
Nucleosome binding by TP53, TP63, and TP73 is determined by the composition, accessibility, and helical orientation of their binding sites
- 1Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, New York 14203, USA;
- 2Department of Biomedical Informatics, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, New York 14203, USA
Abstract
The TP53 family of transcription factors plays key roles in driving development and combating cancer by regulating gene expression. TP53, TP63, and TP73—the three members of the TP53 family—regulate gene expression by binding to their DNA binding sites, many of which are situated within nucleosomes. To thoroughly examine the nucleosome-binding abilities of the TP53 family, we used Pioneer-seq, a technique that assesses a transcription factor's binding affinity to its DNA-binding sites at all possible positions within the nucleosome core particle. Using Pioneer-seq, we analyzed the binding affinities of TP53, TP63, and TP73 to 10 TP53 family binding sites across the nucleosome core particle. We find that the affinities of TP53, TP63, and TP73 for nucleosomes are primarily determined by the positioning of TP53 family binding sites within nucleosomes; TP53 family members bind strongly to the more accessible edges of nucleosomes but weakly to the less accessible centers of nucleosomes. Our results further show that the DNA-helical orientation of TP53 family binding sites within nucleosomal DNA impacts the nucleosome-binding affinities of TP53 family members, with binding-site composition impacting the affinity of each TP53 family member only when the binding-site location is accessible. Taken together, our results show that the accessibility, composition, and helical orientation of TP53 family binding sites collectively determine the nucleosome-binding affinities of TP53, TP63, and TP73. These findings help explain the rules underlying TP53 family-nucleosome binding and thus provide requisite insight into how we may better control gene expression changes involved in development and tumor suppression.
The TP53 family of transcription factors induces the expression of genes that determine the survival, proliferation, and differentiation of cells. In this family are three members: TP53, TP63, and TP73. TP53 has a well-known role in tumor suppression and is mutated in >50% of human cancers (Joerger and Fersht 2007; Baugh et al. 2018; Marei et al. 2021). Upon activation by cell stress signals, TP53 induces the expression of genes involved in cell cycle arrest and apoptosis (Nguyen et al. 2018; Fischer and Sammons 2024). TP63 and TP73 also induce the expression of some of these TP53-regulated genes (Moll and Slade 2004), although both transcription factors play more of a prominent role in the induction of developmental genes. TP63 induces the expression of genes involved in the proliferation, differentiation, and adhesion of epithelial cells (Kouwenhoven et al. 2015a; Sethi et al. 2017; Boughner et al. 2018), whereas TP73 induces the expression of genes involved in neuronal development, multiciliogenesis, germ cell maturation, and angiogenesis (Nemajerova and Moll 2019; Logotheti et al. 2021). The existence of multiple isoforms of each TP53 family member, which can exhibit overlapping or even opposing functions, adds to the regulatory complexity of this family (Wei et al. 2012; Sethi et al. 2015).
Acting in their capacity as transcription factors, TP53 family members induce gene expression through their binding to specific DNA sequences, also known as “binding sites.” TP53 specifically binds to a canonical 20 bp binding site comprising two repeats of RRRCWWGYYY (where R = A or G, W = A or T, Y = C or T) separated by 0–13 bp (el-Deiry et al. 1992; Funk et al. 1992), although TP53 also binds to a large number of sequences that deviate from this binding-site pattern (Tomso et al. 2005; Veprintsev and Fersht 2008). TP63 and TP73 can bind to the same binding sites as TP53 (Osada et al. 2005; Lokshin et al. 2007; Schavolt and Pietenpol 2007), owing to the high similarity of their DNA-binding domains (Levrero et al. 2000). Each TP53 family member binds to these 20 bp binding sites (hereafter termed “TP53 family binding sites”) as a tetramer, specifically a dimer of dimers that are each bound to an RRRCWWGYYY repeat (Brandt et al. 2009).
Eukaryotic DNA is organized into chromatin, which is made up of nucleosomes (Cooper 2000). A nucleosome comprises an octamer of histone proteins (two copies each of H2A, H2B, H3, and H4) around which ∼146 bp of DNA is coiled (Luger et al. 1997). Although nucleosomal DNA is typically inaccessible to transcription factor binding (Zhu et al. 2018), TP53 is able to bind to instances of its binding sites within nucleosomes (Espinosa and Emerson 2001; Lidor Nili et al. 2010; Sammons et al. 2015), specifically when positioned near the nucleosome edges (Laptenko et al. 2011; Yu and Buck 2019; Nishimura et al. 2020). TP63 is also known to bind to instances of its binding sites within nucleosomes (Sethi et al. 2014; Yu et al. 2021), although TP73 has not yet been tested in any nucleosome-binding studies. Despite the remarkable progress in defining the capabilities of the TP53 family to bind to nucleosomes, little is known of the underlying intricacies of TP53 family–nucleosome binding.
In this study, we examined the relative binding affinity of each TP53 family member to 10 different TP53 family binding sites when positioned at every base pair of the Widom-601 nucleosome. Using this comprehensive data set, we investigated how the DNA-sequence composition, nucleosomal accessibility, and DNA-helical orientation of TP53 family binding sites within the nucleosome influence TP53 family binding. Our results highlight the principles governing TP53 family–nucleosome binding and their broader implications for gene regulation in tumor suppression and development.
Results
Pioneer-seq reveals the nucleosome-binding preferences of TP53, TP63, and TP73
To comprehensively examine the nucleosome-binding abilities of the TP53 family, we developed Pioneer-seq, which is an extension of low-throughput nucleosome-binding assays (Yu and Buck 2019). Pioneer-seq is a high-throughput competitive nucleosome-binding assay for gauging the binding affinities of transcription factors with their transcription factor binding sites at every base pair position of the nucleosome core particle and at several positions of the linker DNA. Pioneer-seq is similar to SeEN-seq, which has been successfully used to identify TF-bound nucleosomes for cryo-EM (Michael et al. 2020, 2023). SeEN-seq has been limited to a small nucleosome library, fewer than 200 sequences with a single TFBS variant. As outlined in Figure 1A, the Pioneer-seq protocol starts with the in silico design of a library of Widom-601 nucleosome-positioning sequences that each contain an individual transcription factor binding site at a unique position of the nucleosome core particle or linker DNA. This DNA library is then reconstituted into a nucleosome library, in which the formation of nucleosomes was highly consistent with the Widom-601 crystal structure (Supplemental Fig. 1; Luger et al. 1997). The nucleosome library is then incubated with the transcription factor of interest. Stable transcription factor–nucleosome complexes are then isolated via gel-shift assay and are DNA-sequenced (Supplemental Fig. 2). The resulting sequencing data are then analyzed to gauge the binding affinity of the transcription factor of interest to its binding site at each nucleosomal position (Equation 1). For this study, we performed Pioneer-seq experiments with TP53, TP63, and TP73 using a nucleosome library that contained 10 TP53 family binding sites individually positioned at every base pair of the Widom-601 nucleosome core particle and at several positions of the linker DNA (Supplemental Table 1).
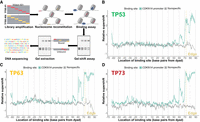
Pioneer-seq maps TP53 family–nucleosome binding. (A) Outline of Pioneer-seq. A library of 217 bp nucleosome-positioning sequences, which are based on the Widom-601 nucleosome-positioning sequence and which have a TP53 family binding site in one of the 146 bp positions of the nucleosome core particle, is reconstituted into nucleosomes and then incubated with TP53, TP63, or TP73. Bound nucleosomes are separated via gel-shift assay, extracted, purified, and sequenced. (B–D) TP53, TP63, or TP73 binding to a CDKN1A-promoter TP53 family binding site at different nucleosomal positions. TP53 family binding to the CDKN1A-promoter binding site is measured relative to TP53 family binding to nonspecific (non-TP53 family) binding sites and represented by relative-supershift values (Equation 1). A nonspecific motif for STAT5A is shown for comparison in gray. (Edge) The right end of the 146 bp nucleosome core particle. Shading indicates SEM. n = 3.
We initially assessed how strongly each TP53 family member binds to a TP53 family binding site from the CDKN1A promoter at different positions of the Widom-601 nucleosome. The predominant pattern of TP53 family–nucleosome binding was that the affinity each TP53 family member has for their CDKN1A-promoter binding site is dependent on how far it is positioned from the dyad of the nucleosome core particle. Notably, though, there were exceptions to this pattern: For example, TP53, TP63, and TP73 bind more strongly to their CDKN1A-promoter binding site when it is 65 bp away from the dyad than when it is 68 bp away (Fig. 1B,C). It is also notable that this pattern varies in extent among TP53 family members. When the CDKN1A-promoter binding site is 61 bp from the dyad, for instance, TP63 and TP73 show a stronger ability to bind compared with that of TP53 (Fig. 1B,C). To control for nonspecific binding to nucleosomal DNA or histone proteins, we also assessed how strongly each TP53 family member binds to a nonspecific STAT5A binding site at different positions of the Widom-601 nucleosome, represented by the gray lines in Figures 1 and 2. Our results underscore the critical role of binding-site positioning within nucleosomes in determining TP53 family binding strength.
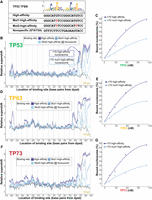
Binding site composition modulates TP53 family–nucleosome binding affinity. (A) A TP53 sequence logo above binding sites: The high-affinity site is based on the TP53 sequence logo; the Mut1-high-affinity site, on the TP53 sequence logo but with one base mutated; the Mut2-high-affinity binding site, on the TP53 sequence logo but with two bases mutated; and the nonspecific STAT5A site. (B,D,F) TP53, TP63, or TP73 binding to the four binding sites in A at different nucleosomal positions, measured relative to TP53 family binding to nonspecific binding sites and represented by relative-supershift values (Equation 1). A nonspecific motif for STAT5A is shown for comparison in gray. (Edge) The right end of the 146 bp nucleosome core particle. Shading indicates SEM. n = 3. (C,E,G) Assessing the binding affinity between TP53, TP63, or TP73 and the +70 high-affinity nucleosome or the +70 mut1-high-affinity nucleosome, both of which are indicated in B. Bound nucleosome (%) was calculated via gel-shift assays featuring Cy5-labeled nucleosomes.
Sequence composition of binding sites modulates TP53-, TP63-, and TP73-nucleosome-binding affinity
Having established where TP53 family members bind to their CDKN1A-promoter binding site within the Widom-601 nucleosome, we then sought to identify the specific factors that underlie where TP53 family members can bind nucleosomes. One potential factor was the sequence composition of TP53 family binding sites, given its known influence on the affinity of TP53 family members for DNA. This led us to investigate how mutations in TP53 family binding sites impact the nucleosome-binding abilities of TP53 family members. We measured how well TP53, TP63, and TP73 bind to unmutated and mutated versions of a high-affinity TP53 family binding site (Fig. 2A) across the Widom-601 nucleosome core particle and linker DNA. At nearly all positions of the Widom-601 nucleosome core particle and linker where TP53 family members could bind to a high-affinity site, they exhibited reduced binding affinity to the single-mutant version and even lower affinity to the double-mutant version. In contrast, at positions where TP53 family members could not bind to the high-affinity TP53 family binding site (about −70 to +50 bp from the dyad), mutations had no measurable effect on binding (Fig. 2B,D,F). These positions appeared intrinsically unfavorable for binding, regardless of sequence composition, highlighting that the nucleosomal positioning of a TP53 family binding site is a crucial determinant of binding. To validate these Pioneer-seq results, we conducted traditional nucleosome-binding assays using two selected nucleosomes from the Pioneer-seq library: one with the high-affinity binding site 70 bp from the dyad and the other with the once-mutated binding site at the same distance (Fig. 2B). These traditional nucleosome-binding assays confirmed that altering a single base pair of a TP53 family binding site significantly decreased the affinity of TP53, TP63, and TP73 for nucleosomes (Fig. 2C,E,G; Supplemental Fig. 4).
Binding-site accessibility is a key determinant of TP53 family–nucleosome binding
Our Pioneer-seq data revealed an inverse correlation between the proximity of TP53 family binding sites to the nucleosome dyad and the nucleosome-binding affinity of TP53 family members (Figs. 1B–D, 2B,D,F). We questioned whether the observed binding pattern was related to the accessibility of binding sites within nucleosomes. To address this question, we digested the nucleosomes in the Pioneer-seq library using micrococcal nuclease (MNase), which is a nonspecific endo- and exonuclease sensitive to the accessibility of nucleosomal DNA (Tsompana and Buck 2014). The susceptibility of library nucleosomes to MNase digestion mirrored the structural organization of the nucleosome core particle, with the tightly wrapped dyad being the most protected from MNase digestion and the more loosely wrapped edges being the least protected from digestion (Fig. 3A). Then we analyzed the MNase-digested Pioneer-seq library to measure how accessible the CDKN1A-promoter and high-affinity TP53 family binding sites are at each base pair position of the Widom-601 nucleosome. Our analysis revealed a strong correlation between the accessibility of the CDKN1A-promoter and high-affinity binding sites and the nucleosome-binding affinities of TP53 family members. Further analysis of the MNase-digested Pioneer-seq library revealed that the accessibility of other TP53 family binding sites—that is, binding sites from the promoters of the PUMA, PPN1, and CHMP4C genes—also correlated with TP53 family–nucleosome binding affinity (Fig. 3B–D). This correlation between binding-site accessibility and TP53 family–nucleosome binding was influenced by the intrinsic affinity of the TP53 family for each site, because the correlation weakened for the mutated sites compared with the high-affinity site (Supplemental Fig. 3).
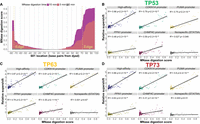
Binding site accessibility is a key determinant of TP53 family–nucleosome binding. (A) Measuring library-nucleosome accessibility via MNase digestion, represented by MNase-digestion scores. (Edge) The left or right end of the canonical 146 bp nucleosome core particle. (B–D) Correlating TP53, TP63, or TP73 binding strength with binding-site accessibility (measured by MNase-digestion scores). Shading around regression lines represents 95% confidence intervals.
Binding-site helical orientation in DNA impacts TP53 family–nucleosome binding
Although the composition and accessibility of binding sites are together strong determinants of TP53 family–nucleosome binding, they still do not fully explain the patterns of TP53 family–nucleosome binding we observed. See, for instance, how TP53 binds with a greater affinity to its high-affinity binding site when positioned 70 bp away from the dyad of the Widom-601 nucleosome core particle than when positioned 73 bp away (Fig. 4A,B), even though this latter position is more accessible (Fig. 3A). This discrepancy suggested to us the involvement of an unrecognized factor in regulating TP53 family–nucleosome binding. To potentially identify this unrecognized factor, we structurally modeled a Widom-601 nucleosome with the high-affinity TP53 family binding site 70 bp away from the dyad of the nucleosome core particle (the “+70 high-affinity nucleosome”) and a Widom-601 nucleosome with this high-affinity binding site 73 bp away (“+73 high-affinity nucleosome”) (Fig. 4A). In the strongly TP53-bound +70 high-affinity nucleosome, the most highly conserved CATG bases (Fig. 2A) are positioned so that their major-groove-facing atoms are exposed at the +70 position. In contrast, in the weakly TP53-bound +73 high-affinity nucleosome, the helical orientation of these CATG bases aligns more of their atoms toward the minor groove (Fig. 4C). To confirm the consistent positioning of the +70 and +73 high-affinity nucleosomes, we performed MNase digestion followed by restriction-enzyme digestion, which maps nucleosome edges relative to a protected restriction site (Thiriet and Hayes 1998). The results show identical band sizes for the +70, +73, and Widom 601 control nucleosomes, confirming their consistent translational settings (Supplemental Fig. 5).
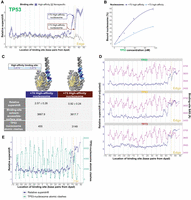
Binding site helical orientation in nucleosomal DNA impacts TP53 family–nucleosome binding. (A) TP53 binding to a high-affinity (HA) TP53 family binding site at different nucleosomal positions, measured relative to nonspecific binding sites and represented by relative-supershift values (Equation 1). A nonspecific motif for STAT5A is shown for comparison in gray. (Edge) The right end of the 146 bp nucleosome core particle. Shading indicates SEM. n = 3. (B) Binding affinity comparison between +70 and +73 high-affinity nucleosomes—both of which are indicated in A. (C) Illustrative models of the +70 and +73 high-affinity nucleosomes. The TP53-relative-supershift value corresponding to each nucleosome is shown. Also shown are the solvent-accessible-surface area of the conserved CATG bases of the high-affinity binding site in each nucleosome and the number of atomic clashes between the TP53 tetramer and nucleosome. (D) TP53 family binding to the HA binding site at different nucleosomal positions is plotted in blue as “control-adjusted” relative-supershift values (i.e., relative-supershift values of nonspecific-binding-site-containing nucleosomes subtracted from relative-supershift values of HA-binding-site-containing nucleosomes). Solvent-accessible surface area (SAS) of the conserved CATG bases of the high-affinity binding site is plotted in purple. (Edge) The right end of the 146 bp nucleosome core particle. (E) Plot of TP53 relative-supershift values versus the number of atomic clashes between a TP53 dimer and the nucleosome at different nucleosomal positions.
To further investigate the role of the helical orientation of binding sites in TP53 family-nucleosome binding, we used ChimeraX to measure the solvent-accessible surface area (SASA) of the CATGs of the high-affinity TP53 family binding site. These measurements provided insights into the relative exposure of the binding-site CATGs to the solvent environment, enabling us to assess their potential accessibility to TP53 family binding. We found that the binding-site CATGs in the strongly TP53-bound +70 high-affinity nucleosome were more exposed than the binding-site CATGs in the weakly bound +73 high-affinity nucleosome (Fig. 4C). Then, to comprehensively assess the role of the helical orientation of binding sites in TP53 family–nucleosome binding, we extended our SASA measurements to encompass every library nucleosome that has a high-affinity TP53 family binding site. At positions with high accessibility but unexpectedly low binding, the CATGs had consistently lower SASA values than those at positions with stronger binding. (Fig. 4D). A similar pattern was observed when we mapped the number of clashes between the TP53 dimer and the nucleosome at each binding position using ChimeraX. High-affinity binding-site positions such as +73, which had lower TP53-binding, exhibited a higher number of clashes compared with positions such as +70 (Fig. 4E), supporting the idea that the helical orientation of TP53 family binding sites influences TP53 family binding.
TP53 family–nucleosome binding is a function of the composition, accessibility, and helical orientation of TP53 family binding sites
Our analysis revealed three factors underlying TP53 family–nucleosome binding: (1) binding-site composition, (2) accessibility, and (3) DNA-helical orientation. Because we had already investigated how each of these factors individually affects binding, we next sought to investigate how their combined effects influence TP53 family–nucleosome binding. To do this, we performed an exploratory stepwise multiple-regression analysis that included both the individual factors and their interaction terms, allowing us to model how these features work together to drive TP53 family binding. The key variables used in the regression model to represent these factors were (1) the binding-site FIMO score, which quantifies how well the binding site matches the TP53, TP63, or TP73 motifs from the JASPAR database; (2) the binding-site MNase-digestion score, which represents how accessible the binding site is in the nucleosome; and (3) the binding-site SASA, which serves as a metric for the binding-site helical orientation by measuring how exposed the binding-site CATG residues are to the solvent environment. We also included interaction terms between these variables in the model to explore the potential combined effects of binding-site composition, accessibility, and orientation. This approach allowed us to assess whether these factors, when acting together, provide a more comprehensive explanation of TP53 family–nucleosome binding than when considered individually.
The final model resulted in an equation that captures the combined effects of binding-site composition, accessibility, and helical orientation on TP53 family binding (Fig. 5A). To determine whether binding-site composition, accessibility, and orientation have a synergistic effect on TP53 family–nucleosome binding, we compared the ability of our predictive model to explain TP53 family–nucleosome binding to models based on either accessibility or helical orientation alone. Although accessibility alone explained 68% of the variance observed in TP53 family–nucleosome binding (Fig. 5B) and helical orientation alone explained only 4% (Fig. 5C), our exploratory model explained a notable 74% of the variance (Fig. 5D). This modest but statistically significant increase from 68% to 74% (P < 2.2 ×10 −16) shows that binding-site composition and helical orientation refine accessibility-driven predictions, underscoring their interconnected roles in collectively influencing TP53 family-nucleosome binding. Our current model focuses on explaining in vitro binding; extending it to predict in vivo binding events would require accounting for additional complexities such as chromatin state and cofactor interactions.
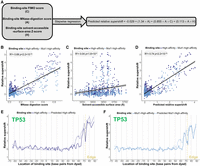
TP53 family–nucleosome binding is collectively determined by the composition, accessibility, and helical orientation of TP53 family binding sites. (A) Outline of regression model predicting TP53 family–nucleosome binding based on binding-site FIMO scores, MNase-digestion scores, and solvent-accessible-surface-area Z-scores. The resulting equation illustrates the predictive relationship between these variables and TP53 family–nucleosome binding. (B–D) Correlating the relative-supershift values (Equation 1) of TP53, TP63, and TP73 to two TP53 family binding sites with either the MNase-digestion score of these binding sites, the solvent-accessible surface area of the CATGs of these binding sites, or the relative-supershift values of TP53, TP63, and TP73 to these binding sites predicted by the multiple regression model outlined in A. (E,F) Comparison of actual versus model-predicted TP53 relative-supershift values for high-affinity and Mut1-high-affinity binding sites.
TP53 family binding in cells: interplay of binding-site affinity and position within nucleosomes
We next examined whether the binding patterns observed in vitro were consistent in a cellular context. To extend our findings, we examined TP53 and TP63 ChIP-seq with MNase-seq to examine nucleosome occupancy at sites bound by these factors. The site of binding was defined by the TP53 (MA0106.3) or TP63 (MA0525.2) motif occurring within each ChIP-seq peak. Locations with multiple motifs were excluded from the analysis. Appropriate MNase-seq data were not available for TP73. To capture nucleosome positioning before TP53 or TP63 binding and any resultant chromatin remodeling, the MNase-seq data sets were from cell lines before TP53 activation (via Nutlin) or TP63 expression. This approach avoided the cofounding effects of transcription factor–driven chromatin remodeling, which can obscure the original nucleosome landscape. Consistent with the methods of previous studies, nucleosome occupancy was enriched at TP53- and TP63-bound sites (Fig. 6A,B; Sammons et al. 2015; Yu and Buck 2019; Yu et al. 2021). To further examine the relationship of TP53 and TP63 with nucleosomes, we separated all TP53 or TP63 sites by their inferred binding-site affinity as defined by the FIMO score and examined the relationship between binding and distance to nucleosome centers. Binding sites were grouped into four categories based on whether they were higher or lower affinity and their distance from the nucleosome dyad (within 10 bp or 60–100 bp). Our analysis revealed a clear ranking of binding levels across different site categories for both TP53 and TP63. On average, lower-affinity near-dyad binding sites exhibited the lowest binding levels, followed by higher-affinity near-dyad sites, lower-affinity outer-nucleosome sites, and high-affinity outer-nucleosome sites (Fig. 6C,D). Lower-affinity outer-nucleosome sites displayed higher binding levels than high-affinity near-dyad sites, emphasizing that binding-site nucleosomal positioning can play a larger role in binding than the site's inherent affinity, consistent with our Pioneer-seq data. This result supports our Pioneer-seq findings, highlighting that the nucleosomal positioning of a TP53 family binding site is critical to understanding TP53 family binding, even more than the inherent affinity of the TP53 family member for that site in some cases.
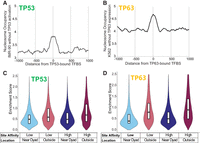
TP53 and TP63 enrichment relative to nucleosome positioning and binding-site affinity. (A) Average nucleosome occupancy at TP53 binding sites before activation with Nutlin in IMR-90 cells, as determined from MNase-seq data. These sites represent the locations where TP53 binds following its activation. (B) Average nucleosome occupancy at TP63 binding sites before expression in K562 cells, as determined from MNase-seq data. These sites correspond to the locations where TP63 binds upon expression. (C,D) Violin plots of TP53 and TP63 ChIP-seq peak enrichment scores, categorized by binding-site affinity and nucleosome position (lower/higher-affinity, near-dyad/outer nucleosome). Binding sites were identified using the MA0106.3 TP53 motif (JASPAR), and nucleosome positioning was determined from MNase-seq data.
Discussion
This study explored the interplay between the TP53 family of transcription factors, their binding sites, and the nucleosome, revealing three critical factors influencing TP53 family–nucleosome binding: (1) the DNA-sequence composition of TP53 family binding sites, (2) the accessibility of these binding sites in the nucleosome, and (3) the helical orientation of these binding sites in the nucleosome. These factors collectively shape the binding landscape of the TP53 family to nucleosomes. This suggests that TP53 family binding is fine-tuned by the nucleosomal context and DNA-sequence composition of TP53 family binding sites; for example, a less-favorable TP53 family binding site could be bound strongly if positioned in a more accessible region of the nucleosome (Fig. 6C,D). Our multiple-regression model (Fig. 5A) further supports this, showing that binding-site accessibility is the most significant factor in predicting TP53 family binding, both independently and in combination with other factors. These insights highlight the importance of considering the positioning of a TP53 family binding site relative to nucleosomes, along with the sequence composition of the binding site itself, when interpreting or predicting in vivo binding patterns of TP53 family members.
Consistent with previous findings on the role of sequence composition in TP53 family affinity for naked DNA (Veprintsev and Fersht 2008; Brandt et al. 2009), our findings show that the composition of TP53 family binding sites significantly impacts the affinity of TP53 family members for nucleosomal DNA. Our findings also show that the position of binding sites within nucleosomes is a key determinant of TP53 family binding. The regions of nucleosomes that are bound most strongly by TP53 family members—the edges—are the most accessible regions of the nucleosomes, which aligns with the prevailing model of dynamic partial DNA unwrapping from histones at the edges of nucleosomes (Kitayner et al. 2006; Beno et al. 2011; Jordan et al. 2012). Our findings also show that the helical orientation of TP53 family binding sites at nucleosomal edges has a considerable effect on the TP53 family binding. This supports the hypothesis that the helical orientation of nucleosomal TP53 binding sites affects binding affinity, potentially influencing the differential induction of TP53-regulated genes (Freewoman et al. 2021).
Although all three TP53 family members follow a similar pattern of binding based on site distance from the dyad, each exhibits distinct affinities at specific positions. When the CDKN1A-promoter binding site is positioned 61 bp right of the dyad, for example, TP63 and TP73 show a stronger ability to bind compared with TP53 (Fig. 1B–D). Similarly, when our high-affinity TP53 family binding site is positioned ∼55–60 bp from the dyad, TP63 shows stronger binding compared with both TP53 and TP73 (Fig. 2B,D,F). Our findings suggest that in certain scenarios TP63 and TP73 may be more effective at binding nucleosomes compared with TP53, perhaps reflecting their specialized roles in gene regulation. This is further supported by the prior observation that TP63 and TP73 are required for TP53 binding at the promoter of apoptotic genes such as BAX, NOXA, and PERP (Murray-Zmijewski et al. 2006), indicating a collaborative function in gene regulation that may involve chromatin dynamics. Additionally, prior evidence shows that TP63 promotes TP53 binding by opening chromatin in epithelial cell types (Karsli Uzunbas et al. 2019; Woodstock et al. 2021).
Pioneer factors are a class of transcription factors that can bind to condensed chromatin and initiate chromatin remodeling, making previously inaccessible DNA regions available for transcription (Zaret 2020; Balsalobre and Drouin 2022). A growing body of evidence suggests that TP53 family members function as pioneer factors (Sammons et al. 2015; Yu and Buck 2019; Yu et al. 2021). ChIP-seq studies suggest that ∼50% of TP53 binding events occur in nucleosome-occupied regions (Espinosa and Emerson 2001; Laptenko et al. 2011). In some cases, these binding events lead to chromatin opening and potential enhancer activation, suggesting TP53 pioneer-factor activity (Younger and Rinn 2017). For the majority of these binding events, though, TP53 is not required for chromatin accessibility (Karsli Uzunbas et al. 2019). TP63, on the other hand, plays an extensive role in establishing and sustaining chromatin accessibility at regulatory sites linked to epithelial-cell maturation (Bao et al. 2015; Kouwenhoven et al. 2015b; Qu et al. 2018). ΔNp63α, the predominant TP63 isoform in epithelial cells, has been repeatedly associated with binding to inaccessible chromatin and driving changes in chromatin accessibility (Kouwenhoven et al. 2015a; Yu et al. 2021; Kuang and Li 2023). Despite these insights, the pioneer factor activities of other TP63 isoforms remain poorly understood, and even less is known about TP73 isoforms.
In our study, we used the full-length isoforms of each TP53 family member: TP53α, TAp63α, and TAp73α. It is thought that TAp63α adopts a “spring-loaded” activation mechanism, remaining in a dimeric state until phosphorylation triggers tetramerization, thereby enhancing its DNA-binding affinity (Gebel et al. 2020). This conformation appears to be unique to TAp63α and does not occur in TP53α or TAp73α. Because all of our binding assays were conducted with unphosphorylated transcription factors, it is possible that the lack of phosphorylation influenced their binding behavior, particularly for TAp63α.
From previous studies, it had already emerged that the affinity TP53 and TP63 have for nucleosomes is dependent on the positioning of their binding sites (Yu and Buck 2019; Nishimura et al. 2020; Yu et al. 2021). This led us to expect TP53 family–nucleosome binding affinity to simply increase as TP53 family binding sites were positioned further from the dyad of the nucleosome. Although our initial expectation mostly held true, our high-throughput Pioneer-seq assay revealed a clear periodic pattern of nucleosome-binding deviations that was not apparent in earlier studies. Previous studies examined TP53 and TP63 binding at only a few nucleosomal positions, which likely limited their ability to detect this periodicity. By analyzing TP53 family binding across multiple nucleosomal positions, we were able to identify subtle but significant deviations at which binding was weaker in specific rotational settings, despite greater distance from the dyad. These findings demonstrate that the helical orientation of TP53 family binding sites is an important factor influencing TP53 family binding at nucleosome edges.
Although TP53's affinity for specific DNA sequences (i.e., its binding sites) in nucleosomes is well established, our previous work showed that TP53 also has a considerable affinity for nonspecific DNA sequences in nucleosomes, mediated by its C-terminal domain (Yu and Buck 2019). These nonspecific TP53-nucleosomal-DNA-binding events were illustrated recently in a set of cryo-electron-microscopy experiments, revealing that the C-terminal domain of TP53 is required for nonspecific binding to the nucleosome dyad and linker (Nishimura et al. 2022). In the present study, however, we did not examine nonspecific binding, on account of Pioneer-seq being a competitive nucleosome-binding assay. Pioneer-seq measures the binding capability of a transcription factor for a specific sequence located within or near a nucleosome, by directly controlling for nonspecific binding to other sequences. To address this limitation, it would be beneficial for future studies to investigate the contributions of individual residues within the domains of TP53 family members to nucleosome-binding affinity and specificity. This could be achieved through mutagenesis studies that introduce specific amino acid substitutions in the TP53 family proteins and then assess their ability to bind to nucleosomes. By systematically mutating key TP53 family residues and evaluating the resulting effects on nucleosome-binding affinity and specificity, a more comprehensive view of the molecular mechanisms underlying TP53 family–nucleosome binding could be achieved.
This study demonstrated that TP53 family nucleosomal binding is shaped by the DNA-sequence composition, accessibility, and helical orientation of their binding sites. This has significant implications for comprehending how TP53 family members regulate gene expression in various cellular processes, including development and tumor suppression. By identifying the key factors influencing TP53 family–nucleosome binding, this research paves the way for deeper investigations into gene regulation and the potential development of therapeutic strategies targeting TP53 family dysfunction.
Methods
Pioneer-seq library design
The nucleosomes in the Pioneer-seq library were based on the widely used Widom-601 nucleosome-positioning sequence (Flaus 2011; Yu and Buck 2020). To ensure the Widom-601 sequence had no preexisting transcription factor binding sites, FIMO (Grant et al. 2011) and JASPAR (Rauluseviciute et al. 2024) were used to scan this sequence for the occurrence of transcription factor binding sites. Matching bases detected by FIMO (P-value < 0.01) were modified to remove detected transcription factor binding sites.
For each of the transcription factor binding sites included in the Pioneer-seq library, a set of Widom-601 sequences was designed, each containing a single copy of this transcription factor binding site positioned at one of the 146 bp positions of the Widom-601 nucleosome core particle or at one of several linker positions.
Pioneer-seq nucleosome library assembly
All library nucleosome sequences were flanked by primer sequences to generate 217 bp sequences. The library of nucleosome sequences, totaling 7500 unique sequences, was acquired from Agilent as a custom oligonucleotide library, which was amplified in 15 PCR cycles using Herculase II fusion DNA polymerase in a 100 μL reaction mixtures (1× Herculase II reaction buffer, 1 mM dNTP, 200 pM Agilent library, 250 nM forward and reverse primers). In a typical Pioneer-seq experiment, the DNA obtained from 11 reactions was purified with a QIAquick PCR purification kit (Qiagen 28104), quantified with a NanoDrop spectrometer, and then visualized with a 2%-agarose ethidium bromide gel.
Nucleosomes were reconstituted by incubating H2A/H2B dimers and H3.1/H4 tetramers (New England Biolabs) with library DNA at a histone/DNA ratio of 1:1.5 in a buffer containing 10 mM DTT and 1.8 M NaCl for 30 min at room temperature. Nucleosomes were then transferred to a Slide-A-Lyzer MINI dialysis unit (10,00 MWCO; Thermo Fisher Scientific 69750) for dialysis performed with 1.2 mL dialysis buffers at 4°C in 1.0 M NaCl for 2 h, 0.8 M NaCl for 2 h, 0.6 NaCl for 2 h, and TE buffer (pH 8.0) overnight. After dialysis, nucleosomes were transferred to a clean 1.5 mL tube pretreated with 0.3 mg/mL bovine serum albumin (BSA), and nucleosome formation was confirmed via 4%-native-polyacrylamide-gel electrophoresis. Free DNA was then removed from nucleosomes via a 7%–20%-sucrose gradient. Purified nucleosomes were then quantified via qPCR and stored for up to 1 month at 4°C.
Nucleosome-binding assay followed by gel-shift assay
Protein–nucleosome binding assays were performed by incubating the abovementioned purified Pioneer-seq nucleosome library with human TP53α (Abcam ab84768), TA-TP63α (Origene TP710041), or TA-TP73α (Origene TP320864) in 7 μL DNA-binding buffer (10 mM Tris-Cl at pH 7.5, 50 nM NaCl, 1 mM DTT, 0.25 mg/mL BSA, 2 mM MgCl2, 0.025% Nonidet P-40, and 5% glycerol) for 10 min on ice and then 30 min at room temperature. Increasing concentrations of transcription factor (0, 15, 30, 60, 120, and 240 nM) were added to 30 nM purified nucleosome library. TP53 family-bound and -unbound nucleosomes were separated via gel-shift assays using 4%-native-polyacrylamide gels (acrylamide/bisacrylamide, 29:1) in 0.5× Tris-borate-EDTA buffer at 100 V at 4°C. Gel-shift assays were initially done using a wide range of TP53 family-protein concentrations to determine the optimal protein amount and to ensure a shifted band is observed on the gel (Supplemental Fig. 2).
DNA isolation and purification
After staining the above-mentioned gel-shift assays with SYBR green (Lonza), all visible gel bands, as well as the corresponding invisible gel bands in the other lanes, were excised from the gel. Excised gel bands were then immersed in diffusion buffer (0.5 M ammonium acetate, 10 mM magnesium acetate, 1 mM EDTA at pH 8.0, 0.1% SDS) overnight at 50°C. The gel-band-diffusion-buffer immersion was then filtered through glass wool to remove polyacrylamide, and DNA from the resultant supernatant was then purified with a QIAquick gel extraction kit (Qiagen 28704).
Library construction and sequencing
Illumina sequencing libraries were prepared using a two-step PCR method. In the first step, DNA was amplified using four sets of primers designed to offset reads and dephase the libraries during sequencing (Handelmann et al. 2023). The number of PCR cycles used in this step was determined by the DNA concentration of each sample, as measured by qPCR. In the second step, each sample was indexed using Nextera dual indices (Nextera XT index primer 1 [N7xx] and Nextera XT index primer 2 [S5xx]). After each PCR step, the reaction mixtures were cleaned up with AMPure XP beads. The concentration of each sample was then determined using the Invitrogen Quant-iT dsDNA assay kit, and equal amounts of DNA from each sample were pooled for paired-end sequencing on an Illumina NextSeq 2 × 150. Sequencing and quality control were performed at the University at Buffalo Genomics and Bioinformatics Core.
Pioneer-seq analysis
FASTQ files of NextSeq reads were processed with an automated Snakemake (Köster and Rahmann 2018) pipeline of applications to refine and identify the sequences present in the sample pool. The 3′ ends of FASTQ reads with low-quality scores were removed using cutadapt (Martin 2011) with a quality cutoff of 30 (-q 30). Using Vsearch (Rognes et al. 2016), forward and reverse FASTQ reads were then merged (‐‐fastq_mergepairs) if they shared at least 20 overlapping nucleotides (‐‐fastq_minovlen 20) and had no more than two mismatched nucleotides between them (‐‐fastq_maxdiffs 2). Primer sequences present at the ends of FASTQ reads were then removed using cutadapt. FASTQ reads more than 220 or less than 174 nucleotides in length (‐‐maximum-length 220 ‐‐minimum-length 174) were then filtered out using cutadapt. Using FASTX-Toolkit (https://github.com/agordon/fastx_toolkit), FASTQ reads were then converted to FASTA reads (FASTQ-to-FASTA). Then, using Vsearch (‐‐dbmatched), each FASTQ read was mapped to a sequence in a database of the 7500 nucleosome sequences in the Pioneer-seq library if the FASTQ read and library sequence had alignment lengths of at least 150 nucleotides (‐‐mincols 150), had at least 98.5% similarity (‐‐id 0.985), and were the query and database sequence pairing with the highest percentage of identity (‐‐top_hits_only).
The relative binding affinities of TP53, TP63, and TP73 to each of the 7500 Pioneer-seq library nucleosomes were calculated
relative to control nucleosomes containing nonspecific binding sites (e.g., FOXA1, KLF4, STAT3) or no binding site: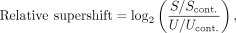 (1)
where, for a given nucleosome in the library, S is the number of reads of this given nucleosome in a shifted gel band, Scont. is the average number of reads of library nucleosomes with TP53 family-nonspecific binding sites in the same shifted gel
band, U is the number of reads of this given nucleosome in the unshifted gel band from the lane of the gel to which no transcription
factor was added, and Ucont. is the average number of reads of library nucleosomes with TP53 family-nonspecific binding sites in the same unshifted gel
band.
(1)
where, for a given nucleosome in the library, S is the number of reads of this given nucleosome in a shifted gel band, Scont. is the average number of reads of library nucleosomes with TP53 family-nonspecific binding sites in the same shifted gel
band, U is the number of reads of this given nucleosome in the unshifted gel band from the lane of the gel to which no transcription
factor was added, and Ucont. is the average number of reads of library nucleosomes with TP53 family-nonspecific binding sites in the same unshifted gel
band.
Because Pioneer-seq determines binding relative to nonspecific binding to the background sequence of the Widom-601 nucleosome-positioning sequence, any background nonspecific binding in our assay would be controlled for in the analysis.
Pioneer-seq experiments were initially performed using multiple concentrations of TP53 family protein in case of differences in inherent binding affinities and protein purity. The lowest TP53 family concentrations that enabled the highest TP53 family-binding-site-specific binding—the concentrations used throughout this paper—were as follows: 15 nM TP53, 60 nM TP63, and 60 nM TP73.
Nucleosome-binding assay using Cy5-labeled nucleosomes
Nucleosomal DNA labeled with the fluorescent cyanine dye Cy5 on its 5′ and 3′ ends was formed into purified nucleosomes as described above. Nucleosomes (30 nM) were incubated with increasing amounts of TP53 family protein (0, 15, 30, 60, 120, 240 nM) in 7 μL DNA-binding buffer for 10 min on ice and then for 30 min at room temperature. TP53 family-bound and -unbound nucleosomes were then separated via gel-shift assays using 4%-native-polyacrylamide gels in 0.5× Tris-borate-EDTA buffer at 100 V at 4°C. After the gel-shift assays, the nucleosomes were visualized and quantified via their Cy5 labels using a ChemiDoc MP imaging system. The intensity of the Cy5 fluorescence was directly proportional to the amount of nucleosomes present, enabling the quantification of the percentage of nucleosome bound.
MNase-digestion of pioneer-seq nucleosome library
MNase was used to measure the accessibility of nucleosomes as previously described (Handelmann et al. 2023). The Pioneer-seq nucleosome library (30 nM) was digested by MNase (0.05 U/μΛ) in nuclease digestion buffer (10 mM Tris-HCL at pH 8.0, 2 mM CaCl2) for 0, 5, or 10 min at 37°C; digestion was stopped with 2% SDS and 40 mM EDTA. Each sample was then incubated with Proteinase K (16 μg) for 1 h at 55°C. DNA from each sample was then purified and concentrated with the QIAquick PCR purification kit. Concentrations of DNA in each sample were then determined with the Invitrogen Quant-iT dsDNA assay kit and equalized. Illumina sequencing libraries were generated using an NEBNext Ultra II DNA library prep kit. Individual samples were multiplexed for paired-end sequencing on an Illumina MiSeq 2 × 150. Sequencing and quality control were performed at the University at Buffalo Genomics and Bioinformatics Core.
FASTQ files of MiSeq reads were quality-filtered (q > 30) and adapter-trimmed using cutadapt. The filtered and trimmed reads were then merged and mapped to a database of the 7500 nucleosome sequences in the Pioneer-seq library using Vsearch. Based on the number of reads and the end positions of these reads, MNase-digestion scores were calculated for each base pair of library nucleosomes as the ratio of the number of reads covering that base pair to the total number of reads mapped to the respective library nucleosome. To define nucleosome populations from the MNase-seq data, we examined the center of each MNase-seq fragment from the 10 min time point. Fragments were first filtered by size (146 to 148 bp), and then, the center was determined. All fragment centers were then used to construct a histogram (Supplemental Fig. 1).
Modeling Pioneer-seq-library nucleosomes and measuring SASA
Structural models for nucleosomes in the Pioneer-seq library were generated using version 1.7 of ChimeraX (Goddard et al. 2018) and based on the crystal structure of a Widom-601 nucleosome (PDB accession no. 4QLC). The linker DNA on one side of the Widom-601 crystal structure was extended to match that of the nucleosomes in the Pioneer-seq library.
The SASA of the two CATGs within the high-affinity TP53 family binding site was calculated using the “measure sasa” ChimeraX command with a “probeRadius” of 0.9 Å.
MNase digestion and restriction-enzyme treatment of nucleosomes
Nucleosomes from the Pioneer-seq library, including a Widom-601 nucleosome and modified Widom-601 nucleosomes with TP53 family binding sites 70 bp and 73 bp to the right of the dyad, were subjected to MNase digestion for 0 or 5 min. Digested products were analyzed on a 5% TBE gel. Nucleosome-sized fragments were purified and subsequently treated with no enzyme, HaeIII, or MfeI and visualized on a 5% TBE gel.
Modeling steric hindrance of TP53 binding throughout the nucleosome
The structures of the TP53 dimer and Widom-601 nucleosome were retrieved from the Protein Data Bank (accession nos. 3EXJ and 50XV, respectively). The Widom-601 nucleosome was edited to a single nucleosome of 180 bp of DNA using ChimeraX (Goddard et al. 2018). The x3DNA software package (Colasanti et al. 2013) was used to change the Widom-601 DNA sequence to include the appropriate TP53 binding site at the desired location within the nucleosome structure. The TP53 structure was then imposed onto the nucleosome using the ChimeraX “matchmaker” command, aligning the DNA chain in the nucleosome as the reference structure and the DNA chain in the TP53 structure as the match structure, using the Needleman–Wunsch algorithm for sequence alignment. Steric hindrance between the TP53 and the nucleosome for each location was determined with the “clashes” command in ChimeraX. The number of clashes between the TP53 protein and the nucleosome are graphed, showing the calculated hindrance at every binding position throughout the nucleosome.
Stepwise regression to model TP53 family–nucleosome binding
For each nucleosome in the data set, we obtained a relative-supershift value for each of the three TP53 family members, representing
their binding affinity. The data set consisted of more than 980 rows, in which each row corresponds to a unique combination
of a TP53 family member and a nucleosome that has a TP53 family binding site. Each row includes the “BS_score,” representing
the binding-site FIMO score, which quantifies how well the binding site matches the TP53 family motifs (TP53, TP63, or TP73)
from the JASPAR database; “MNase,” representing the binding-site MNase-digestion score, which measures the accessibility of
the binding site within the nucleosome; and “SAS,” representing the binding-site SASA, which quantifies the exposure of key
CATG residues, providing a measure of the binding-site helical orientation. As mentioned, each row includes an “RS,” or relative-supershift
value for the TP53 family member being analyzed, which serves as the dependent variable in the regression model. To investigate
how these factors, and their combined effects, influence TP53 family–nucleosome binding, we constructed a stepwise multiple-regression
model. This model selection approach included the individual factors (BS_score, MNase, and SAS) and their interaction terms,
allowing us to assess how these factors work together to affect binding affinity. The regression model was built using the
following formula in R (R Core Team 2020):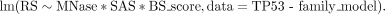
We evaluated the model's performance by comparing the R² values of the full multivariate model, which included all factors and their interaction terms, with models that considered only individual factors.
Analysis of TP53 and TP63 ChIP-seq peaks relative to nucleosome centers and binding-site affinity
ChIP-seq data for TP53 were obtained from Sammons et al. (2015), sourced from the IMR-90 cell line. TP63 ChIP-seq data were sourced from Yu et al. (2021), obtained from the K562 cell line. Binding sites corresponding to the MA0106.3 TP53 and MA0525.2 TP63 motifs (JASPAR) were identified using the UCSC Genome Browser (Raney et al. 2024), which provides occurrences of this motif in the human genome along with a score indicating the favorability of binding. Only ChIP-seq peaks overlapping with a single occurrence of the motif were included. Peaks with multiple motif occurrences were excluded to ensure focus on single binding sites. MNase-seq data from IMR-90 cells and K526 were obtained from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE21823 and GSM2083140 (Kelly et al. 2012; Mieczkowski et al. 2016) and aligned to GRCh38 (hg38) with Bowtie 2 (Langmead and Salzberg 2012). The MNase-seq reads were extracted for the TP53 and TP63 bound motifs, standardized (10 billion reads), and extended (120 bp) as done previously with ArchTEx (Lai et al. 2012; Rizzo et al. 2012).
Nucleosome centers for GSE21823 and GSM2083140 were obtained from the NucMap2 database with iNPS positioning (Chen et al. 2014; Nie et al. 2024). For each motif-containing binding site, the distance to the nearest nucleosome center was calculated. Binding sites within 10 bp of the nucleosome center were categorized as near-dyad, whereas those 60–100 bp from the center were categorized as outer-nucleosome. Nucleosomes with shoulders, doublets, or unclear centers were excluded from the analysis. Motif occurrences were stratified by binding affinity, with lower-affinity binding sites defined as those from the lowest quartile, and higher-affinity sites as those from the top quartile.
Data access
Pioneer-seq data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1048449. The custom scripts used for data analysis are available as Supplemental Code and at GitHub (https://github.com/pwilson97/Pioneer-seq_snakemake).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This study was supported by the National Institute of General Medical Sciences (R01GM132199) to M.J.B. We thank the UB Genomics and Bioinformatics Core for high-throughput sequencing services.
Author contributions: P.D.W., X.Y., and M.J.B. conceptualized the study. P.D.W. and M.J.B. were major contributors to writing the manuscript. C.R.H. modeled TP53-nucleosome steric hindrance. P.D.W. performed wet-laboratory analyses and bioinformatic analyses.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279541.124.
- Received May 3, 2024.
- Accepted February 3, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








