
Diffusion-based generation of gene regulatory networks from scRNA-seq data with DigNet
Abstract
A gene regulatory network (GRN) intricately encodes the interconnectedness of identities and functionalities of genes within cells, ultimately shaping cellular specificity. Despite decades of endeavors, reverse engineering of GRNs from gene expression profiling data remains a profound challenge, particularly when it comes to reconstructing cell-specific GRNs that are tailored to precise cellular and genetic contexts. Here, we propose a discrete diffusion generation model, called DigNet, capable of generating corresponding GRNs from high-throughput single-cell RNA sequencing (scRNA-seq) data. DigNet embeds the network generation process into a multistep recovery procedure with Markov properties. Each intermediate step has a specific model to recover a portion of the gene regulatory architectures. It thus can ensure compatibility between global network structures and regulatory modules through the unique multistep diffusion procedure. Furthermore, through iMetacell integration and non-Euclidean discrete space modeling, DigNet is robust to the presence of noise in scRNA-seq data and the sparsity of GRNs. Benchmark evaluation results against more than a dozen state-of-the-art network inference methods demonstrate that DigNet achieves superior performance across various single-cell GRN reconstruction experiments. Furthermore, DigNet provides unique insights into the immune response in breast cancer, derived from differential gene regulation identified in T cells. As an open-source software, DigNet offers a powerful and effective tool for generating cell-specific GRNs from scRNA-seq data.
The transcriptional state of a cell is intricately controlled by a gene regulatory network (GRN) comprising numerous transcription factors (TFs) and their target genes. Coordinating with other regulatory elements, GRNs modulate the cell's phenotype, identity, and function in a dynamic and specific manner (Levine and Davidson 2005). These complex regulatory architectures are important in unraveling precise gene expression pathways and providing crucial insights for elucidating the physiological processes or disease mechanisms of multicellular organisms (Moris et al. 2016). Currently, single-cell RNA sequencing (scRNA-seq) techniques offer more precise methods for profiling high-resolution transcriptional states and delineating differences among diverse cell types. Given the importance of cell heterogeneity, it is naturally expected that variations in transcriptional states will correspond to changes in cell state–dependent gene regulatory interactions, which cannot be represented as static networks (Huang et al. 2018). Because of the specificity and dynamics of tissue microenvironments, technical noise, and impacts from other sources at single-cell resolution, the transcriptomic gene expression levels may be partially decoupled from TF regulation, posing significant challenges to exploring the complex cellular landscape (Wagner et al. 2016).
Inferring gene regulation from transcriptomic data stands as a significant challenge in computational biology, aiming to reveal the cellular dynamics inherently manipulated by the interplay of genes. Over the past decades, numerous computational methods have emerged to infer GRN from gene expression profiles. These include correlation-based networks (Chan et al. 2017; Specht and Li 2017), Gaussian graphical models (Kotiang and Eslami 2020), tree-based ensemble pipelines (Huynh-Thu et al. 2010; Aibar et al. 2017), dynamic Bayesian models (Liu et al. 2016), and deep learning-based algorithms (Shu et al. 2021). Although these existing methods have achieved some advancements in inferring GRNs from transcriptomic data, they predominantly concentrate on modeling the regulatory relationships between individual genes and their multiple partners. Although this approach can capture the local neighborhood influences, it face challenges in simultaneously modeling the interconnected and compatible regulatory relationships among a vast array of genes. Consequently, the derived networks are predominantly constructed from isolated gene interactions, as well as lack of system-level understanding of complex regulatory mechanisms (Ma et al. 2023; Wang et al. 2023). These limitations undermine the accuracy of reconstructing GRNs from scRNA-seq data within specific contexts and hinder the ability of existing methodologies to decipher complex network structures. The architecture of GRNs, both globally or locally, is crucial in complex biological systems, revealing essential nodes (e.g., highly interconnected TFs) and regulatory modules and elucidating how GRNs adapt to intercellular variations and environmental stimuli. Although cutting-edge algorithms can discern some linear or nonlinear regulatory relationships, they predominantly focus on coupling individual gene pairs, rarely considering the intricate regulatory interplay among multiple gene simultaneously.
To address these challenges, we introduce DigNet, a deep generative model capable of deriving the underlying cell-specific GRN responsible for the transcriptional state directly from gene expression profiling data. The inspiration for DigNet comes from the booming generative techniques (Ho et al. 2020; Guo et al. 2023). Traditional generative models are usually conducted in an easy-to-understand Euclidean space, but it is difficult to accurately capture the complex regulatory relationships between genes. Therefore, DigNet employs a diffusion model framework, leveraging scRNA-seq data to embed the GRN structure into a non-Euclidean space with broader applicability, thereby generating sparse network architectures with unique structural characteristics. To reduce the complexity of embedding the GRN in non-Euclidean space and to enhance the model's interpretability, DigNet further models the network with a binary discrete representation that includes only binary (“on” and “off”) states. This effectively ensures the preservation of both the topological network structure and underlying biological characteristics.
DigNet stands as a generative model that concurrently delves into the intricate regulatory interplay among genes. It emphasizes the global architecture information within a GRN and generates a corresponding network structure from scRNA-seq data. As a highly adaptable and flexible tool, DigNet necessitates merely single-cell gene expression profiles to iteratively generate a GRN from a random starting point. The extracted structure of the GRN allows diverse downstream analyses. A comprehensive benchmark assessment encompassing 13 state-of-the-art GRN inference algorithms underscores the robustness and precision of the proposed network generation algorithm of DigNet. To demonstrate the versatility of DigNet, we applied it to reveal the regulatory landscape of immune responses in human breast cancer (BRCA). We constructed the immune cell–specific GRN and identified the differential networks across breast cancer samples and normal controls. By rediscovering known key regulatory relationships and prioritizing previously unknown candidate regulatory genes, DigNet reveals cellular functional differences in the form of specific network rewiring and proves its utility in exploring network-based biomarkers. These novel differential regulatory associations and interactors offer fresh perspectives on refining the mechanisms underlying breast cancer immune responses and pave the way for the discovery of novel therapeutic targets.
Results
Overview of DigNet
As shown in Figure 1, DigNet generates a cell-specific GRN from scRNA-seq data. Overall, DigNet dissects the network reasoning task into a reversible, multistep recovery process with Markovian properties, including feature extraction, diffusion-based denoising, and backward inference. Consequently, it allows for the delineation of each temporal stage with a distinctive network model, thereby enhancing its capability to discern and reconstruct network structures with increased granularity. Additionally, graph transformer with the self-attention mechanism is employed to learn the complex data distribution in scRNA-seq data and address challenges like experimental noise, high dimensionality, and scalability (see Supplemental Note 1). Once its fully trained model parameters are obtained, DigNet can easily generate a GRN given the gene expression profiles for any cells. Specifically, the initial phase involves optimizing gene expression data to mitigate the impact of single-cell dropout events and elevate data quality (Fig. 1A). Subsequently, DigNet applies a time-step approach to progressively denoise contaminated networks till achieving a clean network (Fig. 1B). During the training phase, DigNet iteratively alternates between the “network contamination” and “noise removal” phases until convergence is achieved. DigNet starts with a random network structure for testing and gradually rectifies it using time-step. Both the training and testing phases engage network encoding and Bayesian inference processes, which are pivotal to its performance (Fig. 1C,D). Finally, DigNet incorporates an ensemble learning strategy to counteract the instability issues stemming from random samplings (Fig. 1E). After being trained on a single-cell GRN and corresponding transcriptomic data, DigNet can generate an appropriate network for new gene expression profiles, facilitating various downstream analytic tasks, such as cellular differential gene expression analysis and biomarker discovery (Fig. 1F).
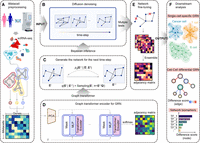
Overview of DigNet. It generates a cell-specific gene regulatory network (GRN) and extracts differential network structures from single-cell gene expression profiles. (A) Data preprocessing of human tissue scRNA-seq data. (B) Network diffusion denoising across time-steps in DigNet. (C) Transforming the adjacency matrix exported by the encoder through Bayesian inference to predict the GRN at the subsequent time-step. (D) Utilizing a transformer to encode GRN for scRNA-seq data based on the current time-step information. The procedures in C and D are repeated with each time-step to progressively denoise the network structure. (E) Correcting the network linkages (removing incorrect regulatory interactions) and integrating multiple diffusion-generated networks to produce the final cell-specific network. (F) Once the generated network structure is obtained from scRNA-seq data using DigNet, multiple downstream analytics can be performed to identify key network features driving cell heterogeneity or biomarker signatures indicative of cancerous states.
Benefiting from the diffusion generative framework, DigNet is one of the few models that directly generates network architectures at the global scale from scRNA-seq data (for details, see Supplemental Fig. S1A,B; Supplemental Note 2). It emphasizes a holistic network generation process for the entire architecture, placing significant emphasis on ensuring compatibility between global regulatory network structure and gene expression profiles, thereby altering the approach to understanding cellular regulatory mechanisms. Moreover, it changes the traditional single-step network inference paradigm into a multistep network generative process. This enables the proposed method to pay more attention to the detailed dynamics in network structure with global architecture corresponding to gene expressions. Moreover, the reversibility of the network generation process allows DigNet to learn precise network architectures, which can be flexibly applied in important reverse operations, underscoring its adaptability and robustness in various analytical contexts.
Extensive benchmark testing on simulation data confirms DigNet efficiency
To evaluate the performance of DigNet in network generation, we develop a simulation scheme for benchmarking gene expression profiles with single-cell GRNs. The rationale for employing simulated gene expression data lies in establishing these predefined GRNs as the ground truths for assessment. Taking one of the given GRNs as an example, Figure 2A illustrates how DigNet starts with a random network wiring and progressively generates a clean network. The gene expression profile serves as the input XT, with the initial adjacency matrix being randomly generated as ET. DigNet relies on the previous time-step network adjacency matrix Et (where t decreases from T to zero) and employs a Markov stochastic process in conjunction with the gene expression profile to iteratively generate a new adjacency matrix Et−1. Upon repeating this process T times, a clean network will be derived as the final output (for details, see Methods).
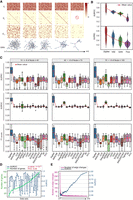
Benchmark evaluations of DigNet on simulation data sets. (A) The intermediate network illustration in progressively generating a clean network from a random network, where each column represents a snapshot in time, including the input data Xt, the predicted clean adjacency matrix E0, and the inferred output for the subsequent time-step Et−1. (B) Performance evaluation of DigNet compared with alternative generative models (diffusion vs. VAE, GAN, and Flow) in terms of AUROC and AUPRC. (C) Performance evaluation of DigNet and 13 other GRN inference algorithms using 100 synthetic data sets. The red line denotes the mean values of these comparison methods. (D) The evaluation for DigNet network generation capability on the size impact of the number of genes, using AUROC value with the calculation of t-test P-values and PCCs between them. (E) During the network generation, the comparison of the number of edges corrected by DigNet at different time points during network generation and its subsequent effect on the overall AUROC value.
Based on the diffusion model, DigNet is a GRN generation method from single-cell gene expression profiles (see Supplemental Note 3). It decomposes the task of network generation into a series of sequential steps, each refining the current network wiring architecture guided by the previous one. To justify the effectiveness of the multistep diffusion strategy, we also introduce three other popular generative models, namely, variational autoencoders (VAEs) (Way et al. 2020), generative adversarial networks (GANs) (Wang et al. 2018), and Flow (Stimper et al. 2022), into our algorithmic framework. These models are commonly used in scRNA-seq data analytics for denoising and generating low-dimensional representations. To our knowledge, they also have not been explored in generating GRNs. Mimicking the network generation pipeline of DigNet, we equip each of them accordingly with a GRN layer by replacing the diffusion strategy of DigNet to fulfill the network generation tasks. The detailed network constructions are described in Supplemental Fig. S1C–E. For a fair comparison, we train and test each generative method on the same simulation data set and evaluate them using the area under the receiver operating characteristic curve (AUROC), the area under the precision-recall curve (AUPRC), and F1-score metrics (Fig. 2B; Supplemental Fig. S1F). Compared with the VAE, GAN, and Flow models, the results demonstrate the superior performance of diffusion-based DigNet, with increasing AUROC values of 16.54%, 23.62%, and 45.81%, respectively, and AUPRC improvements of 18.92%, 31.82%, and 41.90%, respectively. These findings provide direct evidence that DigNet outperforms other generative algorithms in reverse engineering GRNs from scRNA-seq data.
Different from traditional network reconstruction algorithms based on gene pair reasoning, DigNet emerges as a network generation method that focuses on wiring global network structure, demonstrating better performance over other generative models (see Supplemental Note 4; Supplemental Fig. S2). Subsequently, we compare DigNet with 13 state-of-the-art traditional GRN inference algorithms, including ARACNE (Margolin et al. 2006), context likelihood of relatedness (CLR) (Faith et al. 2007), DeepSEM (Shu et al. 2021), GENIE3 (Huynh-Thu et al. 2010), GRISLI (Aubin-Frankowski and Vert 2020), lag-based expression association for pseudotime-series (LEAP) (Specht and Li 2017), PIDC (Chan et al. 2017), SCENIC (Aibar et al. 2017), SCODE (Matsumoto et al. 2017), SINCERITIES (Papili Gao et al. 2018), and Tigress (Haury et al. 2012), along with mutual information (MI)– and Pearson correlation coefficient (PCC)–based methods as baselines (Supplemental Table S7). The modeling basis of these methods is to infer gene regulatory pairs and then assemble them into a network, which is significantly different from the proposed network generation strategy. For completeness, the functionalities of these comparing methods with their implementation details are available in the Supplemental Material (Supplemental Note 5). We run these algorithms individually across 100 synthetic data sets of varying gene/node sizes and assess their performance using AUROC, AUPRC, and F1-score metrics with the gold-standard prior networks. Based on the number of genes, the data sets are divided into three categories for evaluation: nodes 10∼40, 41∼70, and 71∼100. As shown, the results demonstrate that DigNet consistently exhibits exceptional GRN reverse inference capabilities across all three size networks (Fig. 2C). In terms of the AUROC value, DigNet achieves significant improvements of 24.80%, 13.89%, and 12.08% over the second-place algorithm (GRISLI) across three different network sizes. Similarly, for the AUPRC metric, DigNet improves over the second-place algorithm with 31.76%, 23.14%, and 19.67% for the three different-size networks, respectively. Regarding the F1-score, DigNet achieves substantial improvements of 23.24%, 17.49%, and 17.09% over the second-place algorithm, respectively. Except for CLR, LEAP, and SCODE, which performed poorly on certain metrics, the evaluation results for these comparing algorithms are relatively consistent and stable. In general, the performance of the diffusion model–based DigNet is significantly higher than other GRN inference methods.
Furthermore, we investigate the impact of network size of gene numbers on the performance of DigNet (Fig. 2D). As shown, the PCC of 0.09 and the t-test P-value of 1 × 10−60 indicate a lack of significant correlation between network scale and network generation performance via DigNet. The robustness underscores that the generative capabilities of DigNet are not influenced by the number of genes, allowing it to unbiasedly generate networks of varying size from gene expression data. Moreover, using one gold-standard network as an illustration, we examine the network wiring information corrected by DigNet at each time-step and assess the corresponding individual AUROC values (Fig. 2E). Starting from a randomly initialized network, the early diffusion denoising process alters a significant number of regulatory links, yet without a significant improvement in AUROC values. As the diffusion progresses, DigNet gradually captures the core architecture of the underlying network from the data, in which minor edge modifications lead to significant AUROC improvements. Toward the end of the time-steps, the structure of the network generated by DigNet almost cease to change, and corresponding AUROC values stabilize accordingly.
DigNet generates reliable GRNs in specific single cells
Subsequently, we verify the GRN generation capabilities of DigNet in real single-cell contexts of breast cancer. We compile the gene expression profiles of T cells, B cells, and cancer cells from five breast cancer patients (S33, S39, S42, S53, and S60, as specified in Supplemental Table S1; Qian et al. 2020). Given the availability limitation of the gold-standard networks, benchmarking GRN generation under real single-cell settings remains a significant challenge. Consequently, we first develop specific gene reference networks for each cell type within every individual sample based on prior knowledge bases and gene expression profiles. To reduce computational demands and topological complexity, we further divide the complete gene network into numerous subnetworks, each representing distinct biological pathways and functions according to the KEGG database. To comprehensively evaluate the model performance, the proposed DigNet method is rigorously compared against the former 10 baselines, including ARACNE, CLR, DeepSEM, GENIE3, GRISLI, PIDC, SCENIC, SCODE, SINCERITIES, and Tigress. We conduct a detailed evaluation of GRN in the gene set of the breast cancer KEGG pathway (Pathway ID hsa05224).
Overall, DigNet outperforms the other GRN inference algorithms with the highest AUROC and AUPRC values (Fig. 3A; Supplemental Fig. S1G). Regarding AUROC evaluation, DigNet achieves optimal performance in eight out of 14 evaluations, ranking within the top five in 13 evaluations, and only one exception in the cancer cell context. Moreover, DigNet exhibits the best average performance across all three cell types. In benchmark tests, the performance of DeepSEM, GRISLI, GENIE3, SCODE, and SINCERITIES (which DigNet surpasses by 6.5% to 11%) is under random predictions, suggesting their inability to accurately infer gene networks from breast cancer scRNA-seq data. The ARACNE, CLR, PIDC, GRNboost2, and Tigress algorithms can infer some proper regulatory relationships within single-cell environments. However, their performance is still slightly inferior to DigNet, with a decrease ranging from 1.7% to 3.4%. We observe that several better-performing algorithms are mostly based on correlation or regression methods, which may be effective in navigating network inference in intricate cellular environments. When considering AUPRC values, DigNet achieves optimal performance in six out of 14 evaluations and ranks among the top five in 12 evaluations, sharing a comparable optimal average performance with SCENIC. Generally, DigNet consistently demonstrates remarkable and stable capabilities in directly generating appropriate network architectures for single-cell gene expression profiles.
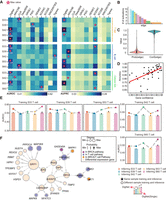
Performance evaluation and network analysis of DigNet in breast cancer case study. (A) AUROC and AUPRC results for DigNet applied to breast cancer single-cell data. The horizontal axis denotes individual cell types, and the vertical axis compares DigNet with 10 other alternative algorithms. (B) DigNet generates specific GRNs for T cells of patient S42 10 times. We have conducted a statistical summary of all the activated regulatory links, for which the horizontal axis represents the existing regulatory relationships, and the vertical axis indicates their frequency occurrence. (C) Prob and Conf represent the probability and confidence scores of all regulatory relationships across the 10-time generated networks, respectively. (D) Visualization of the relationship between accuracy and gene confidence, in which gene confidence is the aggregated confidences of generation edges associated with each gene. (E) Benchmark testing of DigNet across diverse individual cell environments. The dotted line shows the AUROC evaluation results for DigNet on different individual cells after a single training session, and the bar graph shows results from a single run of DigNet (excluding the ensemble network aggregation module). (F) Specific GRN generated for T cells of patients S33–S60, showcasing only subnetworks with relatively high node degrees for clarity.
Furthermore, we explore the stability of network generation by DigNet. Utilizing T cells from patient S42 as a case study,
we execute DigNet 10 times and count the number of activations for each regulatory relationship (Fig. 3B, displaying only edges activated more than once for clarity). To facilitate the observation of edge permutations, we define
Prob and Conf to represent the probability and confidence of Gene a regulating Gene b, respectively.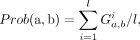 (1)
(1)
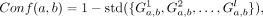 (2)
where
(2)
where 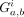 denotes the regulatory relationship between Gene a and Gene b in the ith iteration's network, and “std” represents the standard deviation. Figure 3C compiles the Prob and Conf for all regulatory relationships, indicating that most gene pairs do not have a regulatory relationship and that only a few
gene pairs have regulatory information transmission. This result is consistent with the expected distribution of biological
network regulations. We observe that the confidence of the majority of edges remains high across multiple repetitions, indicating
that the networks generated by DigNet are quite stable. Furthermore, we calculate the node confidence (the cumulative confidence
of incoming and outgoing edges) and the accuracy of the corresponding edges (Fig. 3D). Based on the results of regression analysis, we find that nodes with higher confidence within the network also exhibit
higher accuracy. Therefore, we have reason to conclude that the networks generated by multiple DigNet repetitions are not
only high confidence but also facilitate the selection of more credible gene regulatory relationships based on confidence
values.
denotes the regulatory relationship between Gene a and Gene b in the ith iteration's network, and “std” represents the standard deviation. Figure 3C compiles the Prob and Conf for all regulatory relationships, indicating that most gene pairs do not have a regulatory relationship and that only a few
gene pairs have regulatory information transmission. This result is consistent with the expected distribution of biological
network regulations. We observe that the confidence of the majority of edges remains high across multiple repetitions, indicating
that the networks generated by DigNet are quite stable. Furthermore, we calculate the node confidence (the cumulative confidence
of incoming and outgoing edges) and the accuracy of the corresponding edges (Fig. 3D). Based on the results of regression analysis, we find that nodes with higher confidence within the network also exhibit
higher accuracy. Therefore, we have reason to conclude that the networks generated by multiple DigNet repetitions are not
only high confidence but also facilitate the selection of more credible gene regulatory relationships based on confidence
values.
DigNet demonstrates notable generalization capabilities in network generation performance across novel environments for models trained under varying conditions. To address the scenario of network generation with limited training data sets, DigNet can be trained on samples from the same type of cells across different individual tissues and generate a network structure for new samples. To align the new samples with the feature distribution of the training environment, we utilize PCA to project the new sample data into the principal component space defined by the training data, significantly enhancing the model's generalizability. Taking T cells as an illustration, DigNet uses multiple trained models to generate a network structure for five distinct samples (Fig. 3E). Additionally, under a cross-sample testing scenario, the AUROC values for samples S33, S39, and S53 demonstrate improvements compared with when the same models are trained and tested on identical data sets: 0.5491 (vs. 0.5759), 0.5318 (vs. 0.5470), and 0.5630 (vs. 0.5643), respectively. In cross-sample testing experiments, networks generated by DigNet exhibit considerable and promising performance across most data sets, with network structures generated across different samples outperforming single iterations on certain data sets. Furthermore, the performance of an ensemble DigNet approach has been proven to surpass that of a single run.
GRN reveals key regulatory pathways in breast cancer T cells
To further verify the efficiency of DigNet in the real application scenario, we apply it to generate an appropriate cell-specific GRN using the gene expression profiles of breast cancer T cells. Initially, we merge the gene count matrices from breast cancer patients S33 to S60 into a comprehensive data set. Then, we construct a curated gene set of differentially expressed genes, T cell signaling pathways, and KEGG breast cancer pathways (Supplemental Note 6; Supplemental Fig. S3; Supplemental Tables S8–S11). DigNet is executed to generate a T cell–specific GRN on the integrated data (Fig. 3F; Supplemental Fig. S4).
DigNet reveals unprecedented regulatory connections among multiple key genes, providing a new perspective for our in-depth understanding of the T cell immune response in breast cancer. Moreover, our analysis using DigNet reveals that SIRT1, a member of the Sirtuin protein family renowned for bridging transcriptional regulation with intracellular energy metabolism, exerts its influence on T cell immune responses via its downstream targets HIPK1 and MAP3K8. This discovery underscores a novel, yet underappreciated, function of SIRT1 in modulating breast cancer immunity. Although SIRT1 is not explicitly listed as a key player in the breast cancer pathway according to the KEGG database (absent from hsa05224), it is recognized as a tumor suppressor owing to its protective role against DNA damage and oxidative stress, safeguarding genomic stability during tumor progression (Sung et al. 2010; Elangovan et al. 2011; Bajrami et al. 2021). Moreover, HIPK1, another gene in our analysis, has been implicated in aberrant states during inflammatory responses and tumor development (Liu et al. 2018; Zhang et al. 2021). Although there is no direct evidence of HIPK1 being regulated by SIRT1, its homolog HIPK2 has shown potential cross talk with SIRT1 in DNA damage response, leading to the phosphorylation of SIRT1 at serine 682 after lethal damage (Puca et al. 2009; Conrad et al. 2016; Choi et al. 2017). Moreover, SIRT1’s inhibitory effect on the proinflammatory cytokine TNF during immune responses is well documented, and MAP3K8, another key player in our findings, is crucial for TNF production (Gantke et al. 2011; Huang et al. 2012; Chen et al. 2020; Wang et al. 2020). MAP3K8 participates in T helper cell differentiation and intracellular gene regulation, and under certain conditions, it activates the MAPK/ERK pathway, leading to TNF production, thereby playing a key role in immune responses (Tsatsanis et al. 2008).
Moreover, an important downstream application of DigNet lies in prioritizing key regulatory genes to elucidate the mechanisms
underlying disease progression when cellular processes become dysregulated. Specifically, by applying DigNet, we meticulously
compare the regulatory networks of T cells between normal and cancerous breast tissue samples, pinpointing crucial nodes that
underlie the regulatory discrepancies driving disease progression. As a reference, we construct a knowledge-based GRN for
normal breast T cell samples obtained from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE195665. Based on the breast cancer–specific T cell GRN and the normal breast T cell GRN generated
by DigNet, we quantify the regulatory rewiring differences for each gene within the normal/cancer networks (Fig. 4A). To this end, we introduce a gene difference score (GDS) to evaluate the variation in node connectivity between two gene
networks: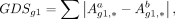 (3)
where Aa and Ab represent the adjacency matrices of the respective GRN, and g1 denotes the gene node. A GDS greater than zero suggests an active regulatory function of the gene in normal breast T cells.
Consequently, we select the top three genes with the highest GDS in both normal and cancer contexts (normal: JUN, MYC, SP1; cancer: IGFBP4, FABP5, ZC3H12A) and assemble a differential GRN for breast cancer (Fig. 4B). Among these genes, MYC and SP1 belong to the KEGG breast cancer pathway, whereas JUN is featured in both the breast cancer and T cell receptor pathways. The differential regulatory network analysis also highlights
substantial variation in the regulatory interactions mediated by these genes in normal versus cancerous T cell environments.
Furthermore, most downstream target genes of these prioritized genes exhibit abnormal activation in cancerous conditions,
evidenced by their tendency toward magenta color in Fig. 4B.
(3)
where Aa and Ab represent the adjacency matrices of the respective GRN, and g1 denotes the gene node. A GDS greater than zero suggests an active regulatory function of the gene in normal breast T cells.
Consequently, we select the top three genes with the highest GDS in both normal and cancer contexts (normal: JUN, MYC, SP1; cancer: IGFBP4, FABP5, ZC3H12A) and assemble a differential GRN for breast cancer (Fig. 4B). Among these genes, MYC and SP1 belong to the KEGG breast cancer pathway, whereas JUN is featured in both the breast cancer and T cell receptor pathways. The differential regulatory network analysis also highlights
substantial variation in the regulatory interactions mediated by these genes in normal versus cancerous T cell environments.
Furthermore, most downstream target genes of these prioritized genes exhibit abnormal activation in cancerous conditions,
evidenced by their tendency toward magenta color in Fig. 4B.
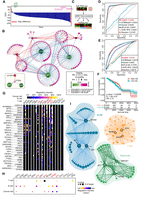
Breast cancer cell–specific GRN reveals key genes and regulatory architectures. (A) Gene differential scores are calculated based on the difference between T cell GRN in normal breast tissue and cancerous samples, highlighting the top three genes with the highest scores. (B) Extraction and analysis of T cell differential gene regulations associated with high differential genes, with a focus on the regulatory heterogeneity of the PTEN gene under normal conditions (bottom left). (C) Schematic illustration of deconvolving TCGA breast cancer bulk RNA-seq data into scRNA-seq data using the ARIC method. (D) Classification AUROC values for the top 10 biomarker genes identified by DigNet and other methods in TCGA BRCA T cells, with optimal values highlighted in red. (E) Classification AUROC values for the top 20 biomarker genes selected by DigNet and alternative methods in normal/cancerous breast T cells. (F) Kaplan–Meier (K-M) curves for the top 10 biomarker genes chosen by DigNet in TCGA breast cancer T cells, analyzed through a multifactorial Cox survival analysis, where CI represents the confidence interval, and HR stands for the hazard ratio. (G) Regulatory landscape of transcription factor (TF)–target gene interactions in T cells, B cells, and cancer cells of patient S53, with regulatory strength inferred from DigNet probability values. (H) Cell type–specific summary of each TF's target genes in subfigure G, where dot size reflects target gene frequency, and color indicates the average probability regulatory strength). (I) Top three genes (or TFs) ranked by “Var-score” for each cell type, along with their associated regulatory relationships, with TF genes highlighted in red in G and H.
In addition to these three hotspot genes, we focus on IGFBP4, FABP5, and ZC3H12A, which exhibit associations with cell proliferation, inflammatory responses, or immune regulation. For instance, IGFBP4, swiftly induced by estrogen and exhibiting abnormal expression across diverse tumor tissues, emerges as a crucial prognostic indicator for breast cancer patients (Ryan et al. 2009; Flynn and Houston 2022; Chen et al. 2023). Moreover, IGFBP4 plays a pivotal signaling role in the differentiation of select T cell subtypes, maintaining a delicate balance between T helper 17 and regulatory T cells (Miyagawa et al. 2017; DiToro et al. 2020). Similarly, FABP5 and ZC3H12A genes are intriguing in breast cancer progression and T cell immune responses, potentially representing novel therapeutic targets and prognostic markers (Matsushita et al. 2009; Liu et al. 2011; Levi et al. 2013; Lu et al. 2016; Senga et al. 2018; Li et al. 2022). Furthermore, the derived differential GRN implies regulatory functions in both normal and cancerous tissues. For instance, PTEN is known to be regulated by MYC and SP1 in normal conditions, yet DigNet suggests an additional regulatory link with IGFBP4 in cancerous tissue. This novel regulatory interaction is not documented in existing knowledge bases, and studies suggest that they could affect tumor proliferation through signaling pathways (such as the AKT pathway), with other members of the IGFBP family also having some interactions with PTEN (Baxter 2014; Lee et al. 2018; Ruan et al. 2023). In normal tissues, estrogen or progesterone receptors regulate IGFBP4 levels via SP1, but in breast cancer T cells, DigNet found that this regulatory relationship is not active. Although no studies support or refute the latter, we propose this hypothesis from the differential network.
To validate the differential GRN generated by DigNet from multiple perspectives, we attempt to distinguish T cell data from breast cancer samples in TCGA using the top 10 key genes identified by GDS (Supplemental Table S2). The T cell data from TCGA breast cancer cases is obtained through ARIC deconvolution (Fig. 4C; Supplemental Note 7; Supplemental Table S12; Zhang et al. 2022, 2023). We benchmark several prominent feature selection algorithms, DUBSTepR (Ranjan et al. 2021), Seurat (Hao et al. 2024), sPLS-DA (Lê Cao et al. 2011), and SVM-RFE (Duan et al. 2005), by comparing their performance against a randomly selected gene set. These algorithms are employed to select key biomarker signature genes within our breast cancer/normal single-cell data set, which are then evaluated on the TCGA deconvoluted T cell data for their effectiveness. We conduct T cell classification experiments under an SVM classifier based on fivefold cross-validation to ensure robustness (Fig. 4D). To further strengthen the validation of these biomarker genes, we replicate the experiments on a single-cell data set of normal/cancerous breast tissue sourced from the GEO database (accession number GSE114725) (Fig. 4E), in which all algorithms use the top 20 key genes by GDS (Supplemental Table S2). Both sets of results underscore the remarkable discriminative capacity of the key network nodes identified by DigNet. Furthermore, survival analysis of TCGA patients affirms that the biomarkers pinpointed by DigNet exhibit statistically significant differences (Fig. 4F). Furthermore, we perform survival analysis specifically on the top 10 genes with high GDS in the TCGA breast cancer cohort (Fig. 4F). Multivariable Cox regression analysis is utilized to estimate coefficients, alongside confidence intervals (CIs) and hazard ratios (HRs) for these genes (Supplemental Note 8). Based on the derived coefficients, we assigned weighted scores to the top 10 genes and classified patients into high/low-risk groups accordingly. Subsequently, we estimated the survival probability for these groups using the Kaplan–Meier (K-M) method, which revealed a statistically significance difference (P-value = 0.00056). These findings indicate that the biomarkers identified by DigNet exhibit significant differences in survival time estimation for breast cancer.
Overall, the unique network structure generation approach of DigNet enables us to construct cell-specific regulatory networks, discern normal–disease differential networks, and uncover important network-based biomarkers. The essential modules, regulatory interactions, and biomarkers embedded within these networks reflect the internal carcinogenic mechanisms within cells, holding immense potential to revolutionize cancer diagnosis, prognosis, and therapy.
Differential analysis of single-cell gene networks reveals the heterogeneity among breast cancer cells
Cell-specific GRNs serve as potent instruments to decipher variation in cellular functions, as they orchestrate the expression of gene products and shape the unique developmental pathways of individual cells (Karlebach and Shamir 2008). In contrast, public, nonspecific reference networks, rooted in general knowledge bases, do not adequately capture the intricacies of these phenomena. However, DigNet offers a solution by generating cell-specific GRNs that facilitate analyses of the nodal interactions and network architectures responsible for functional disparities among diverse cell types. Specifically, we harness DigNet to construct cell-specific networks for T cells, B cells, and cancer cells from breast cancer patient sample S53 (Supplemental Tables S3–S5). Furthermore, we quantify the regulatory influence from TFs to target genes across these three cell types to reflect changes in TF activity within each cellular context (Fig. 4G). Moreover, significant disparities in TF activity are observed among these cells (Fig. 4H). For instance, TFs such as E2F3, ESR1, and ESR2, which influence tissue growth and development, exhibit heightened activity in B cells, regulating multiple target genes (Zhu et al. 2001; Xiao et al. 2008; Langendonk et al. 2022). In contrast, the TF MYC is markedly silent in B cells but active in T cells, with extensive prior research underscoring the consequences of MYC overexpression in T cells (Zimmerli et al. 2022). These findings demonstrate the superiority of specific GRNs over public networks and elucidate the paramount importance of prioritizing TFs associated with immune cells in breast cancer progression.
Furthermore, we aimed to delineate cell type–specific genes and network architectures to address the heterogeneity in cellular
functions. To accomplish this, we devised a multicellular gene differential score metric to evaluate the gene quality specific
to each cell type. Specifically, we transformed the adjacency matrices generated by DigNet into binary (zero–one) matrices,
denoted as A. Subsequently, we formulated the following equation to capture the differential score of gene g1 in cell type a: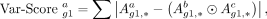 (4)
where
(4)
where 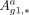 signifies the comprehensive set of target regulatory interactions involving gene g1 in cell type a. ⊙ is the Hadamard product. Furthermore, we quantified the Var-Score for genes across the specific networks of all cells
(see Supplemental Table S6), and extracted the network structures corresponding to the top three gene with the highest “Var-Score” (Fig. 4I). The key gene sets for the three groups of cells are as follows: MYC, BRAF, and KRAS for T cells; SP1, NFKB2, and JUN for B cells, and SP1, MAP2K1, and NFKB2 for cancer cells. Although B cells and cancer cells share two key genes, their target genes are entirely different. For example,
the gene MTOR, likely regulated by SP1, controls the growth, development, and proliferation of B cells (Astrinidis et al. 2010; Iwata et al. 2017). In cancer cells, SP1 robustly regulates genes such as SOS1, NFKB2, HRAS, and AKT2, and elucidating these aberrant regulatory mechanisms can provide insights into cancer cell invasion and metastasis. It is
noteworthy that the key genes in T cells exhibit marked differences from those of other cell types. Besides the previously
discussed MYC, the specific responses generated by mutant KRAS and BRAF in T cells are also prime targets for antitumor immune therapy (Wilmott et al. 2012; Tran et al. 2016). The partial regulatory actions of these genes demonstrate heterogeneity across diverse cell types, facilitating the understanding
of cell functions within the tumor microenvironment and identifying crucial therapeutic targets. In essence, the single-cell
GRN generated using DigNet from data enables the discovery of significant cell-specific regulatory relationships and network
nodes via downstream analyses. This heightened resolution and specificity in gene regulatory dynamics, alongside with the
intricate molecular details, surpass the limitations of average gene expression profiles and publicly available gene networks.
signifies the comprehensive set of target regulatory interactions involving gene g1 in cell type a. ⊙ is the Hadamard product. Furthermore, we quantified the Var-Score for genes across the specific networks of all cells
(see Supplemental Table S6), and extracted the network structures corresponding to the top three gene with the highest “Var-Score” (Fig. 4I). The key gene sets for the three groups of cells are as follows: MYC, BRAF, and KRAS for T cells; SP1, NFKB2, and JUN for B cells, and SP1, MAP2K1, and NFKB2 for cancer cells. Although B cells and cancer cells share two key genes, their target genes are entirely different. For example,
the gene MTOR, likely regulated by SP1, controls the growth, development, and proliferation of B cells (Astrinidis et al. 2010; Iwata et al. 2017). In cancer cells, SP1 robustly regulates genes such as SOS1, NFKB2, HRAS, and AKT2, and elucidating these aberrant regulatory mechanisms can provide insights into cancer cell invasion and metastasis. It is
noteworthy that the key genes in T cells exhibit marked differences from those of other cell types. Besides the previously
discussed MYC, the specific responses generated by mutant KRAS and BRAF in T cells are also prime targets for antitumor immune therapy (Wilmott et al. 2012; Tran et al. 2016). The partial regulatory actions of these genes demonstrate heterogeneity across diverse cell types, facilitating the understanding
of cell functions within the tumor microenvironment and identifying crucial therapeutic targets. In essence, the single-cell
GRN generated using DigNet from data enables the discovery of significant cell-specific regulatory relationships and network
nodes via downstream analyses. This heightened resolution and specificity in gene regulatory dynamics, alongside with the
intricate molecular details, surpass the limitations of average gene expression profiles and publicly available gene networks.
Discussion
In this paper, we introduce a network generation method called DigNet for deriving cell-specific GRN from scRNA-seq data. DigNet uses Bayesian inference and graph transformer techniques by iteratively refining an initial random network to construct a comprehensive and detailed GRN for individual cells. The non-Euclidean discrete diffusion modeling enables DigNet to generate a global network architecture rich in structural features. Meanwhile, the progressive generation process and reversibility enable DigNet to capture structural details within the entire network, ensuring that the overall structure of the generated network remains consistent with the input gene expression profiles. The uniqueness of DigNet can be summarized by three important aspects: the generation of GRN from gene expression data with discrete diffusion models, multi-time-step diffusion techniques for noise reduction and network refinement, and the integration of generative deep learning with a hybrid model architecture. Through rigorous benchmark tests across diverse biological contexts and data sets, we demonstrate the efficiency, robustness, and superiority of DigNet, particularly in terms of reproducing cell type gene regulatory specificity. Moreover, DigNet achieves single-cell-specific GRN inference from scRNA-seq data, identifying crucial regulatory network nodes and causal modules leading to cell type specificities. DigNet introduces a novel generation network model for GRN reverse engineering, enabling it to respond to single-cell gene expression profiles with a more suitable network architecture through a progressive denoising procedure rather than assembling isolated regulatory signals.
Recovering GRN architectures through generative models offers a novel reverse engineering paradigm and alternative for gene expression data, presenting multiple challenges. A critical challenge for DigNet is that simple random sampling can result in slight variations in outcomes at the same time-step, which may inadvertently introduce novelty-driven rewiring and unwarranted randomness into the network. Unlike conventional diffusion models, DigNet incorporates no specific conditional controller to determine which networks are more suitable, primarily owing to the absence of clear criteria or justifications for filtering specific network architectures across diverse cellular environments. To address this, our solution strategy revolves around statistically estimating the probability of regulatory events by counting the activation frequencies of regulatory signals across multiple networks, offering a straightforward yet effective learning approach. Compared to other graph neural network (GNN)-based methods, DigNet eliminates the requirement for preconstructed initial graphs by utilizing a diffusion model–based generation strategy (Supplemental Note 9). This approach enhances both the adaptability and accuracy of GRN inference. A potential future direction for DigNet involves incorporating cell developmental trajectories to model dynamic GRN throughout continuous cellular developmental stages. Furthermore, the integration of multiomic data, which encompasses genomic sequence information, chromatin accessibility data, TF activity, and protein–protein interaction networks (Badia-i-Mompel et al. 2023), emerges as a crucial future direction for advancing the capabilities of DigNet. By utilizing these diverse multiomic data, we foresee a significant enhancement in the accuracy and precision of reconstructing dynamic GRNs from complex data sets. Moreover, the involvement of TF information will be substantially increased in this comprehensive integration. For more detailed expansions and limitations, please refer to Supplemental Note 10.
Methods
Framework
The emergence of generative techniques, such as the GAN, VAE, Flow, and diffusion models, has revolutionized data generation and diversification (Kingma and Welling 2013; Goodfellow et al. 2014; Ho et al. 2020; Austin et al. 2021; Ramesh et al. 2022). Currently, the diffusion model, a typical generative model, has demonstrated its powerful generative capabilities across text, image, and video domains (Ho et al. 2022; Saharia et al. 2022). In this work, DigNet leverages the diffusion model framework for discrete space modeling to capture complex gene regulatory interactions within non-Euclidean biological systems. It transforms the input gene expression profiling data into vector representations in a high-dimensional space, which is then utilized to refine gene regulatory relationships in wiring-contaminated networks. Given a GRN G = (V, E), where V denotes genes and E signifies their regulatory relationships between regulators and targets, we consider the GRN to contain binary attributes, one or zero, corresponding to the activation or inhabitation of gene switches, respectively. DigNet primarily comprises two components: the forward diffusion (noise addition) of gene network E and the backward denoising stage (employing a neural network). The relevant parameter settings are discussed in Supplemental Note 11, Supplemental Table S13, and Supplemental Figure S5, A–C. To ensure the efficiency and effectiveness of the diffusion model, DigNet embodies the following three properties for both forward diffusion and backward denoising processes:
-
q(Et|E0) possess a closed-form solution to ensure its stability across varying time-step t.
-
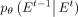 is an expression with a closed-form solution, which empowers the neural network with parameters θ in learning and targeting
the original network E0.
is an expression with a closed-form solution, which empowers the neural network with parameters θ in learning and targeting
the original network E0.
-
As the diffusion time T approaches infinity, the network structure should converge to a marginal distribution related solely to noise values, independent of E0, denoted as q(ET) ∝ q(ET|E0).
Forward diffusion
The noise diffusion process for GRN is based on a Markov chain framework, in which the generation of subsequent networks with
noise values progresses step by step along a predetermined direction and noise level. For the network Et at time t, its derivation solely depends on Et−1, and it can further induce Et+1 based on the predetermined noise values. Because of the Markov property, given the preset noise parameters and the initial
network E0, we can derive the joint prior distribution of networks at any given time as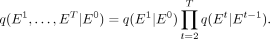 (5)
When T is sufficiently large, the model aligns with a Markov jump process under a discrete space distribution. The forward noise
levels are predetermined as a state transition matrix Q, which contains two states, zero and one, to map the GRN. At t = 0, Q is initialized such that the probability of self-transition is one. As t progresses from one to T, the state transition probabilities evolve, gradually transforming the original GRN toward a random network. Let α denote
the network noise coefficient, ranging from zero to one. Then,
(5)
When T is sufficiently large, the model aligns with a Markov jump process under a discrete space distribution. The forward noise
levels are predetermined as a state transition matrix Q, which contains two states, zero and one, to map the GRN. At t = 0, Q is initialized such that the probability of self-transition is one. As t progresses from one to T, the state transition probabilities evolve, gradually transforming the original GRN toward a random network. Let α denote
the network noise coefficient, ranging from zero to one. Then, 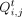 represents the probability of transitioning from state i to state j at time t, which can be mathematically described as follows:
represents the probability of transitioning from state i to state j at time t, which can be mathematically described as follows: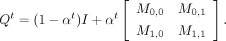 (6)
As the noise coefficient α approaches one, the network converges to a random distribution based on a fixed value Mt.
(6)
As the noise coefficient α approaches one, the network converges to a random distribution based on a fixed value Mt.
Furthermore, based on the noise values, we can infer the network for the subsequent time-step as follows: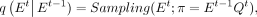 (7)
where Sampling(E, π) refers to a state distribution encoded in a one-hot scheme, derived from simple random sampling with a probability value
of π. Furthermore, because the noise values are predetermined in a closed form, we can combine and simplify the probability
distributions up to t − 1 similar to the approach in DDPM (Ho et al. 2020), to derive the marginal distribution network at time t conditioned on the initial network E0:
(7)
where Sampling(E, π) refers to a state distribution encoded in a one-hot scheme, derived from simple random sampling with a probability value
of π. Furthermore, because the noise values are predetermined in a closed form, we can combine and simplify the probability
distributions up to t − 1 similar to the approach in DDPM (Ho et al. 2020), to derive the marginal distribution network at time t conditioned on the initial network E0: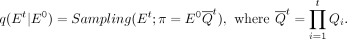 (8)
Given that the noise value Q is fixed, Et can be directly derived from E0, enabling the diffusion model to be trained from any arbitrary time-step. Based on Equations 6 and 8, we can generate the noised network at any specified time-step.
(8)
Given that the noise value Q is fixed, Et can be directly derived from E0, enabling the diffusion model to be trained from any arbitrary time-step. Based on Equations 6 and 8, we can generate the noised network at any specified time-step.
Backward denoising
During the reverse process, DigNet accomplishes the task of regenerating network linkages from Et to Et−1 through trained neural networks. By training the deep neural network under specific parameters, the clean network can be
iteratively denoised and obtained from the noisy network at time t (Supplemental Note 12). Intuitively, Et−1 can be inferred by decoding Et through the neural network. However, this iterative process may lead to error accumulation, making model training extremely
challenging. Based on the Bayesian theorem, we derive a posterior probability inference related to Et, E0, and Q: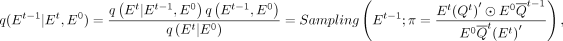 (9)
where Et is known, and E0 and Q are constants. Thus, Equation 9 admits a closed-form solution. Analogous to the likelihood estimation in continuous spaces, the integral of the evidence
lower bound (ELBO) in discrete space is formulated as
(9)
where Et is known, and E0 and Q are constants. Thus, Equation 9 admits a closed-form solution. Analogous to the likelihood estimation in continuous spaces, the integral of the evidence
lower bound (ELBO) in discrete space is formulated as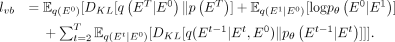 (10)
Each component can be estimated as follows: DKL[q(Et|E0)||p(E0)] (prior loss) does not require optimization because it contains no trainable parameters, and Et is predefined as a stochastic distribution network when T is sufficiently large;
(10)
Each component can be estimated as follows: DKL[q(Et|E0)||p(E0)] (prior loss) does not require optimization because it contains no trainable parameters, and Et is predefined as a stochastic distribution network when T is sufficiently large; 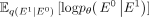 (reconstruction loss) is derived from the clean gene network based on the final noise-free network; and
(reconstruction loss) is derived from the clean gene network based on the final noise-free network; and 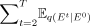
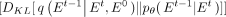 (diffusion loss) enforces consistency matching between predictions and noise-adding processes at each intermediate step,
ensuring that the prediction network align with the noise-adding network. Our optimization objective is to train
(diffusion loss) enforces consistency matching between predictions and noise-adding processes at each intermediate step,
ensuring that the prediction network align with the noise-adding network. Our optimization objective is to train 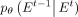 to closely match q(Et−1|Et, E0).
to closely match q(Et−1|Et, E0).
Although 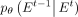 can be predicted directly, according to the experience of Ho et al. (2020), the prediction of
can be predicted directly, according to the experience of Ho et al. (2020), the prediction of 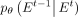 with inherent noise may lead to model instability owing to the noise level of uncertainty (Austin et al. 2021). Recognizing that the neural network model can learn the intrinsic data distribution, a feasible solution is to predict
with inherent noise may lead to model instability owing to the noise level of uncertainty (Austin et al. 2021). Recognizing that the neural network model can learn the intrinsic data distribution, a feasible solution is to predict
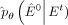 and subsequently derive
and subsequently derive 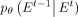 based on the Bayesian theorem:
based on the Bayesian theorem: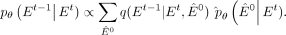 (11)
When the estimated
(11)
When the estimated 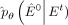 perfectly aligns with the data distribution of E0, the Kullback–Leibler (KL) divergence
perfectly aligns with the data distribution of E0, the Kullback–Leibler (KL) divergence 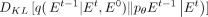 approaches zero. The parameterization of E0 not only enhances the stability and performance of model training but also simplifies the learning task of deep neural network
model.
approaches zero. The parameterization of E0 not only enhances the stability and performance of model training but also simplifies the learning task of deep neural network
model.
Therefore, optimizing the ELBO problem essentially involves training a neural network to predict clean GRN from an arbitrarily
contaminated GRN. To train the neural network 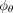 , we optimize the cross-entropy loss lossCE between the predicted probability edges
, we optimize the cross-entropy loss lossCE between the predicted probability edges 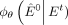 and the true network E0:
and the true network E0: (12)
Distinct from other generative models, lossCE is exclusively focuses on generating cleaner network architectures and does not encompass other tasks. The training process
framework for our proposed diffusion model is illustrated in Supplemental Figure S6.
(12)
Distinct from other generative models, lossCE is exclusively focuses on generating cleaner network architectures and does not encompass other tasks. The training process
framework for our proposed diffusion model is illustrated in Supplemental Figure S6.
Denoising transformer network
Our task is to predict the distribution of clean networks conditioned on noisy networks and gene expression profiles, which involves detailed scoring of potential interactions between TF and target gene pairs. To achieve this, we employ the graph transformer network with a self-attention mechanism (AGTN) (Dwivedi and Bresson 2020), capable of leveraging existing regulatory edge features to amplify the scores of the implicit attention mechanism and infer richer feature information.
To better learn the complex distributions of scRNA-seq data and accurately capture the intricate network structures within
GRN, we improved the original AGTN method. Specifically, we substituted the Laplacian features and positional embeddings in
AGTN with dimensionality-reduced features obtained through PCA. Furthermore, we integrated two feature learning modules, FiLM
and PNA, to refine the modulation of nodes and edges features, respectively (Perez et al. 2018). The definitions of these two feature learning layers are detailed below: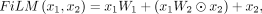 (13)
(13)
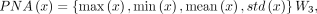 (14)
where W1, W2, and W3 are learnable weight matrices. ⊙ denotes element-by-element multiplication. {max(x), min(x), mean(x), std(x)} represents the operations of taking the maximum, minimum, mean, and standard deviation of matrix x by rows, respectively, and horizontally concatenating the results. Recognizing that the core of the diffusion model lies
in the denoising of networks across different time-steps, we incorporated a time-step module into AGTN. This module is conditioned
equally by node information, edge information, and time information, enabling it to effectively capture temporal dynamics
during the denoising process.
(14)
where W1, W2, and W3 are learnable weight matrices. ⊙ denotes element-by-element multiplication. {max(x), min(x), mean(x), std(x)} represents the operations of taking the maximum, minimum, mean, and standard deviation of matrix x by rows, respectively, and horizontally concatenating the results. Recognizing that the core of the diffusion model lies
in the denoising of networks across different time-steps, we incorporated a time-step module into AGTN. This module is conditioned
equally by node information, edge information, and time information, enabling it to effectively capture temporal dynamics
during the denoising process.
In addition, we defined and weighted the self-attention module for edge learning. Specifically, DigNet obtains the regulatory
scores between TF and target gene pairs through self-attention learning of low-dimensional embeddings of the gene expression
matrix. This is formulated as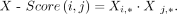 (15)
Subsequently, the current network Et is modified by the regulatory scores X-Score, and the network information evolves along the time dimension, namely,
(15)
Subsequently, the current network Et is modified by the regulatory scores X-Score, and the network information evolves along the time dimension, namely,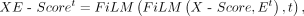 (16)
where the XE-Scoret represents the final outcome obtained through the weighted self-attention mechanism. It seamlessly integrates gene expression
profiling, input network topology, and temporal information, fully utilizing multidimensional information for the learning
of network edge weights. For details on the algorithm runtime environment and initialization, refer to Supplemental Notes 13 and 14 and Supplemental Figure S5, D and E.
(16)
where the XE-Scoret represents the final outcome obtained through the weighted self-attention mechanism. It seamlessly integrates gene expression
profiling, input network topology, and temporal information, fully utilizing multidimensional information for the learning
of network edge weights. For details on the algorithm runtime environment and initialization, refer to Supplemental Notes 13 and 14 and Supplemental Figure S5, D and E.
Marginal probability and noise presets
The selection of the Markov transition matrix that defines the network regulatory probabilities within the network is inherently
subjective, and there is often uncertainty regarding which noise model would optimally capture the dynamics of the diffusion
process. Under most conditions, the state transition probabilities are assigned indiscriminately, adhering to a uniform probability
distribution. However, given that GRN are inherently sparse, it becomes evident that a uniform distribution model inadequately
represents the natural state of GRN. To address this limitation and enhance the realism of transition, we propose reducing
the probability of regulatory relationship activation in random networks. In our experiments, the probabilities for M0,1 and M0,0 are constrained using the number of nodes Nnode and edges Nedge, namely, max(Nedge/(Nnode(Nnode − 1)), 0.1). We empirically set a lower limit of 0.1 on the probability values to maintain the stability of model training.
Furthermore, the noise coefficient in the transition matrix is set to be the commonly used cosine schedule, and the values
are determined according to the following formula with 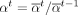 (Fig. 5A):
(Fig. 5A):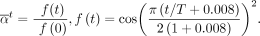 (17)
(17)
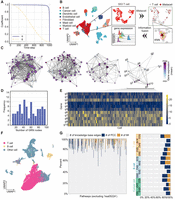
Details of the DigNet method and benchmark data sets. (A) The decay curve of the noise coefficient alpha as the time-step increases within DigNet. (B) Cell UMAP plots of breast cancer patients S33–S60 (left) and the detailed process of constructing iMetacells. (C) Partially synthesized gene network structures (with varying gene numbers of 80, 51, 28, and 11, respectively), with colors indicating their mean gene expression values. (E) Statistics on the number of genes in the synthetic networks. (D) Generating gene expression profiles for networks of different sizes using SERGIO. (F) UMAP of single-cell data for normal and cancerous breast tissues, with merged and labeled distributions of T cells and B cells. (G) Network composition information for a subset of benchmark data used in the DigNet training set (left; the benchmark network for B cells of patient S53). The KEGG breast cancer pathway hsa05224 is used for performance testing (right; the benchmark network for all cells).
Feature enhancement by iMetacell
Currently, the scRNA-seq techniques, despite their capability of revealing high-resolution biological landscapes that traditional bulk sequencing cannot achieve, inherently introduce noise and other stochastic effects owing to cellular heterogeneity. This phenomenon frequently manifests as dropouts in gene expression measurement. Even among cells of the same type, the capture of low-abundance mRNA can be compromised by technical limitations, resulting in incomplete data. To reduce technical noise and optimize cell representation, we devised a Bagging-based cell ensemble algorithm, called iMetacell (Fig. 5B, Algorithm 1). Ideally, cells of the same type would conform to a uniform distribution, but this premise is often challenged by the different stages of differentiation and functional states exhibited by most tissue cells (Baran et al. 2019; Morabito et al. 2023). Consequently, we modeled the cell population as a dynamic ensemble of distinct cell subsets, each representing a unique state (Supplemental Note 15, Algorithm 1; Supplemental Fig. S7A,B). In addition, unlike the MetaCell method proposed by Baran et al. (2019), iMetacell employs a neighborhood-based cell aggregation strategy, allowing cells to be shared across multiple cell sets. Methodologically, iMetacell is more aligned with the “neighborhood” framework proposed by Bilous et al. (2024), where cells can exist across multiple local neighborhoods, thereby capturing the continuity and complexity of cell states more accurately.
Data sets and preprocessing
Simulated single-cell gene expression profiles are used to evaluate model performance
In the simulation-data experiments, SERGIO (Dibaeinia and Sinha 2020) is used to generate scRNA-seq gene expression profiles for synthetic GRN. Specifically, we constructed 100 random GRN, each containing 10 to 100 genes, for performance testing. Additionally, we synthesized 200 further random GRN for model training. Utilizing the stochastic differential equation algorithm embedded in SERGIO, we simulated the gene expressions of 100 cells for each GRN, ensuring that the GRN in these simulation data sets are cell specific, as they correspond to a homogeneous cell type. Representative networks and their corresponding gene expression profiles are displayed in Figure 5, C and D. A comprehensive overview of our simulation framework is outlined in Supplemental Files (Supplemental Note 16; Supplemental Figs. S7C, S8). Furthermore, we conducted an analysis of the distribution of gene nodes within these networks (Fig. 5E). The simulated data and evaluation results from the DREAM challenge are also discussed in the Supplemental Files (Supplemental Note 17; Supplemental Fig. S9).
Breast cancer single-cell gene expression profiling data
To demonstrate the capability of DigNet, we use breast cancer as a representative example and expand our case study. We employ real BRCA scRNA-seq data sourced from Qian et al. (2020). For detailed processing steps, please refer to Supplemental Note 18.
Immune cell sequencing data of human breast cancer tumor and normal tissues
We downloaded the scRNA-seq data curated by Azizi et al. (2018), comprising both tumor and control samples, to validate the efficacy of DigNet. Unlike the data provided by Qian et al. (2020), this collection includes original sequencing information about both normal human breast tissue and cancerous conditions. The gene expression profiles are normalized using the “LogNormalize” method with a default scale factor based on Seurat v4 (Hao et al. 2021). Additionally, we performed SAVER data imputation to enhance data quality. This comprehensive data set encompasses sequencing results for diverse immune cell subtypes. To facilitate a broader analysis, we merged cell subtypes according to the annotations provided by the processed data (Fig. 5F). This approach enables a more streamlined comparison and interpretation of the immune landscape across normal and cancerous tissues.
Constructing single-cell-specific reference GRNs
For constructing gold-standard GRNs, we combined universally recognized reference networks with data-driven specific regulatory relationships recorded in knowledge bases. Specifically, we utilized an updated version of RegNetwork (Liu et al. 2015) to build the original reference network and extracted edges with high PCC and high MI values from the gene expression profiles, thereby forming the cell-specific gene reference networks (Supplemental Note 19). Moreover, DigNet was tested on the hsa05224 breast cancer pathway, while utilizing the remaining pathways for training (Fig. 5G).
Data sets
The public data sets used in this paper are freely available. The scRNA-seq data of breast cancer can be downloaded from EMBL-EBI ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) under accession number E-MTAB-8107. The scRNA-seq data of normal breast tissue can be downloaded from NCBI's GEO database under accession number GSE195665. Sequencing data of normal and diseased breast immune cells can be downloaded from GEO under the accession number GSE114725. Furthermore, the TCGA breast cancer RNA-seq data (level 3) was downloaded from the UCSC Xena public database (https://xena.ucsc.edu), along with the corresponding clinical information.
Software availability
The source code and pretrained models used in this study have been distributed across multiple platforms for easy access. The comprehensive source code is included in the Supplemental Code file, which has been uploaded as Supplemental Material and can also be found at GitHub (https://github.com/zpliulab/DigNet) and Zenodo (https://doi.org/10.5281/zenodo.10907470). Additionally, the data sets and pretrained models are available as Supplemental Data, as well as at GitHub and Zenodo.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was partially supported by the key program of National Natural Science Foundation of China (nos. 92374107, 62373216), the National Key Research and Development Program of China (no. 2020YFA0712402), and the Fundamental Research Funds for the Central Universities (no. 2022JC008).
Author contributions: Z.-P.L. conceived the project. Z.-P.L. and C.W. designed the framework. C.W. collected the data and conducted the experiments. C.W. and Z.-P.L. wrote the manuscript. Both authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279551.124.
-
Freely available online through the Genome Research Open Access option.
- Received May 6, 2024.
- Accepted December 10, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








