
Ultraviolet damage and repair maps in Drosophila reveal the impact of domain-specific changes in nucleosome repeat length on repair efficiency
- Benjamin Morledge-Hampton1,3,
- Kathiresan Selvam1,3,4,
- Manish Chauhan1,
- Alan G. Goodman1,2 and
- John J. Wyrick1
- 1School of Molecular Biosciences, Washington State University, Pullman, Washington 99164, USA;
- 2Paul G. Allen School for Global Health, Washington State University, Pullman, Washington 99164, USA
-
↵3 These authors contributed equally to this work.
Abstract
Cyclobutane pyrimidine dimers (CPDs) are formed in DNA following exposure to ultraviolet (UV) light and are mutagenic unless repaired by nucleotide excision repair (NER). It is known that CPD repair rates vary in different genome regions owing to transcription-coupled NER and differences in chromatin accessibility; however, the impact of regional chromatin organization on CPD formation remains unclear. Furthermore, nucleosomes are known to modulate UV damage and repair activity, but how these damage and repair patterns are affected by the overarching chromatin domains in which these nucleosomes are located is not understood. Here, we generated a new CPD damage map in Drosophila S2 cells using CPD-seq and analyzed it alongside existing excision repair-sequencing (XR-seq) data to compare CPD damage formation and repair rates across five previously established chromatin types in Drosophila. This analysis revealed that repair activity varies substantially across different chromatin types, whereas CPD formation is relatively unaffected. Moreover, we observe distinct patterns of repair activity in nucleosomes located in different chromatin types, which we show is owing to domain-specific differences in nucleosome repeat length (NRL). These findings indicate that NRL is altered in different chromatin types in Drosophila and that changes in NRL modulate the repair of UV lesions.
Cells are equipped with multiple DNA repair mechanisms to repair various types of DNA damage that would otherwise compromise the integrity of the genome. Nucleotide excision repair (NER) is a conserved DNA repair pathway that is important for removing bulky, helix-distorting DNA lesions, such as ultraviolet (UV)-induced cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs) (Schärer 2013). Global genomic NER (GG-NER) is an NER subpathway that removes lesions throughout the genome (Sugasawa et al. 2001; Gillet and Schärer 2006), whereas transcription coupled NER (TC-NER) is the other NER subpathway that is dedicated to the rapid removal of lesions in the transcribed strand (TS) of active genes (Hanawalt and Spivak 2008). The two NER subpathways differ at the damage-recognition step but utilize the same mechanisms for incision of the DNA on both sides of the lesion, excision of an oligonucleotide containing the lesion, repair synthesis, and ligation. The relative efficiency of these two subpathways, along with variations in chromatin organization, drive differential patterns of regional NER activity across the genome (Adar et al. 2016; Hu et al. 2017). However, to what extent variations in chromatin organization cause regional differences in UV damage formation is controversial (García-Nieto et al. 2017; Roberts et al. 2019).
In eukaryotic cells, 147 bp of genomic DNA wrap around a histone octamer (consisting of an H3–H4 tetramer and two H2A–H2B dimers) to form a nucleosome, and groups of nucleosomes are further folded into higher-order chromatin structures (Luger et al. 1997; McGinty and Tan 2015). Although chromatin packaging can protect the DNA from damage (Takata et al. 2013), it also inhibits the accessibility of DNA repair machinery (Rodriguez et al. 2015; Brown et al. 2018; Pich et al. 2018). Some repair factors, such as the UV–DDB complex, can recognize lesions within nucleosomes via a register-shifting mechanism to promote repair in nucleosomal DNA in vitro (Scrima et al. 2008; Matsumoto et al. 2019), whereas lesion recognition by other repair factors is significantly inhibited by nucleosomes (Rodriguez et al. 2015). The accessibility of repair proteins to DNA damage in chromatin is also influenced by histone post-translational modifications (PTMs), which are associated with regional chromatin states (Selvam et al. 2024). For example, previous studies in UV-irradiated human cells indicate that repair rates for CPDs and 6-4PPs are highest in regions with open chromatin states, such as in active promoters or strong enhancers, and are comparatively lower in repressed and heterochromatic states (Adar et al. 2016). Additionally, the wrapping of DNA around the histone octamer in individual nucleosomes significantly modulates UV damage formation and repair, generating nucleosome-associated patterns that correlate with somatic mutation rates in skin cancers (Brown et al. 2018; Pich et al. 2019). For example, the formation of UV-induced CPDs is elevated at rotational settings in which the minor groove of the DNA faces out from the histone octamer (minor-out) (Gale et al. 1987; Mao et al. 2016; Brown et al. 2018; Pich et al. 2018; Stark et al. 2022), resulting in a ∼10 bp rotational periodicity of UV damage in nucleosomal DNA. Previous genome-wide studies in both yeast and human cells indicate that the repair of UV damage is inhibited in nucleosomes relative to adjacent linker DNA, generating a translational periodicity in repair activity that reflects the nucleosome repeat length (NRL) (Mao et al. 2016, 2020; Brown et al. 2018). To what extent the regional chromatin state in which nucleosomes are located affects these patterns of damage formation and repair in nucleosomes is unclear.
The length of linker DNA between nucleosomes determines the NRL, which varies depending upon species as well as between cell types within the same species. This variation can be influenced by a number of factors, including the differential abundance of H1 subtypes in linker DNA and the activity of chromatin remodeling complexes (Fan et al. 2005; Beshnova et al. 2014). Even within the same cell type, transcription-dependent differences in NRL have been reported. Specifically, domains associated with active promoters were found to have the shortest spacing (i.e., shortest NRL), whereas heterochromatin domains had the largest spacing between nucleosomes (Valouev et al. 2011). Such local variation in NRL can depend on the metabolic state of the cell, regional regulatory state, and local gene activity in addition to the DNA sequence (Szerlong and Hansen 2011; Jiang and Zhang 2021; Singh and Mueller-Planitz 2021). However, to what extent variations in NRL impact cellular processes such as DNA repair is unclear.
Drosophila melanogaster (hereafter referred to as Drosophila) has commonly been used as a model organism to study chromatin and its associated biological functions (Schulze and Wallrath 2007; Filion et al. 2010; Kharchenko et al. 2011; Chaouch and Lasko 2021). A study analyzed comprehensive genome-wide binding profiles of 53 chromatin proteins and used this information to divide the Drosophila genome into five chromatin types, each originally labeled with a different color (Filion et al. 2010). These include three heterochromatin-like states (repressive chromatin [BLACK], Polycomb group [PcG] heterochromatin [BLUE], and centromeric chromatin [GREEN]) and two euchromatin states (inducible euchromatin [RED] and constitutive euchromatin [YELLOW]), each defined by a unique combination of chromatin-associated proteins and linked to specific histone PTMs (Table 1). We aimed to use these chromatin states alongside damage and repair maps to study how CPD formation and repair in Drosophila S2 cells are modulated by chromatin organization.
Summary of 5 chromatin types
Results
Nucleosomes modulate UV damage formation in Drosophila
Previous studies have indicated that nucleosomes modulate UV damage formation and repair in humans and yeast (Mao et al. 2016, 2017; Brown et al. 2018; Gonzalez-Perez et al. 2019). To study the influence of nucleosomes on UV damage formation in Drosophila, we used CPD-seq (Mao et al. 2016; Mao and Wyrick 2020) to map CPDs at single-nucleotide resolution in S2 cells immediately after UV irradiation and in genomic DNA from S2 cells that was UV-irradiated in vitro as a naked DNA control (Fig. 1A). Analysis of the resulting sequencing reads revealed that in both UV-irradiated S2 cells and naked DNA, CPD-seq reads are enriched at dipyrimidine sequences, especially those with a 5′ thymine (TT and TC), as expected (Fig. 1B; Brash 2015; Mao et al. 2016). In contrast, no enrichment at dipyrimidine sequences was observed in the unexposed (no UV) control (Fig. 1B). Cellular CPD counts occurring at lesion-forming dipyrimidine sequences were then mapped to nucleosome positions called using Drosophila MNase-seq reads from a previous study (see Methods) (Nalabothula et al. 2014) and normalized to CPD counts in UV-irradiated naked DNA to account for sequence bias. This analysis revealed a strong rotational periodicity of 10.2 bp (signal-to-noise ratio [SNR] = 142), consistent with the helical repeat length of nucleosomal DNA (Fig. 1C; Luger et al. 1997). Normalized CPD formation rates were lower at positions where the DNA minor groove faced inward (minor-in), toward the histone octamer, and higher at minor-out positions (Fig. 1C,D). This rotational periodicity indicates that nucleosomes modulate UV damage formation across the Drosophila genome.
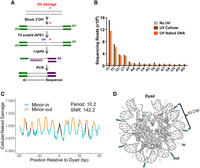
CPD-seq reads are enriched at dipyrimidines and minor-out positions in Drosophila. (A) An outline of the CPD-seq protocol used to generate a map of UV-induced CPDs in Drosophila. (B) Counts of CPD-seq reads from Drosophila S2 cells (with and without UV exposure) and UV-irradiated naked DNA, stratified by the dinucleotide sequence at the putative CPD positions. (C) Damage enrichment with respect to rotational positioning within nucleosomes. Data are normalized by a UV-exposed, naked DNA control. (D) Model of how rotational positioning impacts CPD formation in Drosophila nucleosomes. The nucleosome structure corresponds to Protein Data Bank (PDB; https://www.rcsb.org) entry 2PYO (Clapier et al. 2008). Minor-in positions are labeled with transparent lightning bolts to represent low rates of CPD formation, and minor-out positions are labeled with opaque lightning bolts to represent high rates of CPD formation.
Chromatin type influences repair, but not formation, of CPDs
To determine whether the regional, higher-order chromatin organization across the Drosophila genome impacts CPD formation, as has been reported in human cells (García-Nieto et al. 2017), we analyzed cellular CPD levels in UV-irradiated S2 cells that had been normalized to a naked DNA control in 10,000 bp bins across each chromosome. The resulting data showed that, after normalizing for DNA sequence content using the naked DNA control, cellular CPD formation was relatively constant across the genome (Fig. 2A; Supplemental Fig. S1). In parallel, we also analyzed CPD repair activity using excision repair-sequencing (XR-seq) data from a previous study (Deger et al. 2019). CPD repair reads obtained from Drosophila S2 cells 30 min after UV exposure (30 min repair time point) were normalized to cellular CPD levels measured using CPD-seq and used to determine genome-wide repair patterns. Compared with damage, normalized repair activity was much more variable across the genome (Fig. 2B; Supplemental Fig. S2).
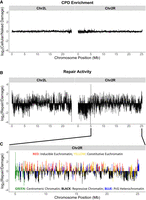
Regional differences in chromatin type impact CPD repair activity. (A,B) Relative CPD enrichment (A) and repair activity (30 min; B) binned in 10,000 bp regions across Chromosome 2 of the Drosophila genome. (A) CPD enrichment is normalized to a UV-exposed, naked DNA control. (B) Repair data are normalized to cellular damage data. (C) Chromatin domains superimposed on repair data from B. Data are zoomed in on positions between 5 Mb and the end of Chromosome 2R for increased resolution of bins and exclusion of the centromere-proximal region, which does not have defined domains. Chromatin type was assigned when >50% of the bin was covered by regions assigned to that type. Bins that failed to meet this threshold are colored gray.
We hypothesized that differences in chromatin state may be responsible for the observed variability in CPD repair activity. To test this hypothesis, we leveraged data from a previous study that divided the Drosophila genome into five distinct chromatin types based on differential protein binding in Drosophila Kc167 cells (Table 1; Filion et al. 2010). Cross-referencing the domains for each of these chromatin types with CPD repair data revealed that inducible euchromatin generally displayed high repair activity at the 30 min time point, whereas repressive chromatin had low repair activity (Fig. 2C). Repair activity in constitutive euchromatin, PcG heterochromatin, and centromeric chromatin was more variable, with associated domains showing both elevated and reduced repair activity (Fig. 2C). These observations were supported by statistical tests using ANOVA, which revealed significant differences in repair activity between nearly all of the chromatin types, in both Chromosome 2R and genome-wide (P < 0.01) (Supplemental Fig. S3), with inducible euchromatin showing the highest repair activity and repressive chromatin showing the lowest.
To test if these patterns were consistent as repair progressed, we performed similar analyses with data at later repair time points from the same XR-seq data set (Deger et al. 2019). At the latest repair time point (24 h), the opposite pattern was seen, with lower CPD repair activity in both euchromatin types compared with repressive chromatin (Supplemental Fig. S4). To better characterize this trend, we generated a time course of genome-wide CPD repair activity in each domain, starting at 10 min of repair and ending at 24 h (Supplemental Fig. S5). The results indicated that relative repair activity in both euchromatin types decreased over time, following initially higher repair activity. In contrast, repair activity in repressive chromatin increased over time until it surpassed all other chromatin types at the 12 h and 24 h time points. These data are consistent with a model in which euchromatin domains are repaired more rapidly (i.e., higher repair activity at earlier time points) than domains with low transcriptional activity.
Because a major distinction between the euchromatin types and the repressive chromatin type is their transcriptional activity, we suspected TC-NER could play a role in the observed differences in repair activity between the associated domains. This pathway repairs lesions more quickly than GG-NER but operates only in actively transcribed regions of the genome (Hu et al. 2015; Adar et al. 2016; Selvam et al. 2022). As a result, CPD lesions in these regions may be preferentially repaired early on and become scarce at later time points. To test this hypothesis, we analyzed repair on the TS versus nontranscribed strand (NTS) for the 30 min and 24 h time points. To compare across genes of different lengths, repair data were divided into six bins for each gene (each with one-sixth the length of the gene), with three additional bins before the TSS and three after the TES (each 356 bp long, the median length of coding bins). Repair activity was higher on the TS at the 30 min time point but lower at 24 h (Supplemental Fig. S6A,B), indicating more rapid repair of the TS relative to the NTS. These results are consistent with a previous analysis of these XR-seq data indicating efficient TC-NER of the TS in Drosophila genes (Deger et al. 2019). We then further stratified the repair data by chromatin type. This stratification showed accentuated repair asymmetry in the euchromatin and centromeric chromatin domains (i.e., these domains exhibited greater repair activity on the TS at the 30 min time point and lower repair activity at the 24 h time point), whereas repressive chromatin and PcG heterochromatin domains showed little to no repair asymmetry at either time point (Supplemental Fig. S6C,D). These findings suggest that differential TC-NER activity is at least in part responsible for the differences in repair kinetics across the five chromatin types (Supplemental Table S1). In addition, these results emphasize the heterogenous nature of the centromeric chromatin type, which harbors significant transcriptional activity despite also being associated with markers of heterochromatin (Filion et al. 2010).
Nucleosomal DNA exhibits reduced repair of CPDs compared with linker DNA across all chromatin types
Next, we investigated repair activity in nucleosomal DNA relative to adjacent linker DNA. Analysis of CPD formation using our CPD-seq data indicates that the rotational positioning of nucleosomes modulates CPD formation, but there is no significant difference in CPD enrichment in nucleosomal DNA relative to adjacent linker DNA (Fig. 3A). In contrast, analysis of repair activity at the 30 min time point indicated elevated CPD repair in linker DNA compared with nucleosomal DNA (Fig. 3B). These results are consistent with previous genome-wide studies in yeast cells showing that repair of CPDs is more efficient in linker DNA (Mao et al. 2016). Notably, this variation in repair activity is greater than the variation in CPD enrichment seen in the corresponding damage patterns (Fig. 3A). Although these data represent the combined repair activity across both DNA strands, analysis of each strand individually revealed higher repair activity on their 5′ ends in nucleosome core regions (i.e., −60 to +60 bp from the nucleosome dyad) (see Supplemental Fig. S7), consistent with previous work in yeast and human cells indicating strand-specific repair asymmetry in nucleosomal DNA (Mao et al. 2020).
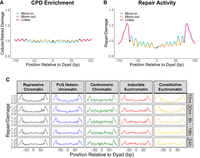
Repair of CPDs is faster in linker DNA regardless of chromatin state. (A,B) Relative CPD enrichment (A) and repair activity (30 min; B) with respect to rotational nucleosome positioning. The CPD enrichment data used to generate plot A are the same as in Fig. 1C, but both axes have been expanded to include linker DNA and highlight differences in amplitude compared with repair data (B). (A) CPD enrichment is normalized to a UV-exposed, naked DNA control. (B) Repair data are normalized to cellular damage data. (C) CPD repair activity within 103 bp of nucleosome dyads, stratified by chromatin type and time point. The data in C were scaled for each subplot so that they have a uniform mean of one.
Having established these general CPD repair patterns in nucleosomal and linker DNA, we proceeded to analyze the repair data across five time points (spanning from 10 min to 24 h), stratified by chromatin type, in order to determine if chromatin state impacts nucleosomal repair patterns. The results across all of these stratifications showed clear peaks of repair activity in the linker regions, with magnitudes similar to the aggregate data (∼20%) (Fig. 3B,C), suggesting that faster repair in the linker region occurs in all chromatin states and persists for at least the first 24 h of repair, irrespective of differences in higher-order chromatin organization and histone H1 occupancy between these chromatin types (Table 1). Additionally, repair activity at earlier time points in constitutive euchromatin domains decreased near the center of the nucleosome dyad, resulting in a concave shape (Fig. 3C). This could be indicative of preferential repair at the edges of the nucleosome, possibly owing to transient nucleosomal DNA unwrapping (Thoma 1999; Kono and Ishida 2020). Altogether, these results show that in Drosophila, CPD repair activity is increased in linker regions regardless of chromatin type or repair time point.
NRL varies across chromatin types and impacts CPD repair
In addition to the rotational periodicity in nucleosomes caused by the orientation of DNA with respect to the histone octamer, a translational periodicity can arise from differences between nucleosomal and linker DNA across groups of nucleosomes. The higher CPD repair activity we observed in linker DNA compared with nucleosomal DNA indicated a translational periodicity for CPD repair, which we aimed to characterize further. Because translational periodicities are directly influenced by the spacing between nucleosomes, we first characterized the NRL of the Drosophila nucleosome map (the same map used for the analysis of rotational periodicity) using the mutperiod software package (Morledge-Hampton and Wyrick 2021). This analysis estimated a NRL of 181.65 bp, which is consistent with the median NRL given by the researchers who produced the original MNase-seq map (181.4 ± 0.4 bp) (Nalabothula et al. 2014). Using the NRL from mutperiod, we first mapped CPD enrichment across a 1000 bp window centered on each nucleosome and found that there was no clear translational periodicity (Fig. 4A). In contrast, CPD repair activity at the 30 min time point was consistently higher in linker DNA and lower in nucleosomes (Fig. 4B). Quantifying these periodicities using a Lomb–Scargle periodogram revealed a peak period in the repair data at 180.98 bp but no concurrent peak in the damage data (Fig. 4C).
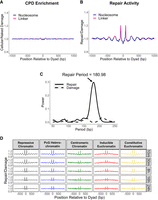
CPD repair activity exhibits a translational nucleosome periodicity that is inconsistent across different chromatin types. (A,B) Relative CPD enrichment (A) and repair (30 min; B) trends with respect to translational nucleosome positioning. (A) CPD enrichment is normalized to a UV-exposed, naked DNA control. (B) Repair data are normalized to cellular damage data. (C) Lomb–Scargle periodogram of translational nucleosome periodicities for CPD damage and repair. The damage and repair periodicities were derived from the data plotted in A and B, respectively. (D) CPD repair activity within 1000 bp of nucleosome dyads, stratified by chromatin type and time point. The data in C were scaled for each subplot so that they have a uniform mean of one.
Subsequently, we analyzed the CPD repair data in a 1000 bp window across the five chromatin types and repair time points in order to determine how they affected the observed translational periodicity. These data revealed that the translational periodicity is strongest at earlier repair time points (e.g., 10 or 30 min) and weaker at later repair time points (e.g., 8–24 h), with visibly diminished oscillations in the data (Fig. 4D). These data are consistent with the rapid repair of linker DNA associated with early repair time points. Notably, these data are scaled so that the normalized repair activities for all subplots have a mean value of one and are easier to compare to one another. Plots scaled uniformly by differences in sequencing depth between the damage and repair data can be found in Supplemental Figure S8. A closer examination of the repair data revealed that the translational periodicity was not visible for all chromatin types. Although repressive chromatin, PcG heterochromatin, and constitutive euchromatin display regular peaks in repair activity associated with linker regions, inducible euchromatin and centromeric chromatin do not show this pattern, except for in linker regions immediately flanking the central nucleosome (Fig. 4D). Curiously, both of these cohorts (i.e., chromatin types with clear translational periodicities and chromatin types without) contain transcriptionally active and inactive domains, suggesting that transcriptional activity alone is not responsible for the presence or absence of translational periodicity in CPD repair data.
To better understand these differences in translational repair patterns across chromatin types, we quantified the translational repair periodicity of each chromatin type at the 30 min time point using Lomb–Scargle periodograms. For each chromatin type, we identified the translational period with the maximum power for the CPD repair data and calculated an SNR as a measure of its clarity. Consistent with our previous observations (Fig. 4D), repressive chromatin, PcG heterochromatin, and constitutive euchromatin chromatin had clear periodicities, with the strongest signal in constitutive euchromatin (SNR = 78.3) (Fig. 5A), followed by repressive chromatin (SNR = 41.0) (Fig. 5B) and then PcG heterochromatin (SNR = 24.8) (Fig. 5C). Centromeric chromatin (Fig. 5D) and inducible euchromatin (Fig. 5E) had no clear periodicities and relatively low SNR values (5.3 and 6.4, respectively). Next, we compared the translational periods associated with these SNR values (i.e., the average distance between repair peaks associated with linker regions in each domain) against the genome-wide translational period (180.98 bp) (Fig. 4C). This analysis revealed that repressive chromatin and PcG heterochromatin had slightly higher translational periods at 184.8 bp and 183.8 bp, respectively, whereas the translational period for constitutive euchromatin was much lower at 173.7 bp (Fig. 5A–C). Similar results were obtained for the 10 min time point (<1 bp difference in translational repair periods for each of the chromatin types compared with the 30 min time point for those with clear translational periods) (Supplemental Fig. S9). These results indicate that CPD repair patterns with respect to nucleosomes are significantly altered by the overarching chromatin domain in which the nucleosomes are located.
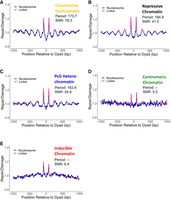
Translational CPD repair periodicities vary in strength and period across chromatin types. (A–E) CPD repair activity (30 min) with respect to translational nucleosome positioning for constitutive euchromatin (A), repressive chromatin (B), PcG heterochromatin (C), centromeric chromatin (D), and inducible euchromatin (E). All repair data are normalized to cellular damage data. Periods were not assigned to centromeric chromatin (D) and inducible euchromatin (E) owing to the diminished SNR values.
Given that translational repair periodicities typically follow patterns of nucleosome positioning, we hypothesized that the differences in translational periods for repair activity between chromatin types were owing to differences in the underlying NRL. To test this hypothesis, we quantified the NRL of each chromatin type using a Lomb–Scargle periodogram of relative nucleosome positions based on the MNase-seq data (Nalabothula et al. 2014), similar to the quantification of the translational repair periodicity. This analysis revealed that constitutive euchromatin, repressive chromatin, and PcG Heterochromatin had NRLs of 173.9 bp, 184.8 bp, and 183.2 bp, respectively (Fig. 6A). These NRLs almost exactly matched their corresponding translational repair periods at the 30 min time point, supporting our hypothesis (Fig. 6A,B; Supplemental Table S1). Additionally, both the centromeric chromatin and inducible euchromatin types have clear and quantifiable NRLs, indicating that their lack of translational repair periodicity is not merely owing to a lack of regular nucleosome positioning (Fig. 6B; Supplemental Table S1). The differences in repair patterns between repressive chromatin and constitutive euchromatin and the relationship between NRL and repair periodicity can be observed more clearly when the translational data for constitutive euchromatin and repressive chromatin are superimposed on one another. Over a distance of 1000 bp, the two waveforms become completely out of phase with one another, and this phenomenon is clearly observed in both the nucleosome occupancy (Fig. 6C) and repair data (Fig. 6D). These data indicate that NRL varies across chromatin types, resulting in altered CPD repair patterns.
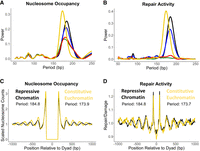
Translational CPD repair periods and nucleosome repeat length vary in tandem across chromatin types. (A,B) Lomb–Scargle periodograms showing nucleosome occupancy (A) and CPD repair (B) periodicities for each chromatin type. Black trend lines represent repressive chromatin; blue trend lines, PcG heterochromatin, green trend lines, centromeric chromatin; red trend lines, inducible euchromatin; and yellow trend lines, constitutive euchromatin. (C,D) Overlaid constitutive euchromatin and repressive chromatin translational patterns for nucleosome occupancy (C) and CPD repair activity (30 min; D). Nucleosome dyads were never called within 147 bp of one another, so the region from −147 to 147 bp in the nucleosome occupancy data (C) was excluded from the periodicity analysis.
Discussion
Nucleosomes are ubiquitous throughout the genomes of higher eukaryotes and have indispensable roles in genome organization (McGinty and Tan 2015). Exploring the influences of nucleosomes on DNA repair systems such as NER is critical to our understanding of genome maintenance and stability. Here, we produced the first CPD damage map in Drosophila and combined it with previously published data on CPD repair (Deger et al. 2019), nucleosomes (Nalabothula et al. 2014), and chromatin domains (Filion et al. 2010) to explore how chromatin organization modulates CPD damage formation and repair throughout the Drosophila genome. Our study indicates that regional differences in chromatin state influence repair of CPDs but do not substantially impact damage formation. Furthermore, inhibition of CPD repair by nucleosomes relative to fast-repairing linker DNA produces a translational repair periodicity throughout the Drosophila genome. However, this translational periodicity is modulated across different chromatin domains, suggesting that pre-existing differences in NRL in distinct chromatin types significantly affect the repair of UV-induced CPD lesions.
As a key component of this study, we produced a map of CPD lesions in Drosophila S2 cells using CPD-seq. This damage map displayed a clear rotational periodicity within nucleosomes, with CPD enrichment being elevated at minor-out rotation settings, consistent with previous findings (Gale et al. 1987; Mao et al. 2016; Brown et al. 2018; Pich et al. 2018; Stark et al. 2022). Previous studies have suggested that regional variations in chromatin state, which are associated with differences in spatial positioning within the nucleus, can also affect DNA damage formation (García-Nieto et al. 2017). However, our data indicate chromatin state does not significantly modulate CPD enrichment in UV-irradiated Drosophila S2 cells relative to a naked DNA control. In contrast, analysis of published XR-seq data generated from the same cell type showed significant regional variations in CPD repair activity (Deger et al. 2019). This pattern was apparent even after normalizing these data with initial damage levels based on our CPD-seq data in order to account for the impact of differential CPD formation on repair (e.g., owing to sequence context or chromatin interactions, such as in nucleosomes) (Mao et al. 2016, 2017; Brown et al. 2018; Gonzalez-Perez et al. 2019).
To explore the source of these regional variations in repair activity, we leveraged existing data on five distinct chromatin types in the Drosophila genome (Filion et al. 2010). This analysis showed clear correlations between chromatin domains and areas of higher/lower repair activity. Areas of higher repair activity were associated with inducible euchromatin at early repair time points, whereas areas of lower repair activity were generally associated with repressive chromatin domains. These differences seemed to be broadly influenced by TC-NER, which was especially active in both euchromatin types as well as in centromeric chromatin. Curiously, the centromeric chromatin type is also characterized by the heterochromatin marker H3K9me2, (Filion et al. 2010), possibly indicating a composite of highly transcribed and inactive regions within this chromatin type. Additionally, despite constitutive euchromatin boasting the highest transcriptional activity of the five chromatin types (in undamaged cells), its genome-wide repair activity was lower than inducible euchromatin at early repair time points and displayed less transcriptional repair asymmetry. Lower repair activity in constitutive euchromatin may be explained in part by its more compact nucleosome organization, as reflected in the shorter NRL of constitutive euchromatin relative to other chromatin types. The lower transcriptional asymmetry in constitutive euchromatin might arise from the phenomena of domain-associated repair observed in human cells, whereby active transcription relaxes chromatin structure to allow for easier access to the NTS of genes (Bohr et al. 1985; Nouspikel et al. 2006; Zheng et al. 2014). Alternatively, it is possible that the expression patterns in undamaged cells do not accurately reflect expression patterns in cells exposed to UV light, consistent with previous studies indicating that the expression of individual genes is altered in flies exposed to UV light (Ujfaludi et al. 2007; Zhou et al. 2013). To what extent transcriptional changes occur throughout the Drosophila genome in response to UV exposure will be important to address in future studies. Ultimately, these results indicate that regional NER activity in Drosophila is significantly influenced by chromatin state, at least in part owing to differences in transcriptional activity.
Incorporating a nucleosome map derived from published MNase-seq data (Nalabothula et al. 2014) allowed us to analyze the impact of the overarching chromatin type on repair patterns in nucleosomes. All five of the chromatin types in Drosophila exhibit enhanced repair activity at early repair time points in linker DNA, as well as correspondingly lower repair activity in nucleosomal DNA, consistent with previous genome-wide studies indicating that nucleosomes inhibit NER (Mao et al. 2016, 2020; Brown et al. 2018; Pich et al. 2018). However, only three of these chromatin types (repressive chromatin, PcG heterochromatin, and constitutive euchromatin) exhibit translational periodicities spanning multiple nucleosomes. Notably, repressive chromatin displays this translational periodicity, reflecting elevated repair activity in linker DNA, despite high levels of histone H1 binding (Filion et al. 2010). Previous biochemical studies have indicated that histone H1 can inhibit base excision repair of DNA base damage in linker DNA in vitro (Cole et al. 2010), but our results suggest that histone H1 binding does not significantly impair NER of UV damage in linker DNA in Drosophila cells. Curiously, the three chromatin types with clear translational periodicities include both heterochromatin and euchromatin domains, as do the two chromatin types without translational periodicities (centromeric chromatin and inducible euchromatin), indicating that general characteristics of such chromatin domains (e.g., euchromatin vs. heterochromatin or high vs. low transcriptional activity) are not necessarily accurate predictors of repair patterns in nucleosomes.
Even within the three chromatin types that exhibit clear translational periodicities, the exact repair periodicities were inconsistent. Repressive chromatin and PcG heterochromatin had relatively similar repair periods of ∼185 bp and ∼184 bp, respectively, but constitutive euchromatin had a much shorter period of ∼174 bp. This 11 bp difference in NRL reflects a >25% decrease in the length of linker DNA, presumably reflecting a more compact chromatin structure. Despite this, elevated repair activity in linker DNA was still apparent in nucleosomes within constitutive euchromatin domains. Importantly, all of these periods were highly similar to calculated NRLs for each of the domains based on published MNase-seq data (Nalabothula et al. 2014), indicating that underlying and pre-existing differences in nucleosome organization are driving these distinct repair patterns. Although previous studies in mouse (Teif et al. 2012) and human (Valouev et al. 2011) cells have reported different NRLs across cell types and chromatin domains, respectively, the potential impact of these differences in NRL on cellular processes such as DNA repair was previously unclear. Our data indicate that variations in NRL across different chromatin domains in Drosophila significantly affect DNA repair.
A key question is what molecular mechanism(s) is responsible for the shorter NRL in constitutive euchromatin relative to other chromatin types. Previous studies have shown that transcriptional activity can reduce the spacing between nucleosomes as RNA polymerase II transcribes through them (Studitsky et al. 1994, 1997; Weiner et al. 2010). Given that constitutive euchromatin exhibits the highest transcriptional activity of the five chromatin types (Filion et al. 2010), this may help explain its shorter NRL. Additionally, this chromatin type is enriched in H3K36me3 (Filion et al. 2010), which has been reported to regulate nucleosome spacing in yeast (Eriksson and Clark 2021). Further research is required to determine whether these or other mechanisms reduce nucleosome spacing in constitutive euchromatin.
Although the differences in translational repair periodicity between repressive chromatin, PcG heterochromatin, and constitutive euchromatin can be explained by differences in NRL, the lack of a translational repair periodicity in centromeric chromatin and inducible euchromatin cannot. Analysis of MNase-seq data with respect to these chromatin types revealed periodic nucleosome organization consistent with NRLs of ∼179 and ∼181 bp, respectively, yet this translational periodicity was not reflected in the corresponding repair data. One possible explanation for these findings is that the nucleosome organization in these chromatin types may be more susceptible to disruption or repositioning in response to the cellular stress and ongoing repair activity induced by UV damage. For example, a previous study has shown that the SWI/SNF complex can remodel nucleosomes to accommodate the repair of UV damage (Gaillard et al. 2003). Such a mechanism may be particularly important for inducible euchromatin, because this chromatin state contains many stress-responsive genes (Filion et al. 2010). It will be important in future studies to investigate to what extent UV irradiation induces nucleosome disruption or repositioning in these chromatin states.
In summary, we have generated a CPD damage map in Drosophila and analyzed it alongside XR-seq data (Deger et al. 2019), chromatin domains (Filion et al. 2010), and nucleosome positions (Nalabothula et al. 2014) in order to investigate regional and nucleosomal damage and repair patterns across the Drosophila genome. Our analysis indicates that although nucleosomes modulate both UV damage formation and repair, regional variations in chromatin state only affect repair activity. Importantly, our findings indicate that repair patterns in and around nucleosomes are significantly modulated by the chromatin type in which the nucleosomes are located. This effect is primarily owing to domain-specific changes in NRL, which we show significantly impacts repair activity. DNA–protein interactions dominate the genome landscape and can modulate DNA damage and repair in numerous ways, impacting genome stability and shaping the mutational landscape of genetic diseases such as skin cancer (Pich et al. 2018, 2019; Selvam et al. 2023). These results in Drosophila inform these interactions by showing how variable nucleosome organization across chromatin domains impacts NER.
Methods
Culture and UV treatment of the cell lines
Drosophila S2 cells were maintained at 28°C in tissue culture flasks containing Schneider's Drosophila medium (GIBCO) supplemented with 10% heat-inactivated fetal bovine serum (FBS; HyClone), 100 U/mL of penicillin, 100 g/mL of streptomycin, and 0.25 g/mL of amphotericin B (Fungizone) antimycotic (Life Technologies). Upon ∼80% confluence, the culture medium was removed, washed once with 1× phosphate buffered saline (PBS), and layered with 2 mL of sterile PBS. The cells were exposed to 100 J/m2 UVC. Following irradiation, PBS was removed, and cells were harvested with vigorous pipetting and pelleted for the “UV cellular” sample. Cells without treatment were pelleted for the “no UV” and “UV naked DNA” control samples. All the cell pellets were stored at −80°C till genomic DNA isolation.
Genomic DNA isolation and UV irradiation of naked DNA
Genomic DNA was isolated from the frozen cell pellets using GenElute mammalian genomic DNA miniprep kits (Sigma-Aldrich G1N70). One fraction of “no UV” DNA was spotted on clean cover glass and exposed to 80 J/m2 UVC. After irradiation, the DNA samples from the cover glass were collected and processed as the “UV naked DNA” sample.
CPD-seq library preparation
All the DNA samples were processed according to published protocols (Mao et al. 2016; Mao and Wyrick 2020). The genomic DNA samples were sonicated, end-repaired (NEB E6050L) and dA-tailed (NEB E6053L), ligated to F1 adapter, and treated with terminal transferase (NEB M0315L). The DNA was then digested with T4 endonuclease V (T4 PDG, NEB M0308S) and AP endonuclease (NEB M0282S) to create 3′-OH groups immediately upstream of the CPD lesions, and 5′ phosphate groups were removed using shrimp alkaline phosphatase (Affymetrix AF78390500). The samples were subsequently denatured for 5 min at 95°C and snap-cooled on ice. The biotin-labeled second adapter, the A adapter with unique barcodes for different samples, was then ligated to the 3′-OH groups. The single-stranded DNA was eluted with streptavidin beads (Thermo Fisher Scientific 11205D), and final PCR was done with trP1 and A primers. The final PCR product was sequenced using Ion Proton sequencing (Life Technologies).
Calling CPD lesions
CPD lesions were called as previously described (Mao et al. 2016). Samples were differentiated by barcodes on the 5′ ends of sequencing reads, and the barcodes were then removed along with one base on the 3′ end. These trimmed reads were aligned to the dm6 genome using default Bowtie 2 parameters and converted to BED files using SAMtools (Li et al. 2009) and BEDTools (Quinlan and Hall 2010). Putative lesions were called at the positions complementary to the two bases 5′ of each aligned read. Only putative lesions at dipyrimidines were retained for further analysis.
Calling nucleosome dyads
Nucleosome dyads were called from existing MNase-seq data (Nalabothula et al. 2014) by using a pipeline developed for analyzing nucleosome periodicities (Pich et al. 2018). The pipeline was modified slightly so that nucleosomes were called throughout the entire genome instead of solely from mappable genic regions. Dyad positions were converted to the dm6 genome assembly from dm3 using the UCSC Genome Browser liftOver tool (Hinrichs et al. 2006). Nucleosomes were assigned to specific chromatin domains by intersection of the dyad centers with the domain positions (Filion et al. 2010).
Calling XR-seq lesion positions
XR-seq reads (Deger et al. 2019) were aligned to the dm6 reference genome using Bowtie 2 (Langmead and Salzberg 2012) with default parameters. Trimmomatic (Bolger et al. 2014) v0.3.9 was used with the trimming parameters “ILLUMINACLIP:[XR-seq_adapter_file]:2:30:10” to remove adapter sequences from XR-seq reads prior to alignment. The resulting SAM files were converted to BED files using SAMtools (Li et al. 2009) and BEDTools (Quinlan and Hall 2010). Lesions were called at the seventh and eighth nucleotides relative to the 3′ end (regardless of nucleotide identity) for reads 24–32 bp in length. All other reads were omitted. These parameters were determined by searching for the most common read lengths that still maintained high dipyrimidine enrichment at 3′ proximal positions, based on known NER excision patterns (Huang et al. 1992; Kemp et al. 2012; Hu et al. 2015).
Quantification of nucleosome periodicities
Nucleosome periodicities were quantified using the mutperiod software package (Morledge-Hampton and Wyrick 2021). Damage and repair events were counted relative to nucleosome dyads and normalized to a naked DNA control and damage data, respectively. Periodicities were quantified using a Lomb–Scargle periodogram, and SNR values were calculated as previously described (Pich et al. 2018; Morledge-Hampton and Wyrick 2021). Expected linker and nucleosomal regions were derived from the NRL for each nucleosome map, calculated using a Lomb–Scargle periodogram. Minor-in and minor-out positions were derived from structural models of rotational positioning throughout the nucleosome (Cui and Zhurkin 2010).
Classifying genome-wide bins and genes by chromatin type
Genome-wide bins and genes were assigned a chromatin type when >50% of the bin/gene fell within domains belonging to that chromatin type (Filion et al. 2010). If this threshold was not met, the bin/gene was classified as “gray” (in reference to the original “color” names for the chromatin types) to indicate that no chromatin type encompassed the majority of the region.
Transcriptional asymmetry analysis
Each gene was split into six bins equal to one-sixth of that gene's length so that the bins were comparable across all genes. As an internal control, six flanking bins (three on either side) were established around each genic region. The length of each flanking bin was set to the median genic bin length: 356 bp. Repair events were counted inside each bin and assigned to the TS or NTS. For intergenic (flanking) bins, strand designation was determined by the flanked genic region.
Previously published data
The MNase-seq map used to call dyad positions can be found through the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under the accession ID GSE49526 (Nalabothula et al. 2014).
The chromatin domain designations can be accessed through GEO under the accession ID GSE22069 (Filion et al. 2010).
The CPD XR-seq data can be accessed through GEO under the accession ID GSE138846 (Deger et al. 2019).
The RPKM values used in Supplemental Table S1 can be accessed through GEO under the accession ID GSE19325 (Gan et al. 2010).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE267055. The called nucleosomes dyads used in this analysis are available at GitHub (https://github.com/bmorledge-hampton19/Chromatin_Features_Analysis/blob/main/maintained_data/S2_nucleosome_map.bed). The gene designations used in this analysis are available at GitHub (https://github.com/bmorledge-hampton19/Chromatin_Features_Analysis/blob/main/maintained_data/dm6_BDGP6_89_named_genes.bed). The code used to perform these analyses can be found at GitHub (https://github.com/bmorledge-hampton19/Chromatin_Features_Analysis), and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was supported by the following grants from the National Institute of Environmental Health Sciences (NIEHS): R01ES028698 (J.J.W.), R01ES032814 (J.J.W.), and R21ES035888 (A.G.G., J.J.W., and K.S.).
Author contributions: A.G.G. and J.J.W. conceived and supervised the study. K.S. and M.C. performed the experimental work. B.M-H. and J.J.W. performed the computational analysis. B.M-H., J.J.W., and K.S. compiled the figures and wrote the manuscript. All authors edited and approved of the final draft of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279605.124.
- Received May 20, 2024.
- Accepted December 19, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








