evolution of sociality (Hamilton 1964).
As only a subset of individuals accounts for all reproduction in the social group, cooperative breeding invariably implies a reduction in effective population size (Nomura and Takahashi 2012; Romiguier et al. 2014; Settepani et al. 2017). Most cooperatively breeding species maintain a predominantly outcrossing mating system governed by premating or sex-biased dispersal that takes place prior to mating. However, in a subset of species, the transition to sociality is accompanied by a switch from outcrossing to strict inbreeding through the elimination of premating dispersal and reproduction among highly related group members (Settepani et al. 2017). These taxa show a “social syndrome” characterized by convergent independent transitions to cooperative breeding, regular inbreeding, reproductive skew (i.e., a subset of adult females reproduce), and a primary female-biased sex ratio (Bilde and Lubin 2011; Settepani et al. 2017; Vanthournout et al. 2018). This combination of traits has evolved convergently multiple times in spiders (Lubin and Bilde 2007), thrips (Chapman et al. 2000), aphids (Johnson et al. 2000), beetles (Keller et al. 2011), and possibly also mole rats (Reeve et al. 1990). In concert, these traits further act to reduce effective population sizes, and this is predicted to have negative consequences for population genetic diversity and the efficacy of selection through elevated genetic drift (Charlesworth and Wright 2001; Charlesworth 2003; Bechsgaard et al. 2019).
Spiders are an evolutionarily old group (>300 million years) (Garwood et al. 2016), and signs of social behavior were reported from fossil evidence in 100 million-year-old amber (Poinar and Buckley 2012). Although sociality in spiders is rare, the combination of cooperative breeding and an inbreeding mating system has evolved independently at least 23 times (Lubin and Bilde 2007) in six (seven) spider families, constituting about 23 of more than 52,000 extant spider species (https://wsc.nmbe.ch/). Phylogenetic analyses show that social spider lineages are evolutionarily young as they are found at the terminal branches. Furthermore, there are no described cases of speciation following transitions to sociality (Agnarsson et al. 2006; Lubin and Bilde 2007), in contrast to the social insects, in which relatively few transitions to sociality are followed by rich diversifications (Danforth 2002; Moreau et al. 2006). These phylogenetic patterns substantiate the hypothesis that the joint transitions to sociality and inbreeding may represent an “evolutionary dead-end” (Takebayashi and Morrell 2001; Settepani et al. 2017), in which social transitions are driven by ecological benefits, whereas long-term costs of repeated inbreeding cause the demise of social lineages.
Sociality in spiders involves living in communal nests where they cooperate in web building, prey capture, and offspring care (Lubin and Bilde 2007; Avilés 2021). Although there is a degree of behavioral differentiation (Settepani et al. 2013), they do not show eusociality like in ants and bees (Crespi and Yanega 1995), as there are no casts or strict division of labor (Lubin and Bilde 2007). Phylogenetic relationships indicate that sociality in spiders evolves from subsocial ancestors (Kullmann 1972; Agnarsson et al. 2006; Avilés 2010). In subsocial spider species, females provide extensive maternal care for their offspring over a prolonged period after hatching (Lubin and Bilde 2007; Grinsted et al. 2014), and the juveniles of subsocial spider species spend the first weeks in the natal nest before they disperse to establish a solitary life (Bilde et al. 2005). Although both social and subsocial spider species show elaborate maternal care of the offspring, subsocial species maintain premating dispersal and, consequently, an outcrossing mating system (Bilde et al. 2005). In contrast, social spider species are associated with the complete elimination of premating dispersal, which results in obligatory inbreeding as siblings reproduce within the group (Lubin and Bilde 2007). This is accompanied by the evolution of female-biased sex ratios (Vanthournout et al. 2018; Bechsgaard et al. 2019), cooperative breeding in which female helpers provide extensive allomaternal care (Junghanns et al. 2017, 2019), and reproductive skew (Lubin and Bilde 2007). The convergent evolution of traits that constitute this “social syndrome” and the phylogenetic relationships between social and subsocial spider species make this system ideal for understanding causes and consequences of transitions to sociality and inbreeding.
In the spider genus Stegodyphus, which contains more than 25 species, sociality has evolved three times independently (Johannesen et al. 2007; Settepani et al. 2016), and each social species has a closely related subsocial sister-species (see Fig. 1A,B) suggesting that sociality evolved recently (Settepani et al. 2016; Bechsgaard et al. 2019). The social Stegodyphus species express the “social syndrome” characteristics, which are predicted to reduce their effective population sizes largely (Bechsgaard et al. 2019), causing increases in the strength of drift and reducing the efficacy of natural selection (Kimura et al. 1963). This substantiates the question of whether consequences of low Ne have impact on the evolutionary persistence of social lineages. Reports of substantially reduced neutral genetic diversity (Settepani et al. 2017) and elevated rates of nonsynonymous-to-synonymous substitution rates (Mattila et al. 2012; Settepani et al. 2016; Bechsgaard et al. 2019, 2022) in social Stegodyphus species are consistent with reduced effective population sizes. Repeated inbreeding over evolutionary time is likely to have resulted in purging but also elevated rates of fixation of segregating recessive deleterious alleles; this is supported by the absence of inbreeding depression and evidence for outbreeding depression (lower hatching success when individuals from isolated populations were mated) in one of the social species Stegodyphus dumicola (Berger-Tal et al. 2014).
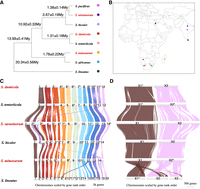
Overview of the genomic data collection. (A) Phylogeny of the spider genus Stegodpyhus with social species in red. The divergence time is estimated from the dS of autosomal single-copy orthologous genes and a yearly mutation rate of 5 × 10−9 per site per year. (B) Sampling locations of the species included in our study. The gene synteny plots among the six de novo chromosome-level assemblies are shown using GENESPACE, separately for autosomes (C) and X Chromosomes (D). The length of chromosomes in the gene synteny plot represents the number of genes. The IDs of chromosomes in C and D are labeled as the number we assigned in the assembled genome of each species. Asterisks mark chromosomes that are complementary-reversed in the synteny analysis.
Here we performed whole-genome assembly and comprehensive comparative analyses of the three social Stegodyphus species and their subsocial sister-species to assess the timing of evolutionary transitions and genomic consequences of social evolution. Based on analyses of Ne trajectories and purifying selection, we determined the divergence time of lineages and the age of individual transitions to sociality to assess whether speciation coincides with the evolution of sociality. This information further serves to investigate the progression of genomic consequences caused by the transition to sociality via comparisons of social spider lineages of different ages. These data sets enable us to assess the hypothesis that transitions to sociality in spiders are evolutionary dead-ends.
Results
De novo assembly and genome annotation
Six species of Stegodyphus (three social, three subsocial) were sequenced to ∼30× coverage using Pacific Biosciences (PacBio) HiFi long reads. Combining this with Hi-C contact maps based on 30× Illumina sequencing, we generated chromosome-level, de novo assemblies (Table 1; Supplemental Fig. S1). The sex chromosomes of each species were identified by the read coverage of male individual alignments. We identified two sex chromosomes in all species except for the outgroup species Stegodyphus lineatus, which has three X Chromosomes (Supplemental Fig. S8). For each species, the inferred number of linkage groups matched the number of chromosomes found by cytological visualization (Avilés et al. 1999; Král et al. 2011). We evaluated possible assembly errors by comparing the independent assemblies by dot plots using dotPlotly (Supplemental Figs. S4–S6). For the most closely related species pairs (1%–3% divergent at the genome level), assemblies were essentially syntenic, suggesting that the rate of assembly error is very low and that the synteny breaks for more divergent species pairs indicate real chromosomal rearrangements and/or inversions. Annotation using BRAKER2 (Brůna et al. 2021) independently for each assembly revealed similar numbers of protein-coding genes ranging from 32,093 to 35,724, except for Stegodyphus sarasinorum, in which 47,066 protein-coding genes were annotated. This recent gene number expansion in S. sarasinorum is not an artefact of annotation and is the focus of a separate study. BUSCO (Simão et al. 2015) analyses show high completeness of the genomes ranging from 91.1% to 93.3%. The GC content ranges from 33.26% to 34.18%, and the proportion of the genomes constituting repeats ranges from 57.17% to 65.20%. We found 10,065 single-copy orthologous genes present in all six species using our de novo assemblies, synteny information, and annotations (Table 1; Supplemental Table S2). Merqury (Rhie et al. 2020) estimation on our genomes reveal k-mer completeness ranging from 97.65% to 99.41% and a consensus quality QV ranging from 62.54 to 73.86, which is higher than the Vertebrate Genomes Project (VGP) benchmarking recommendations of QV > 40 and k-mer completeness > 95% (Rhie et al. 2021). When incorporating the de novo assembled transcriptome from Stegodyphus africanus and short reads DNA sequences from Stegodyphus pacificus, our final data set ends up with 2303 and 348 reliable single-copy orthologs for autosomes and X Chromosomes, respectively, across all eight species.
Assembly and annotation statistics of social and subsocial Stegodyphus spiders
High gene synteny and few chromosomal rearrangements in the Stegodyphus genus
To investigate genome rearrangements across Stegodyphus from the accurate de novo assemblies, we used GENESPACE (Lovell et al. 2022) to retrieve the pair-wise synteny blocks based on the gene order of orthologs found in each de novo annotation (for an outline of the results, see Fig. 1C,D). In general, synteny blocks suggest few chromosomal translocations and further support that the assemblies are accurate.
We identified a number of large-scale structural rearrangements, including splits and joins of chromosomes unique to certain lineages of the phylogeny. For example, the outgroup species S. lineatus has 23 chromosomes including three X Chromosomes, whereas the other species contain 14 to 16 chromosomes and only two X Chromosomes, suggesting that multiple chromosome splits or joins have happened in the S. lineatus lineage or that chromosome joins have occurred after the split with S. lineatus in the common ancestor of the other species (Table 1). We found that the third X Chromosome (Chr X3) in S. lineatus appears to originate from a “cut-and-paste” nonhomologous recombination process, which joins a part of the ancestral X Chromosome X1 and a part of the ancestral X Chromosome X2 (Fig. 1D). Thus, the three X Chromosomes in the S. lineatus lineage have almost the same gene complement as the two X Chromosomes in the other species. In addition, we identified three chromosome join events assigned to the ancestral branch of S. dumicola and Stegodyphus tentoriicola (Chromosomes 1, 2, and 3 are homologous to six separate chromosomes in other species), as well as one chromosome join event (Chromosome 2 is homologous to two separate chromosomes in other species) and a nonhomologous recombination event (Chromosome 3 contains extra components that are homologous to another chromosome in other species) assigned to the ancestral branch of Stegodyphus bicolor and S. sarasinorum.
Estimating the beginning and end of the transitions to sociality
We first estimated divergence times at each node of the Stegodyphus phylogeny (Fig. 1A) using the resampled branch-wise dS from autosomal branches and assuming a mutation rate of 5 × 10−9 per site per generation within the 95% confidence interval range of Arthropods mutation rate (Wang and Obbard 2023). Among the three pairs of social and subsocial sister species, the estimated divergence times are 1.78 ± 0.22 million years between Stegodyphus mimosarum and S. africanus, 1.31 ± 0.18 million years between S. dumicola and S. tentoriicola, and 1.38 ± 0.14 million years between S. sarasinorum and S. pacificus.
Transitions to sociality in spiders are associated with a shift to inbreeding and reduced effective population size (Ne) causing natural selection to be less efficient. We assume that a substantial less-efficient selection and Ne reduction in the social spider species are more likely caused by evolution of sociality combined with a shift to inbreeding than by other factors. We then used our genome data to estimate the time when these transitions to sociality occurred from the expected signals in purifying selection and Ne. We did this in two separate ways: first, from the estimated changes in purifying selection estimated from dN/dS ratios, and second, from the inferred changes in effective population size through time estimated from coalescent analyses.
Intensity of purifying selection
We estimated the transition time to social behavior from dN/dS ratios on the terminal branches of each social/subsocial species pair based on the following assumptions: (1) dN/dS ratios of the social species were the same as for the subsocial species until social transitions on the terminal branches leading to the social species, (2) after transition to sociality, the dN/dS ratio increased owing to less efficient purifying selection, and (3) this “social dN/dS” can be approximated by the piN/piS ratio estimated between divergent and isolated extant populations of each social species (see Methods; for justification, see Supplemental Materials). This dN/dS analysis suggests that the effects of a transition to sociality on purifying selection are very recent (Fig. 2A–D). For example, the dN/dS over the entire S. mimosarum branch is 0.1259 ± 0.0044, and for its subsocial sister-species S. africanus, dN/dS is 0.0927 ± 0.0007. The dN/dS ratio since sociality evolved is estimated at 0.3043 ± 0.0018 comparing two S. mimosarum individuals from Madagascar and mainland Africa. From the divergence time estimates for this species pair, this generates an estimated time since transition to sociality of S. mimosarum of 279 kya (95% CI: 230 kya–332 kya). Using the same calculation (Fig. 3), the social dN/dS ratio of S. dumicola is estimated to be 0.3366 ± 0.0037 by comparing individuals from Otavi, North Namibia, and Karasburg, South Namibia. Combining the dN/dS ratio with 0.1776 ± 0.0039 and 0.1615 ± 0.0054 for the S. dumicola and S. tentoriicola branches, respectively, the estimation of the transition to sociality is 119 kya (95% CI: 72 kya–170 kya) in S. dumicola.
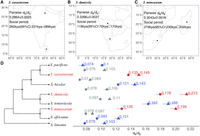
The intensity of purifying selection was assessed using dN/dS from the coding sequences of 2302 single-copy orthologous genes. (A) The locations of the Sri Lanka S. sarasinorum and Himalayas S. sarasinorum samples, along with their pairwise dN/dS estimations for the social period. (B) The locations of the Namibia South S. dumicola and Namibia North S. dumicola samples, with corresponding pairwise dN/dS estimations for the social period. (C) The locations of the South Africa S. mimosarum and Madagascar S. mimosarum samples and their pairwise dN/dS estimations for the social period. (D) Branch-wise dN/dS estimations using PAML, with the 95% confidence interval from resampling. Red, blue, and gray denote social species terminal branches, subsocial species terminal branches, and internal branches, respectively. The dN/dS estimation, indicated by points with circles and triangles denoting autosomes and X Chromosomes, respectively, is aligned to the phylogeny that corresponds to the respective branches. The phylogeny in D provides an indication of the topology without branch length information.
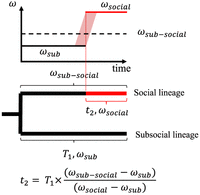
Illustration for calculating the social period length for current social species as an instant process. t2 denotes the length of the social period. T1 denotes the divergence time between social species and its sister subsocial species. ωsub denotes the dN/dS estimated from the subsocial species branch in branch-wise PAML estimations. ωsub–social denotes the dN/dS estimated from the social species branch in branch-wise PAML estimations. ωsocial denotes the dN/dS (in practice, piN/piS) estimated from the pairwise PAML estimations of two separate social species populations. The red shadow region represents the scenario in which the dN/dS may increase as a continuous process.
For the social S. sarasinorum, we performed two separate estimations by comparing S. sarasinorum to the subsocial species S. pacificus, which diverged 1.38 ± 0.14 million years ago, and the subsocial species S. bicolor, which diverged 2.67 ± 0.19 million years ago. In S. sarasinorum, we estimated the social dN/dS ratio to be 0.2864 ± 0.0023 based on two individuals from Himalaya and Sri Lanka. The dN/dS ratio on the terminal branch of S. sarasinorum is 0.1349 ± 0.0009. Compared with the 0.0996 ± 0.0016 of the S. pacificus branch and the 0.1211 ± 0.0007 of the S. bicolor branch, the final estimation for the transition to sociality of S. sarasinorum is 260 kya (95% CI: 231 kya–289 kya) and 223 kya (95% CI: 199 kya–247 kya), respectively.
Additionally, we note that our implication of using piN/piS between divergent populations of the same species could potentially lead to a systematically overestimated “social dN/dS” owing to the deleterious alleles that are not yet removed by purifying selection, rather than reflect the actual selection strength when the selection strength acts differently on synonymous polymorphism sites and nonsynonymous polymorphism sites within species. In S. dumicola and S. mimosarum, we benchmarked that there is minimal difference between piN/piS estimated between one individual from two isolated populations and piN/piS estimated only on sites fixed differently in two isolated populations (Supplemental Fig. S12). No benchmarking has been made on S. sarasinorum owing to the lack of population data in the two isolated regions, but the isolation status of the S. sarasinorum Himalaya population and Sri Lanka population is expected to resemble that of S. mimosarum. Thus, we conclude that piN/piS to a good approximation can be used as a proxy for the dN/dS ratio after sociality (“social dN/dS”; Methods) (for detailed justifications, see Supplemental Materials).
We have attributed the observed higher dN/dS ratio in the terminal branches of social species to a decrease in purifying selection. However, increased adaptive selection acting on social species could have the same effect. RELAX analyses (Wertheim et al. 2015) suggest that the genome-wide elevated dN/dS ratios in social species are driven by relaxed selection in autosomes (Methods) (Supplemental Fig. S3; for explanations, see Supplemental Materials), as has previously been suggested (Tong et al. 2022). We also show elevated dN/dS ratios on X Chromosomes compared with autosomes across all branches in the phylogeny, except for the terminal branches of S. pacificus. Such elevated dN/dS ratios can be attributed to a decrease in purifying selection acting on X Chromosomes, which is consistent with the expected outcome of reduced Ne in X Chromosomes compared with autosomes (Ne_X/Ne_Auto is expected to be 0.75 in diploid species with equal sex ratio). Alternatively, but not mutually exclusive, an adaptive explanation for elevated dN/dS ratios on the X Chromosomes is the so-called faster-X effect (Charlesworth et al. 1987). Recessive variants on the X Chromosomes are directly exposed to selection in males that are haploid for X Chromosomes, and under certain conditions, this will lead to more adaptive substitutions on X Chromosomes compared with autosomes.
Coalescent analysis
Next, we used pairwise sequentially Markovian coalescent (PSMC) (Li and Durbin 2011) analysis to reconstruct the effective population sizes and the sex ratio in the populations over time by analyzing autosomes and X Chromosomes separately in each species. For the social species, which are all highly inbred, a diploid genome was created by merging two haploid genomes sampled from different divergent and isolated populations, namely, an artificial diploid. This allows coalescent events further back in time to be inferred. The transition time to sociality is then estimated by graphically determining the time period when the effective population size of the social species became severely reduced (Fig. 4A–D, left). Compared with subsocial species, the population trajectories of all the social species share a gradual reduction of Ne from more than 300,000 to fewer than 50,000, but starting and ending at different time points within the past 900,000 years. In the social species S. dumicola, Ne is reduced from 300,000 around 420 kya to around 50,000 at 300 kya and is even lower today. In the social species S. mimosarum, Ne is reduced from 1.4 million to 57,500 in the time interval between 900 kya and 350 kya. The reduction of Ne in the social species S. sarasinorum starts 600 kya at 800,000 to fewer than 30,000 around 300 kya.
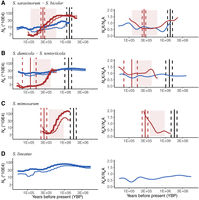
The historical effective population size inferred from the pairwise sequentially Markovian coalescent (PSMC) model, for different Stegodyphus species with chromosome-level assembly. In each panel, the left figure depicts PSMC results, and the right figure displays the effective population size ratio between X Chromosomes and autosomes. Each species’ trajectory is shown with thicker lines for autosomes and thinner lines for X Chromosomes. The types of species are distinguished by color: red for social species and blue for subsocial species. Red shadows mark the estimated social transition period based on the decreasing effective population size in social species. Vertical lines indicate estimated time points by comparing the social species with its closest sister subsocial counterpart. Red vertical lines correspond to the estimated social transition time, derived from relaxed selection in social species, and black vertical lines represent the estimated divergence time between a social species and its closest subsocial sister species. Solid lines provide point estimates, and dashed lines delineate the 95% confidence intervals. The PSMC results are truncated in the near recent past for better comparison across species. Bootstrapped PSMC results without time truncation are provided in Supplemental Figure S9. (A) Data for the social species S. sarasinorum and the subsocial species S. bicolor. Estimates for social transition and divergence time are based on a comparison between S. sarasinorum and its subsocial sister species, S. pacificus. (B) Findings from a comparison between the social species S. dumicola and its subsocial sister species S. tentoriicola. (C) Data for the social species S. mimosarum, with social transition and divergence time estimates based on a comparison with its subsocial sister species, S. africanus. Note that we do not have an effective population size estimation for S. africanus owing to missing whole-genome data. (D) Historical effective population size for the outgroup subsocial species S. lineatus.
Because the PSMC analyses of the social species are made from pseudodiploid genomes combined from separate genetically isolated populations, they can additionally provide an estimate of the time of separation of these populations, because coalescence cannot occur when they are separate, and the Ne estimate will thus be very large (Supplemental Fig. S9). We observe such an uptick in Ne in recent times in the three social species and estimate that the two S. mimosarum populations from Madagascar and South Africa diverged around 200 kya, the two S. dumicola populations in North and South Namibia diverged 20 kya, and the two S. sarasinorum populations from Himalaya and Sri Lanka diverged ∼110 kya.
The pseudodiploid of social species collapsed two heavily inbred diploid individuals. We estimate the run out of heterozygosity (ROH) regions takes 59.83%–86.27% of their genomes (Supplemental Table S4), but the individuals are still not 100% homozygous. Hence, they are not identical as a haploid in practice. Thus, we cannot rule out the fact that pseudodiploid actually capture more than two haploids for part of the genomes outside the ROH region. However, we expect this will only increase a local coalescence rate that is randomly distributed in the genome, resulting in a lower estimation of Ne overall, without biassing the Ne reduction information retrieved from the Ne trajectories.
Evolution of sex ratio bias
The sex determination system of spiders is X0, and as there are four autosomes for each three X Chromosomes in the population with one-to-one sex ratio, the effective population size of X Chromosomes is expected to be 75% of the autosomes. In subsocial species, PSMC analyses of X Chromosomes and autosomes showed that although the estimated Ne for X Chromosomes is indeed lower, the ratio of X Chromosomes to autosome effective population size is ∼50%–60% instead of the expected 75%. This can be because of a lower mutation rate and/or stronger selection on the X Chromosomes, as was previously suggested based on diversity estimates on X Chromosomes and autosomes in S. africanus (Bechsgaard et al. 2019).
In contrast to the subsocial species, the social species show strongly female-biased primary sex ratios (Lubin and Bilde 2007), and this has been shown to be genetically determined (Vanthournout et al. 2018). When a species evolves from an unbiased to a female-biased sex ratio, the effective population size of X Chromosomes should approach that of autosomes. In concordance with this, the PSMC analyses reveal that X Chromosome and autosome Ne are almost identical in the most recent time intervals of the PSMC, as expected when sociality has fully evolved. The evolution of a female-biased sex ratio can therefore also be timed using PSMC analyses, under the assumption that it occurred after the transition to sociality, by comparing the Ne of X Chromosomes and autosomes back in time. We found elevated Ne(X)/Ne(autosomes) ratios in all social species associated with reductions in Ne (Fig. 4A–D, right), which links the evolution of sex-ratio bias with social transitions. This suggests that the evolution of a female-biased sex ratio in the three social species occurred early in the evolutionary transition to sociality.
The evolution of female bias is predicted under strong continuous inbreeding and lack of premating dispersal, characteristics of social spider species and other social arthropods (Chapman et al. 2000; Johnson et al. 2000; Keller et al. 2011), to reduce competition among brothers for fertilization of their sisters (Bulmer and Taylor 1980; West 2009). Furthermore, allocation to daughters as the helping sex in social species is expected by the local resource enhancement model (Trivers and Willard 1973). Biased sex ratios are often caused by sex chromosome meiotic drive (Lindholm et al. 2016), and the driving loci are selfish genetic elements that would usually trigger adaptations to restore even sex ratio (Lindholm et al. 2016). However, selection against female bias is not expected here, because of the adaptive benefits of female-biased sex allocation (Vanthournout et al. 2018) generating predictions of rapid fixation of the sex-linked meiotic driver. If stronger female bias keeps being adaptive, selection will lead to an increasingly female-biased sex ratio following transition to sociality. The results from the PSMC analyses showing that NeX/NeA continues to increase after the transition to sociality suggest that stronger female bias continues to be adaptive. Furthermore, our estimation for transitions to sociality matches with observations of increasing female bias with age of sociality in the three species. The oldest social species S. mimosarum has the strongest sex ratio bias with 9% males; the younger social species S. dumicola and S. sarasinorum have a male percentage of 12% and 8.5%–33%, respectively (Lubin and Bilde 2007). The correlation between sex ratio bias and age of the social period in the three transitions to sociality therefore indicate that the continued loss of males is genetically determined.
Discussion
Consistency of independent methods in estimating age of sociality
The Ne trajectories from PSMC reveal that the transition to sociality is gradual, rather than occurring instantaneously, by showing a decrease in Ne over a few hundred thousand years. We do not know if the start of reductions in Ne is caused by transitions to sociality or if other life history changes that reduce Ne are responsible. However, the main cause of the reductions in Ne is mostly likely owing to the transition to sociality. At the end of the transition period when sociality is fully established, Ne stabilizes at a low level. We use the current “social dN/dS” value for the social species estimated among two currently isolated populations; however, the dN/dS value has likely been increasing gradually during the phase of transition to sociality instead of an instant increase, and the current “social dN/dS” is consequently higher than the mean dN/dS in the period being a social species. Thus, the point estimation of social transition from dN/dS analysis is expected to not coincide with the beginning of the transition to sociality (Fig. 4A–D, left) but to be somewhere within the transition period estimated from PSMC. There are factors that can contribute to pushing the point estimate of transition to sociality closer to present owing to violation of our assumptions: (1) that by using the dN/dS of the subsocial sister-species to approximate the dN/dS before transition to sociality in social lineage, there is a chance that this approximation is lower than the actual value; (2) that we overestimate the dN/dS after transition to sociality in social lineages, owing to potential segregation of deleterious variants in the current social populations; and (3) that the dN/dS continues to increase after the sociality is established. We observe that the point estimates of social transitions from the analyses of dN/dS match with the ending of the transition process estimated from PSMC in S. dumicola and S. mimosarum, suggesting the existence of the confounding factors mentioned above. Nevertheless, both methods agree with the time estimation that sociality is established (ending of social transition) only within 300 kya in all three social spider species. The two estimations do not conflict with each other in the order of relative age of the sociality among the three social spider species.
Divergence times and transition to sociality
To elucidate genomic consequences of repeated, independent transitions to sociality, we first estimated species divergence times and the time of speciation using different approaches (Fig. 5).
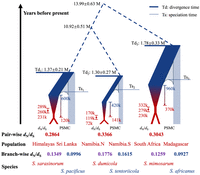
Estimated species divergence (Td) and the time of transition to sociality/cooperative breeding (indicated in red) in three independent social transitions within the Stegodyphus spider genus. Hypothetical speciation time (Ts) between each species pair is marked as the dashed lines. The scale of the plots is symbolic and not proportional. The widths of the terminal branches represent variation in effective population sizes, with red symbolizing social periods and blue indicating subsocial periods. Light blue branches represent species without chromosome-level assemblies for PSMC. For each lineage in which a social transition occurs, point estimates and 95% confidence intervals for social transition times are presented on the left, whereas social transition periods derived from population size history are displayed on the right. In PSMC estimations, the start of social transition periods corresponds to the initial decrease in effective population size, whereas the end coincides with Ne dropping below 50,000.
An obvious question is whether the speciation itself was caused by one of two diverging populations evolving sociality. The large time interval between estimates of species divergence (Td = 1.3 million–1.8 million years) (Fig. 5) and the beginning of social transitions (400,000–900,000) as derived from strong reductions in Ne at first suggests that species separation took place initially with subsequent evolution of sociality and cooperative breeding. However, coalescent theory predicts that it takes 2Ne generations on average for ancestral polymorphisms to find most common ancestry. Because the effective population sizes of subsocial species are very large (300,000–600,000), divergence times (Td in Fig. 5) are expected to be much more ancient than the species separation times (Ts in Fig. 5) owing to long coalescence times in the ancestral species. With a generation time of 1 year, average coalescence time in the ancestral species are between 600,000 and 1.2 million years, and our best estimate of the speciation times (Ts in Fig. 5) is therefore ∼1 million years closer to the present than the divergence times. This corresponds to the beginning of social transitions estimated from PSMC results, thus suggesting that social behavior emerging in a population of a species may facilitate separation of this population into a new species.
There is, however, also some evidence from the social S. dumicola that sociality can arise independently after the speciation process. S. dumicola and S. sarasinorum diverged from their closest subsocial sister species at a similar timescale of 1.30 million and 1.37 million years ago, respectively. The ancestral Ne for S. dumicola (300,000) is smaller than the S. sarasinorum (600,000). Thus, the speciation time of S. dumicola (Ts2 in Fig. 5) is expected to be closer to the species divergence time than the speciation time of S. sarasinorum (Ts1 in Fig. 5) based on coalescence theory (Supplemental Fig. S11). Meanwhile, both analyses from PSMC and dN/dS show that the age of sociality is younger in S. dumicola than in S. sarasinorum. The combination of the earlier speciation time and the younger age of sociality in S. dumicola compared to S. sarasinorum suggests that sociality can arise as a separate event from the initiation of speciation. Nevertheless, evidence for separation of the two events in S. dumicola could be confounded by other factors, including (1) a missing subsocial sister species (unidentified or extinct) to S. dumicola that is not included in the analysis, which can bias the analyses to overestimate the divergence time for S. dumicola lineages, and (2) observed reductions of Ne in the recent past of S. tentoriicola from PSMC (Supplemental Fig. S9) and elevated dN/dS in S. tentoriicola compared with other subsocial species (Fig. 2), which can bias the analyses to underestimate the age of sociality in S. dumicola. Nevertheless, the combination of data presented substantiate that sociality and characteristics of the “social syndrome” evolved much more recently than the divergence estimate.
High extinction risk of social spiders
The genomic consequences of transitions to sociality in spiders are consistent with expectations of strong reductions in Ne when the evolution of sociality is accompanied by a transition from outcrossing to inbreeding and the development of a female-biased sex ratio (Vanthournout et al. 2018). The relatively young age of social lineages indicate that sociality evolves rapidly and frequently in the spider phylogeny and comes with a subsequent strong signal of decreased purifying selection caused by strict inbreeding and much reduced effective population size. Overall, there are at least 23 independent convergent origins of sociality (Agnarsson et al. 2006; Lubin and Bilde 2007). The recurring and rapid evolution of cooperative breeding, coupled with the absence of lineages with a long history of sociality, is strong indirect evidence that sociality in spiders represents an evolutionary impasse, unlikely to persist beyond a timespan of 1 million years once established. Thus, it is plausible that numerous social spider species have emerged and gone extinct throughout evolutionary history, but we can only observe the presence of extant and recently developed social spider species.
The risk of extinction in social spiders is hypothesized to be much higher than in eusocial insects owing to the “social syndrome,” which causes reductions in Ne. Phylogenetic studies show that diversification of species is only associated with a few old transitions to eusociality in the order Hymenoptera, namely, ants, bees, and wasps (Heinze et al. 2017; Hunt and Toth 2017; Wcislo and Fewell 2017), which is different from the repeated and recent convergent evolution of sociality in the spider phylogeny (Agnarsson et al. 2006; Settepani et al. 2016). We propose that a key factor contributing to the difference in longevity of social lineages among major taxa is the elimination of premating dispersal, which results in an obligatory inbreeding mating system. In eusocial insects, new colonies are established by mated queens, and outcrossing is maintained by premating dispersal of reproductive individuals (Heinze et al. 2017; Wcislo and Fewell 2017). Thus, eusocial insects may show reduced Ne and relaxed selection compared with their nonsocial counterparts (Kapheim et al. 2015), but the reduction in Ne is mitigated by the maintenance of outcrossing. In contrast, we here show that transitions to sociality and inbreeding in independently evolved spider lineages are associated with large declines in Ne.
Chromosomal systems, mating systems, and the evolutionary longevity of social lineages
Some of the most successful social lineages have evolved in the Hymenoptera, in which the entire clade is haplodiploid with haploid males and diploid females (Grimaldi and Engel 2005). These lineages are evolutionarily old, for example, in ants in which eusociality is dated at least to the Cretaceous (Table 2), and they have since diversified both in species and in abundance to become one of the most ecologically dominating taxonomic groups in the world (Wilson and Hölldobler 2005). The evolutionary success of haplodiploid social lineages, which is also found in Thysanoptera (thrips) and sporadically in some spider mites, Hemiptera, and Coleoptera, has long pointed to an importance of haplodiploidy in the evolution of sociality (Hamilton 1964; Trivers and Hare 1976; Joshi and Wiens 2023). However, phylogenetic patterns are debated and perhaps inconclusive, as eusociality has evolved repeatedly within the haplodiploid Hymenoptera and Thysanoptera, whereas other successful independently evolved social lineages are diploid, such as in termites and aphids.
Summary of the characteristics of sociality in diploid and haplodiploid organisms with different mating systems
A key question that arises from our study is whether haplodiploidy represents a more robust system for governing the evolutionary longevity of social lineages. Termites and Hymenoptera represent the oldest social lineages and represent both chromosomal systems (Table 2). These taxa also maintain predominantly outcrossing mating systems, which act to maintain higher effective population sizes that are less exposed to genetic drift. In taxa in which social evolution is accompanied by a transition to inbreeding, the diploid social spiders show the most short-lived and recent transitions (Settepani et al. 2017), whereas thrips and beetles with a haplodiploid system may have relatively older social lineages (Table 2). In combination, these patterns suggest that whether or not sociality is associated with a transition from outcrossing to inbreeding is perhaps the most important factor for determining their evolutionary longevity. The taxa that evolve inbreeding mating systems are simultaneously characterized by female-biased sex ratio, which further acts to reduce effective population size (Bechsgaard et al. 2019). Our study provides genomic support for the hypothesis that the independent evolution of sociality accompanied by an inbreeding mating system and female-biased sex ratios results in drastic reductions in Ne and relaxed efficacy of selection, which likely contribute to drive lineage extinctions.
Genomics determinants of evolutionary dead-ends in social spiders
We note that even though the three social transitions we report were initiated at different times, they have followed similar trajectories in population size reduction, increased dN/dS, and female-biased sex ratios showing strong similarities in behavior and life history associated with transitions to sociality in spiders. This almost deterministic genomic response to sociality may also hold the key to understanding why social spider species do not seem to persist for very long. Several possibilities may explain the demise. These include (1) loss of adaptive potential owing to loss of genetic diversity, (2) fixation of deleterious variation, (3) loss of males, and (4) loss of genome integrity. Although possibilities 1 and 2 may definitely make sociality a doomed evolutionary strategy, it is questionable whether these processes can happen within a few hundred thousand years since the fixation of slightly deleterious variants occurs over a very large number of generations (Lynch et al. 1995). Loss of males could carry on until population collapse, and it is intriguing that we see fewer males in the social transition estimated to be the oldest (Fig. 5). Finally, loss of genome integrity by, for example, transposable elements (TEs), increased mutation rate, poorer genomic repair of damage, and double-strand breaks, is a global mechanism that could impact the longevity of the social species. TEs are expected to accumulate in species with reduced Ne. Although the question of whether the TE fraction increases with reduced Ne remains inconsistent in several tested organisms (Glémin et al. 2019), evidence for increased amounts of TEs has been seen in eusocial snapping shrimps with reduced Ne (Chak et al. 2021). Preliminary results of repeat fractions do not differ in each pair of social and subsocial spider species (Table 1; Supplemental Table S7), but this idea warrants future research regarding the dynamics of TE in social transitions (Betancourt et al. 2024).
Methods
Sample collection across the Stegodyphus genus
The spider genus Stegodyphus is distributed widely across Eurasia and Africa, and we collected seven species including three social and four subsocial species in this study (Supplemental Table S1). Samples were collected from multiple regions for social species (Fig. 1A). We collected S. dumicola samples from Otavi and Betta in Namibia (2017), S. mimosarum samples from Antanario in Madagascar and Weenen in South Africa (2011), and S. sarasinorum samples from the Himalaya region and Sri Lanka (2014). For subsocial species, we collected S. lineatus from the Negev Desert in Israel (2020), S. bicolor from Betta in Namibia (2021), and S. tentoriicola from Tierberg in South Africa (2015). S. africanus RNA data were previously published by Bechsgaard et al (2019).
Reference genome: DNA extraction, sequencing, and assembly
DNA was extracted from single individuals using the MagAttract HWM DNA kit from Qiagen. Sequencing was done by PacBio HiFi technology to obtain from 69 to 99 GB data. We assembled haplotype resolved contigs using Hifiasm (Cheng et al. 2021) with PacBio HiFi reads and Hi-C reads. Hi-C reads were aligned to the contigs from the longer haplotype of each species using Juicer pipeline for scaffolding chromosome-level assemblies using 3D-DNA pipeline (Dudchenko et al. 2017). The chromosome-level assemblies were further manually checked and curated using Juicebox Assembly Tools (Durand et al. 2016) based on consistency of Hi-C contact pattern.
Resequencing: DNA extraction and sequencing
DNA was extracted from single individuals using the Qiagen blood and tissue kit, and the DNA was sequenced using DNBSEQ-G400.
Genome annotation
We annotated six species with chromosome-level assemblies using BRAKER2 with RNA-seq (Supplemental Table S3) and protein homology evidence combined. For each species, we used RepeatModeler2 (Flynn et al. 2020) to generate a repeat library and combine it with the repeat library for Arthopoda from Repbase (Bao et al. 2015). The chromosome-level assemblies are masked using RepeatMasker (Tarailo-Graovac and Chen 2009) and their corresponding repeat libraries. We used STAR (Dobin et al. 2013) to align RNA-seq reads to the repeat-masked genome as transcriptome evidence for gene prediction in BRAKER2. For protein homology evidence, we used protein sequence from a previous S. dumicola annotation (Liu et al. 2019) and Arthropoda protein from OrthoDB v10 (Kriventseva et al. 2019). In the end, the completeness of the genome annotations is evaluated with BUSCO together with a set of 1013 Arthropoda genes from OrthoDB v10.
Ortholog groups finding and multiple genome alignments
We performed GENESPACE to check the gene synteny among the six HiFi chromosome-level assemblies. During the GENESPACE analysis, OrthoFinder (Emms and Kelly 2019) was applied to identify orthologous groups across the six species. The 10,065 single-copy orthologous groups out of 48,143 orthologous groups identified from the six species were further used in evolutionary analysis. The incorporation of S. africanus and S. pacificus, for which have no chromosome-level assemblies, into the multiple genome alignment are described in detail in the Supplemental Materials.
Branch-wise dN/dS estimation
We used CodeML in PAML (Yang 2007) to estimate dN and dS in branch-wise mode based on the 2649 single-copy orthologous groups. The 2649 orthologous groups were split up into an autosome set containing 2302 genes and a X Chromosome set containing 347 genes. We randomly sample 500 genes from the 2302 autosomal without replacement in each resampling run, and we run the resampling independently 500 times to have an estimation of dN/dS of each branch in the phylogeny with confidence intervals. For X Chromosomes, we randomly sample 100 genes independently 100 times instead. Please see justification for the number choice in the Supplemental Materials.
We do codon aware alignment in gene-wise for dN/dS estimation. We used alignSequence from MACSE v2 (Ranwez et al. 2018) to align coding sequences of each gene from all the species excluding S. pacificus, which was attached to the alignment according to the S. sarasinorum in the end. We also examined and filtered each gene alignment for local misalignment. The size of each alignment block without gap should have a length for at least 300 nucleotide base pairs as well as a fraction of polymorphism sites under 15% (Supplemental Fig. S2). The passing alignment blocks of the randomly chosen genes were concatenated using GOalign (Lemoine and Gascuel 2021) for dN/dS estimation.
dN/dS (empirical piN/piS) estimation for social species
We used CodeML in PAML to estimate a pairwise dN/dS for each social species with two separate populations. We aligned one individual from different populations to the species reference separately using BWA-MEM2. After calling the SNP using platypus (Rimmer et al. 2014) and filtering for only the SNPs that pass all the default filters, we retrieve the consensus nucleotide to substitute the species reference as the reference genome of each subpopulation. Sequences from the 2649 single-copy orthologous genes were retrieved from the subpopulation references for pairwise dN/dS estimation. We used the same 500 resampled sets of genes as used in the branch-wise dN/dS estimation before to estimate the piN/piS with 95% confidence interval between separate social species populations for autosomes.
dN/dS and estimation of speciation divergence and social transition
For the divergence time between lineages, we used the dS branch length, a constant mutation rate of 5 × 10−9 per site per generation, and a generation time of one year to scale the dS length into years. We use the average dS branch length between a split as an estimation of divergence time except for the split between S. sarasinorum and S. pacificus. The exception is because of the underestimation of dS length of S. pacificus lineage from ignoring of indels; thus, we use the dS length of S. sarasinorum branch alone for divergence time estimation between S. sarasinorum and S. pacificus.
Assuming the transition to sociality is an instant process. The dN/dS estimated for the social species terminal branch is an average of the subsocial period dN/dS and social period dN/dS weighted by the length of each period on that branch (Fig. 3; Supplemental Materials). The same idea has been applied to estimate the age of selfing in Arabidopsis thaliana compared with selfing incompatible Arabidopsis lyrata (Bechsgaard et al. 2006). In our case, the subsocial period can be substituted approximately with the dN/dS for the branch of the subsocial sister species. The social period dN/dS can be estimated by pairwise comparing two individuals from two isolated and divergent populations of the social species (see justifications in Supplemental Materials; Supplemental Fig. S7; Supplemental Tables S5, S6).
For each resampling run of autosomal genes (500 genes out of 2303 genes), we obtained four estimations for each pair of social species and its subsocial counterpart using PAML, including
-
dN/dS of the subsocial lineage,
-
dN/dS of the social lineage,
-
piN/piS between two isolated populations of social species, and
-
dS branch length between social and subsocial species.
The distributions of the dN/dS and piN/piS from the resampling process for each species pair are provided in Supplemental Figure S10. Confidence intervals for mean estimations of dN/dS and piN/piS are retrieved using the 2.5%–97.5% quantile from a normal distribution built according to the mean and standard error. The point estimation for species divergence time is estimated from the mean of value 4 from the 500 rounds of resampling. Thus, the 95% confidence interval of species divergence time is retrieved using the 2.5%–97.5% quantile from a normal distribution built according to the mean and standard deviation of dS. For the time of transition to sociality, we sampled 10,000 sets of observations, each of which consists of a set of values including one observation of 1, 2, 3, and 4 sampled from their distribution built by previous resampling process of 500 random autosomal genes. We then solved the social transition time for each sampled observation set. The point estimation for transition time is obtained from the mean of 10,000 sampled calculations. The 95% confidence interval for transition time is retrieved as 2.5%–97.5% quantile from the empirical distribution of 10,000 rounds. This will account for all uncertainty of the genome-wide selection strength and species divergence time estimated using the resampling process.
PSMC estimation of social transition time
We used PSMC to reconstruct the historic effective population size for all the six species with a chromosome-level genome. The PSMC method relies on a single diploid individual, in which the social species run out of heterozygosity owing to inbreeding. We combine the DNA-seq of two individuals from two separate and diverged populations to retain the loss of heterozygosity.
We benchmarked the fraction of running out of heterozygosity region in the predicted youngest social species S. dumicola. We used the GATK pipeline to genotype nine individuals (five females and four males) from S. dumicola and only kept biallelic SNP variants among the individuals with a genotype quality filter of 30. We did a sliding window heterozygosity with 100 kb window size and 10 kb step size on each individual. Windows of an individual without any variants are further classified as run out of homozygosity regions and summarized into ROH fraction by individual (Supplemental Table S4).
We ran the PSMC method on autosomes and X Chromosomes separately. The generation time is fixed to 1 year per generation. The mutation rate used for autosomes is 5 × 10−9 per site per generation and the rate for X Chromosomes is scaled to 3.8 × 10−9 with a factor of 0.76 based on the piX/piA being 0.57 instead of 0.75 in subsocial species S. africanus (Bechsgaard et al. 2019). To align the estimated historical Ne estimated between autosomes and X Chromosomes, we retrieve all the time points with an estimation of Ne either for autosomal or X Chromosomes; the missing value is filled with the linear interpolate according to the closest values on both sides. Then, we calculated the historical NeX/NeA as an indicator for evolution of sex-ratio bias. In the end, the curve of NeX/NeA was smoothed using a spline fit in R. The period of accomplished social transition by retrieving the time interval in which the decrease of Ne in social species slows down.
Data access
All raw sequencing data generated in this study have been submitted to the NCBI BioProject database under accession number PRJNA994315. The assembled genomes in this study have been submitted to the NCBI Genome database under the following accession numbers: S. dumicola, CP160598-CP160611; S. tentoriicola, CP160584-CP160597; S. bicolor, CP160568-CP160583; S. sarasinorum, CP160513-CP160528; S. mimosarum, CP160529-CP160544; and Stegodyphus lineatus, CP160545-CP160567. All custom scripts required to reproduce the work are available at GitHub (https://github.com/Jilong-Jerome/sociality-in-spiders-dead-end) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the support from Independent Research Fund Denmark (grant no. 0135-00201B). We thank Yael Lubin, Tharina Bird, Virginia Settepani, Lena Grinsted, and Clemence Rose for help in collecting samples included in this study.
Author contributions: J.B., P.V., T.B., and M.H.S. conceptualized the study. J.B. and T.B. designed and collected sample data for the study. J.M. performed bioinformatics analysis. J.M. and M.H.S. were major contributors of the manuscript. A.A., J.B., P.V., and T.B. contributed to designing figures in the manuscript and Supplemental Materials. All authors have read, edited, and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279503.124.
- Received April 22, 2024.
- Accepted February 4, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








