
ScisTree2 enables large-scale inference of cell lineage trees and genotype calling using efficient local search
- 1School of Computing, University of Connecticut, Storrs, Connecticut 06269, USA;
- 2Division of Hematology/Oncology, Boston Children's Hospital, Boston, Massachusetts 02115, USA;
- 3Department of Biomedical Informatics, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 4Institute for Systems Genomics, University of Connecticut, Storrs, Connecticut 06269, USA
Abstract
In a multicellular organism, cell lineages share a common evolutionary history. Knowing this history can facilitate the study of development, aging, and cancer. Cell lineage trees represent the evolutionary history of cells sampled from an organism. Recent developments in single-cell sequencing have greatly facilitated the inference of cell lineage trees. However, single-cell data are sparse and noisy, and the size of single-cell data is increasing rapidly. Accurate inference of cell lineage tree from large single-cell data is computationally challenging. In this paper, we present ScisTree2, a fast and accurate cell lineage tree inference and genotype calling approach based on the infinite-sites model. ScisTree2 relies on an efficient local search approach to find optimal trees. ScisTree2 also calls single-cell genotypes based on the inferred cell lineage tree. Experiments on simulated and real biological data show that ScisTree2 achieves better overall accuracy while being significantly more efficient than existing methods. To the best of our knowledge, ScisTree2 is the first model-based cell lineage tree inference and genotype calling approach that is capable of handling data sets from tens of thousands of cells or more.
An adult human body contains about 30 trillion cells that perform various functions. These cells are interconnected via a shared cellular division history that spans development, aging, and disease. The evolutionary history of cells sampled from an organism can be depicted by a cell lineage tree, a fundamental model integral to the study of tumor evolution and developmental biology (Navin 2014; Bizzotto et al. 2021; Coorens et al. 2021; Simeonov et al. 2021). A cell lineage tree is a rooted tree in which leaves are labeled by the extant cells and internal nodes represent progenitor cells that are ancestral to the extant cells. To simplify our notation, we use the terms “tree” and “cell lineage tree” interchangeably.
Reconstructing cell lineage trees from noisy single-cell DNA data has been actively studied in recent literature. Existing methods differ in their modeling assumptions and also in computational approaches. First, mutational models are essential for single-cell DNA data analysis, as mutations give rise to genomic variants, which constitute the primary data used for inferring cell lineage trees. The primary class of genomic variants studied in this paper is the single nucleotide variant (SNV). We do not consider more complex variants such as copy number variations (CNVs) for cell lineage tree inference (see the Discussion section). The infinite-sites (IS) model is a main mutational model for SNV variants. The IS model assumes that each mutant variant only arises once in the evolutionary history (i.e., there are no recurrent mutations). For example, in the cell lineage tree shown in Figure 1A, there is a single mutation for each SNV site. Many existing approaches assume the IS model (e.g., Jahn et al. 2016; Wu 2020; Kızılkale et al. 2022). An alternative model is the finite-sites model (Zafar et al. 2017; Kozlov et al. 2022), which allows recurrent mutations at a genomic site.

Illustration of cell lineage tree, genotypes with uncertainty, and input of ScisTree2. (A) The true cell lineage tree with five cells and six sites (with mutations labeling branches). Infinite-sites model: one mutation per site. Four internal nodes: a to d. (B) The true (binary) genotypes. There are five cells and six SNV sites. (C) The input of ScisTree2 is the genotype probability matrix of genotypes being the wild type (0). The two boldfaced positions denote genotypes, called using the maximum probability allele, that do not agree with the true genotypes. For example, for the cell C3 and the site S6, M[3, 6] = 0.7, which would wrongly call this genotype the wild type (0).
Existing cell lineage tree reconstruction methods also differ in high-level computational approaches. Several methods are based on probabilistic inference and use Markov chain Monte Carlo (MCMC) (Jahn et al. 2016; Zafar et al. 2017). A major downside of these methods is that they cannot handle large data. A more efficient alternative to MCMC is using local search to find optimal trees on a probabilistic model. Several newer methods including ScisTree (Wu 2020) and CellPhy (Kozlov et al. 2022) adopt this approach. Lastly, there are parsimony-based approaches such as HUNTRESS (Kızılkale et al. 2022) which aim to make the fewest changes to genotypes so that the changed genotypes satisfy the IS model.
We previously developed a cell lineage tree inference method called ScisTree (Wu 2020), which identifies the optimal tree that maximizes the posterior probability under the infinite-sites model through local search in tree space. ScisTree first constructs an initial tree from heuristically called genotypes using the well-known neighbor joining algorithm (Saitou and Nei 1987). It then iteratively evaluates and identifies the tree with the highest posterior probability among those topologically similar to the current tree. Benchmarking on simulated data demonstrated that ScisTree accurately inferred trees and was significantly faster than several existing methods, including SCITE (Jahn et al. 2016).
However, ScisTree becomes inefficient when the number of cells exceeds 1000. The size of single-cell data is rapidly increasing, both in the number of assayed cells and the amount of genomic information captured per cell. Single-cell genomic data with tens of thousands of cells are becoming available. For example, a recently developed variant caller, SComatic, can detect somatic SNVs from scRNA-seq and scATAC-seq, which typically contain 1000s to 10,000s of cells (Muyas et al. 2024). Currently, no existing probabilistic cell lineage tree inference methods are capable of handling data sets of this size. Thus, a new method that can perform accurate cell lineage tree inference for large data is needed.
Results
ScisTree2: a software tool for efficient inference of cell lineage trees from large and noisy single-cell variation data
In this paper, we present ScisTree2, a new cell lineage tree inference approach that improves the original ScisTree. The input for running ScisTree2 is generated by processing single-cell DNA-seq or other types of single-cell sequencing data with genetic variants. Given single-cell sequencing data (reads), the first step is using a genotype caller (e.g., GATK [McKenna et al. 2010]) to call the genetic variants from the single-cell sequence reads. Most genotype callers output a discrete probability distribution over possible genotypes for each cell and variant site. For simplicity, ScisTree2 only considers two genotype states: wild type (allele 0) and mutant (allele 1). ScisTree2 takes the genotype probabilities of multiple single cells at multiple SNV sites and simultaneously infers the cell lineage tree and genotypes.
It is well known that single-cell data are noisy. One of the main sources of noise is allelic dropout (ADO), which is quite common in single-cell data and can lead to fewer or even no sequence reads at some SNV sites and cells. In current single-cell data, the ADO rate can be 50% or higher. ADO may lead to a wrongly called wild-type genotype whereas the true genotype is the mutant (i.e., producing a false negative error). Sequencing errors in single-cell DNA data are also common, which may lead to a wild-type allele being wrongly called a mutant (i.e., producing a false positive error). Noise in single-cell data implies that the called genotypes from single-cell data have significant uncertainty, which poses a major challenge for cell lineage tree inference.
ScisTree and ScisTree2 take a genotype probability matrix M of size n rows by m columns as input. Here, n is the number of cells and m is the number of SNV sites. For each cell c and each site s, M[c, s] is equal to the posterior probability of the cell c having the wild-type genotype at the site s given the sequence reads at s (see Fig. 1C).
The original ScisTree (Wu 2020) finds the tree T* that gives the maximum posterior probability P(T*) and calls genotypes that fit the IS model using local search (Supplemental Methods, Sec. S1). Briefly, local search iteratively searches for an optimal cell lineage tree by finding the tree that gives the maximum posterior probability among all “neighboring” trees that are within a single tree rearrangement move, such as nearest neighbor interchange (NNI) or subtree prune and regraft (SPR), from the current tree. See the Methods section for more details. The original ScisTree performs the NNI search. The running time of the NNI local search is O(Kn2m) for NNI local search where K is the number of iterations, which depends on how far the initial tree is from local optima (Supplemental Methods, Sec. S2). When data size is relatively large (say n = m = 1000), ScisTree becomes slow.
The key technical contribution of the new ScisTree2 approach is an SPR local search algorithm that runs in O(Kn2m) time, which is one order of magnitude faster than a naive alternative. Moreover, the SPR local search is made more efficient by a branch and bound heuristic, which enables ScisTree2 to infer cell lineage trees with 10, 000 or more cells. See the Methods section for the details of the SPR local search algorithms. Experiments show that ScisTree2 runs much faster than all existing cell lineage tree inference methods (except the classic distance-based methods such as neighbor joining). Moreover, ScisTree2 outperforms existing methods in the accuracy of reconstructing the history of mutations and genotype calling.
Simulated data
We compared ScisTree2 with the following methods using simulated data: (i) CellPhy (Kozlov et al. 2022); (ii) HUNTRESS (Kızılkale et al. 2022); (iii) SiFit (Zafar et al. 2017); (iv) the original ScisTree (Wu 2020); and (v) for small data only, SCITE (Jahn et al. 2016). In addition to comparing running time, we used the following metrics (all between 0 and 1, with 1 being the best) for accuracy comparison.
-
Genotype accuracy: the percentage of correctly called genotypes.
-
Tree accuracy: the percentage of the shared clades between the inferred trees and the true trees (which is equal to one minus the normalized Robinson-Foulds distance).
-
Ancestor-descendant (AD) F-score (see, e.g., Kızılkale et al. 2022): for the percentage of the pairs of mutations (ma, mb) where ma is ancestral to mb in the true tree but not so in the inferred tree.
-
Different-lineage (DL) F-score (see, e.g., Kızılkale et al. 2022): for the percentage of pairs of mutations (ma, mb) where ma is not ancestral to mb in the true tree, but ma is ancestral to mb in the inferred tree.
Because CellPhy and SiFit are based on the finite sites model, we used the genotype caller implemented in ScisTree2 to place the mutations on the trees inferred by CellPhy and SiFit. Then, we calculated the AD and DL accuracy based on the placed mutations. Note that this only affected AD and DL results for CellPhy and SiFit. Genotype accuracy of CellPhy and SiFit are based on the genotypes called by these two methods.
We used CellCoal (Posada 2020) under the diploid infinite-sites model to simulate single-cell DNA sequence data. The simulation parameters (along with their default values) were set to: (i) the number of cells n (200); (ii) the number of SNV sites m (five times of n); (iii) the ADO rate (0.2); (iv) sequencing error rate (0.01); and (v) reads coverage (10× for high coverage data). For each setting of parameters, we generated 50 replicates and report the average of these replicates. We ran CellPhy-GL with the simulated VCF files from CellCoal. We ran HUNTRESS and SCITE with the maximum likelihood genotypes extracted from the VCF files. ScisTree2 and ScisTree require posterior probability Pr(G|D) of genotype. Here, the data D are the sequence read counts for different alleles. CellCoal only calculates the likelihood Pr(D|G). We used Bayes’ Theorem to calculate Pr(G|D) from Pr(D|G). The details are given in the Supplemental Methods (Sec. S7).
Simulated high-coverage data
Accuracy
Figure 2 shows a comparison of the inference accuracy for different methods by varying the number of cells. Overall, ScisTree2 and CellPhy had the highest tree accuracy, although CellPhy performed less well in genotype accuracy.
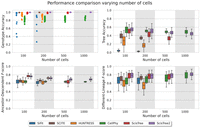
Comparison of inference accuracy of ScisTree2 and five other methods: SiFit, SCITE, HUNTRESS, CellPhy, and the original ScisTree on simulated data with 10× coverage. The number of cells was varied in 100, 200, 500, and 1000, the number of variants was set to 5× the number of cells, and methods that did not complete in one day are omitted. The y-axis denotes four performance metrics: genotype accuracy, tree accuracy, AD F-score, and DL F-score.
Running time
SiFit, SCITE, and the original ScisTree are single-threaded, whereas HUNTRESS, CellPhy, and ScisTree2 support multithreading. The latter three were run using 30 threads. CellPhy was run on its “FAST” mode with the GT10 model that performs fast tree search from a single starting tree. SiFit was run with 10,000 iterations. SCITE was run with 500,000 MCMC iterations. The other methods used their default settings. All methods were tested on a Linux server with an Intel(R) Xeon(R) W-2195 CPU (36 cores). We tested four settings with varying numbers of cells and sites. The results are shown in Figure 3. Results for methods that did not finish within a day are not reported. We report the CPU running time in Supplemental Figure S4.
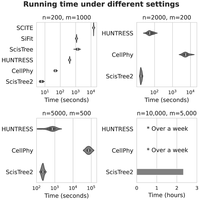
Comparison of the elapsed (user) running time between ScisTree2 and other methods on simulated data with a varying number of cells (n) and sites (m); 30 threads were used for methods supporting multithreading. Methods that are too slow are not reported.
Overall, ScisTree2 demonstrated the highest computational efficiency, particularly for data sets with a large number of cells. CellPhy exhibited good scalability with respect to the number of sites but became computationally expensive as the number of cells increased. In contrast, HUNTRESS efficiently handled a large number of cells but experienced a substantial increase in run time for data sets with many sites.
Performance evaluation of ScisTree2 and other methods by varying parameters on simulated data
We evaluated the effects of ADO rates and read coverage on each method using simulated data. Here, the number of cells was fixed at 200 and the number of sites is 1000. The results are shown in Figures 4 and 5. ScisTree2 still outperformed other methods for data with high dropout rates or low coverage. These results demonstrated that ScisTree2 performs well on data generated with various settings.
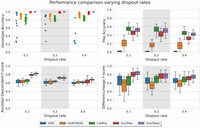
Performance comparison for ScisTree2 and four other methods on various dropout rates. x-axis: dropout rates at 0.1, 0.2, and 0.4. y-axis: performance metric.
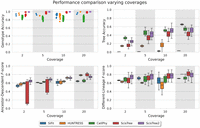
Performance comparison on various coverage (per site per cell). x-axis: coverage at 2×, 5×, 10×, and 20×). y-axis: performance metric.
Accuracy of initial trees
We evaluated the accuracy of the initial trees constructed by the neighbor joining algorithm (Saitou and Nei 1987). The results on the initial tree accuracy for fixed genotypes versus uncertain genotypes are shown in Supplemental Figure S1. Initial trees are reasonably accurate with high-quality data but are less accurate than the trees inferred by ScisTree2 for data with lower quality.
Simulated low-coverage and low-quality data
Some existing single-cell DNA sequence data have coverage lower than what we have simulated so far. For example, the 10× whole genome single-cell DNA sequence data can have as low as 0.01× coverage. Thus, it is useful to evaluate the performance of ScisTree2 on data with coverage that is lower than 1×. Because CellCoal can only simulate data whose coverage is greater than 1×, to obtain data with very low coverage, we first simulated data with coverage 1× using CellCoal. Then, we randomly dropped simulated reads with a certain probability. In this way, we simulated data sets with read coverages lower than 1×. The results are shown in Figure 6. Again, ScisTree2 was highly accurate on data with very low coverage.
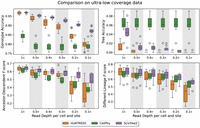
Performance comparison on data with lower than 1× coverage; 200 cells and 1000 sites were used. ScisTree2 outperformed other methods in genotype accuracy and AD and DL F-score: it called genotypes with more than 85% accuracy at 0.1× coverage. No methods achieved high tree accuracy.
In addition, we also simulated reads by manually adding noise (Supplemental Methods, Sec. S10). The results in Supplemental Figure S2 show that ScisTree2 is robust to a certain level of noise.
Simulation for targeted sequencing
Current single-cell targeted DNA sequencing usually generates data with a large number of cells but relatively small number of SNVs. To evaluate the performance of ScisTree2 on data with similar settings as targeted single-cell sequencing, we simulated reads using CellCoal with 1000, 2000, and 50,000 cells while keeping the number of SNVs at 500. ScisTree2 clearly outperformed both HUNTRESS and CellPhy for genotype accuracy and AD/DL F-scores for data with more cells than sites (Fig. 7). No method performed well in tree topology inference when the number of sites was small.
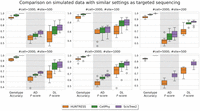
Comparison on simulated data with settings similar to targeted single-cell sequencing data with more cells and fewer sites. Tree accuracy is very low for all methods and is not shown.
Simulation for data where the IS model does not strictly hold
ScisTree2 along with SCITE, HUNTRESS, and the original ScisTree assume the IS model. In real data, it is possible that the IS model does not strictly hold. That is, the data may contain both the sites that follow the IS model and the sites that follow the finite sites (FS) model. To evaluate the performance of methods on data that do not strictly fit the IS model, we performed simulations on data that are generated using different proportions of FS model sites (Supplemental Methods, Sec. S9). As shown in Figure 8, the performance of all methods declined as the proportion of FS model sites increased. However, ScisTree2 consistently outperformed other methods, including SiFit and CellPhy that are specifically designed for the FS model. This suggests that ScisTree2 is robust against minor violations of the IS model.
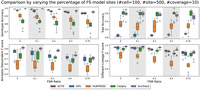
Comparison of simulated data with varying proportions of the FS model sites from 0 to 0.75. The data set consists of 100 cells and 500 sites, with other parameters set to default. Note that AD and DL F-scores are calculated for sites that fit the IS model only.
High-grade serous ovarian cancer single-cell DNA sequencing data
We evaluated the performance of ScisTree2, CellPhy, and HUNTRESS on a low-coverage (with about 0.15× coverage) targeted sequencing data set with 891 cells from three clonally related high-grade serous ovarian cancer (HGSOC) cell lines from the same patient that were processed by rigorous quality control (Laks et al. 2019; downloaded from Zenodo [https://doi.org/10.5281/zenodo.5725635]). The clonal tree of nine clones for the HGSOC data in Laks et al. (2019) is shown in Figure 9. The labels on the branches of the clonal tree are the genes where called mutations occur. Six genes, including three ancestral genes (TP53, FOXP2, and SUGCT), and three clade genes (HTR1D, INSL4, and ZHX1), are reported in Laks et al. (2019). Here, ancestral genes refer to the mutated genes shared by all tumor cells, and clade genes are those shared by a subset of clones. The true cell lineage tree for the HGSOC data is unknown, so we used the ordering of these six genes on the clonal tree (likely the most confident ones reported in Laks et al. 2019) as the ground truth to test some aspects of inference accuracy. After dimensionality reduction and clustering, 891 cells were split into nine clones based on copy number profiles, where the median clonal coverage was 15× (Laks et al. 2019). The resulting data contained the sequence read counts of 14, 068 SNVs. We calculated the likelihood and posterior probabilities of the genotypes from the read counts (see the Supplemental Methods, Sec. S8). We only ran ScisTree2 on the complete HGSOC data (with 14, 068 SNVs), because HUNTRESS was slow on data with a large number of sites, and CellPhy was also slow, which may be due to the large number of missing values in the data.
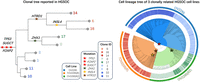
Analysis of the HGSOC data by ScisTree2. Left: The clonal tree of HGSOC with six mutated genes considered important for cancer development in the original study (Laks et al. 2019). Number at a leaf: clone ID (with a distinct color). Right: Reconstructed cell lineage tree of tumor cells (colored w.r.t. their clones) with the same genes mapped on. Outer ring: three cell lines OV2295, TOV2295(R), and OV2295(R2). Inner ring: (colored) clones from the original study. Genes marked as red stars are ancestral genes, whereas others are clade genes.
Tree inference and mutated gene calling
We ran ScisTree2 to infer the cell lineage tree and call genotypes for the complete HGSOC data. The called genotypes contained the 2337 (16%) ancestral mutations that were shared by all 891 cells, 1891 (14%) clonal mutations that were within a single clone, and 9840 (70%) clade mutations that were located along branches of the clonal tree. Because genes affected by ancestral mutations may have a significant impact on early development of tumors, we used ANNOVAR (Wang et al. 2010) to annotate these ancestral mutations and found 48 exonic genes that may potentially be the key driver genes in ovarian cancer oncological pathways.
Evaluation of the called mutated genes and the inferred cell lineage tree
We used the inferred cell lineage tree to obtain the ordering of the six highlighted genes in Laks et al. (2019) and then compared our ordering with the gene ordering from the reported clonal tree (Fig. 9, left). The orderings of these six genes matched perfectly in the two studies. As expected, TP53, a commonly recognized cancer driver gene, appeared before all other mutated genes. In addition, we extracted 49 ancestral genes including the six reported genes based on the provided clonal tree in Laks et al. (2019) from the original study. All 48 genes found by ScisTree2 were among them, with only one gene, XXYLT1, that was not identified as an ancestral gene in ScisTree2. We also investigated the topological concordance between the inferred cell lineage tree (Fig. 9, right) and the clonal tree in Laks et al. (2019). Each of the three cell lines formed a distinct clade in the cell lineage tree as expected. Within each cell line, the inferred cell lineage tree agreed with the clonal tree for the cell line OV2295 and OV2295(R2) but differed in many parts for cell line TOV2295(R). The clonal tree in Laks et al. (2019) was constructed by first clustering the cells into clones using copy number variations and then building trees using SNVs and breakpoints. Our results show that ScisTree2 may be useful in clonal analysis with only SNV variants.
Analysis of reduced HGSOC data
To compare with HUNTRESS and CellPhy, we constructed a smaller subset from the HGSOC data. We followed the same filtering approach used in Kızılkale et al. (2022). We filtered out sites with more than 650 missing values, which led to a reduced-size data set with only 789 SNV sites. The original study in Laks et al. (2019) mapped these SNVs to the branches of the clonal tree, which we used as the ground truth for comparing different methods. ScisTree2 needs two hyperparameters—the ADO rate and genotype priors, which were not known for the HGSOC data. To test the effects of these parameters, we show results with multiple settings of these parameters (Supplemental Methods, Sec. S10). We collected the AD/DL F-scores and the running time of ScisTree2, the neighbor joining algorithm (implemented in ScisTree2), HUNTRESS, and CellPhy. The results are shown in Figure 10. In general, ScisTree2 outperforms the other methods for most settings in these data. Moreover, our results indicate that ScisTree2 is robust to the ADO rate settings, and an appropriately chosen prior improves overall inference accuracy (Supplemental Fig. S3). Therefore, we recommend using genotype priors computed from allele frequencies as the default setting.
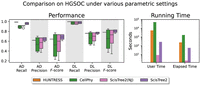
A comparison of ScisTree2 with HUNTRESS, CellPhy, and neighbor joining on reduced HGSOC data. Left: AD and DL F-scores of four methods for mutation ordering. Results from runs with different settings of parameters (e.g., ADO rates) are reported. Right: Running time (in seconds) for user (CPU) time and elapsed (wall clock) time.
Discussion
Integrated analysis of single cells from multiple clones versus analysis of single clones
A commonly used approach for analyzing large single-cell data is first clustering the cells into clones and then analyzing the cells from a single clone (Leung et al. 2017). This approach takes advantage of existing single-cell clustering methods and avoids the computational burden of analyzing large single-cell data. However, a disadvantage of this approach is that errors can potentially be introduced in the clustering step. Because single-cell data usually contain significant noise, clustering single cells is not trivial. Copy number variations, when available, are certainly useful for clustering cells. However, CNVs are rare in normal tissues. SNV-based lineage tracing can be applied to contexts beyond cancer, such as development, aging, and immunology. ScisTree2 provides an alternative way of reconstructing trees directly from large numbers of cells without the need of clustering and can potentially avoid clustering errors. As shown in the Results section, the inferred cell lineage tree can provide the clustering of cells from different cell lines. More experiments will be needed to compare the performance of integrated analysis and single-clone analysis.
The infinite-sites model
One reason for the efficiency of ScisTree2 is its assumption of the infinite-sites model. The IS model greatly simplifies the underlying probabilistic model of ScisTree2. Whereas the IS model is popular in the cell lineage tree inference literature, the IS model may not hold for some data. As we show in the Results section (Fig. 8), ScisTree2 is still reasonably accurate when the deviation from the IS model in the data is moderate. Nonetheless, it is useful to consider extending the model of ScisTree2 to allow violations of the IS model. Because the IS model appears to perform reasonably well in practice, it may be prudent to consider using models that allow moderate deviations from the IS model. In the literature, Kuipers et al. (2022) explored extensions of the IS model to allow some recurrent mutations as well as mutation losses. We note that such extensions can lead to more complex probabilistic models and less efficient inference approaches. Thus, there may be a trade-off between accuracy and efficiency for inference methods.
Beyond binary single nucleotide variants
In this paper, we assume that SNV genotypes are binary. In contrast, ternary SNV genotypes are allowed in Jahn et al. (2016) and Wu (2020). We note that if the IS model holds strictly, SNV genotypes should be binary when recombination is absent. Ternary genotypes are only possible with additional mutational events, for example, recurrent mutations or deletions of wild-type alleles at SNV sites. More generally, when copy number aberrations occur, single-cell genotypes of SNVs may no longer be limited to being binary or ternary. We note that the efficiency of ScisTree2 is based on the simplicity of binary SNV data and the underlying mutation model. Some generalization is possible. For example, Wu (2020) shows that probabilities of ternary genotypes can be calculated efficiently with additional assumptions on the mutation model. Extending the ScisTree2 approach to support more general single-cell genotypes and mutation models remains a research question.
Applying ScisTree2 in studies of cancer biology and stem cell biology
Inference of the cell lineage tree is very relevant to single-cell cancer genomics. For example, finding clonal populations is a common problem in cancer genomics. A clonal population corresponds to a clade (subtree) in the cell lineage tree. Whereas ScisTree2 does not find clonal populations at present, ScisTree2 calls the genotypes and also places mutations on the tree. Such information may be useful for developing applications for downstream analyses in cancer genomics.
The ability of ScisTree2 to analyze a large number of cells may be useful in large-scale cancer genomics analyses. One such analysis is identifying rare cancer subclones, which can drive disease recurrence and therapy resistance. For example, relapse of acute myeloid leukemia can be seeded by a minor subclone that represents ∼1% of the primary tumor (Wong et al. 2015). Such clinically relevant subpopulations can only be identified by analyzing thousands of cells in the tumor.
ScisTree2 may also be useful in the study of stem cell evolution because data in stem cell biology can have a large number of cells. For example, Weng et al. (2024) used large-scale single-cell lineage trees to understand the differentiation dynamics and clonal output of hematopoietic stem cells. Their benchmark data contain mtDNA mutations from 7104 cells.
New software tools for interpreting large trees for practical genomics analyses are needed in order to obtain useful biological insights into single cell evolution. Such tools are emerging. For example, scPhyloX (Wang et al. 2025) is specially designed to infer population dynamics and evolutionary trajectories from cell lineage trees with large numbers of cells.
Speeding up SPR local search
Because local search is a common strategy for phylogenetic inference, speeding up SPR local search has been previously studied in the literature. For example, Hordijk and Gascuel (2005) applied various heuristics to speed up the local SPR search for general phylogenetic inference. Whereas such heuristics can achieve significant speedup in practice, there is little theoretical guarantee about the achieved speedup. In contrast, the SPR local search algorithm in ScisTree2 has a proven theoretical speedup of one order of magnitude. Such a speedup is made possible by the ScisTree2's simple probabilistic model. This simplified model leads to a much simpler structure in its probability function than the one used in the general maximum likelihood tree inference. Working with simpler but still reasonably accurate probabilistic models may be the key for speeding up cell lineage tree inference for large amounts of data.
Methods
The original ScisTree method
To present the new ScisTree2 method, we start by briefly explaining the key aspects of the original ScisTree method. See the Supplemental Methods (Secs. S1 and S2) for a more detailed discussion of the ScisTree method.
Compared to CellPhy, ScisTree uses a simpler probabilistic model, which is designed for the IS model. ScisTree aims at maximizing the posterior probability Pr(G|D) for the given sequence reads D and some genotypes G among all genotypes G that satisfy the IS model. We say that G satisfies the IS model if there exists a potentially multifurcating phylogeny T (called a perfect phylogeny [Gusfield 1991]) where leaves are labeled by the cells (rows) in G, and each column (site) c of G labels a single branch of T such that exactly the cells below this branch are the mutants of c. See Figure 1, A and B, for an illustration. Note that ScisTree uses an approximation to the assumed maximum posterior probability model, although this approximation is an empirically good one as demonstrated by the practical performance of the method.
The posterior probability Pr(G|D) is calculated as follows. We let G(c, s) be the (binary) genotype of the individual cell c at the site s. Note that we also use the term genotype to refer to the list of genotypes at multiple sites or for multiple cells. Then,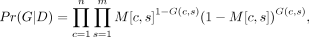 where G satisfies the IS model, n is the number of cells, and m is the number of SNV sites. Here, we assume that each genotype at a site s for a cell c is independent. Recall that M is the genotype probability matrix of size n by m that is obtained from D during genotype calling. See the Supplemental Methods (Sec. S1) for a more detailed discussion of this probabilistic model.
where G satisfies the IS model, n is the number of cells, and m is the number of SNV sites. Here, we assume that each genotype at a site s for a cell c is independent. Recall that M is the genotype probability matrix of size n by m that is obtained from D during genotype calling. See the Supplemental Methods (Sec. S1) for a more detailed discussion of this probabilistic model.
Finding 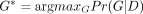 for the given genotype probability matrix M is known to be NP complete (Wu 2020). A key insight made in ScisTree is that, if the underlying rooted and binary tree T is given, then G* can be found in O(nm) time by finding the best branches to place mutations on the given T as follows. Recall that the set of mutants of a site s must form a single clade (subtree) in T. Then, for each branch ending at node v in T, we define Qs(v) as
for the given genotype probability matrix M is known to be NP complete (Wu 2020). A key insight made in ScisTree is that, if the underlying rooted and binary tree T is given, then G* can be found in O(nm) time by finding the best branches to place mutations on the given T as follows. Recall that the set of mutants of a site s must form a single clade (subtree) in T. Then, for each branch ending at node v in T, we define Qs(v) as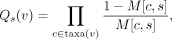 (1)
where taxa(v) is the set of leaves below v. Recall that M[c, s] denotes the probability of the wild-type genotype of the cell c and the site s. Qs(v) only concerns the cells within the subtree rooted at v and has a simple product form. Let v1 and v2 be the two children of v. Then, Qs(v) = Qs(v1)Qs(v2). Thus, we can calculate all Qs(v) in O(n) time for each site in a bottom-up way using dynamic programming. We denote the set of nodes of T as nodes(T). To obtain the maximum posterior probability at a site s, we place the mutation for s on the branch whose destination node is v* where
(1)
where taxa(v) is the set of leaves below v. Recall that M[c, s] denotes the probability of the wild-type genotype of the cell c and the site s. Qs(v) only concerns the cells within the subtree rooted at v and has a simple product form. Let v1 and v2 be the two children of v. Then, Qs(v) = Qs(v1)Qs(v2). Thus, we can calculate all Qs(v) in O(n) time for each site in a bottom-up way using dynamic programming. We denote the set of nodes of T as nodes(T). To obtain the maximum posterior probability at a site s, we place the mutation for s on the branch whose destination node is v* where 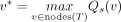 . The maximum posterior probability Pr(G*|T, D) of genotypes conditional on T is then
. The maximum posterior probability Pr(G*|T, D) of genotypes conditional on T is then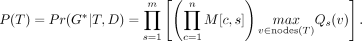 (2)
(2)
Example: Note that when placing mutations on a given tree, each site is treated independently of other sites. So, we use the site
S1 in Figure 1C to show how to find where its mutation is located on the tree in Figure 1A. We write Q1(v) as Q(v) here to simplify the notation. At leaves, 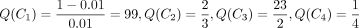 and
and 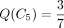 . For internal nodes,
. For internal nodes, 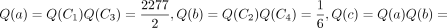
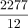 , and
, and 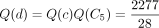 . We place the mutation for S1 at the branch right above the node a because Q(a) is the largest among all the Q values for S1.
. We place the mutation for S1 at the branch right above the node a because Q(a) is the largest among all the Q values for S1.
Local search
Local search is a popular phylogenetic inference approach. At a high level, local search implemented in ScisTree and ScisTree2 works as follows.
-
Construct an initial rooted binary tree T0 based on M. Initialize Topt ← T0, Popt ← P(T0) and Gopt ← G0.
-
Find rooted binary trees
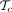 that are within one tree arrangement operation (NNI or SPR) from Topt.
that are within one tree arrangement operation (NNI or SPR) from Topt.
-
Let
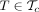 that maximizes the posterior probability P(T) for some genotypes G. If P(T) > Popt, set Topt ← T, Popt ← P(T), Gopt ← G, and go to step 2. Otherwise, stop.
that maximizes the posterior probability P(T) for some genotypes G. If P(T) > Popt, set Topt ← T, Popt ← P(T), Gopt ← G, and go to step 2. Otherwise, stop.
See the Supplemental Methods (Sec. S2) for more details on the original ScisTree method. ScisTree2 constructs the initial tree using an approach that is slightly different from that in the original ScisTree. See the Supplemental Methods (Sec. S6) for more details.
Efficient SPR local search
We call the set of trees that can be obtained by a single tree rearrangement operation (e.g., NNI or SPR) as the 1-operation neighborhood of the current tree. The original ScisTree only implements the NNI local search. The size of 1-NNI neighborhood of a tree T with n taxa is O(n). A main disadvantage of the NNI local search is that the 1-NNI neighborhood is rather restricted. This makes NNI local search easily get trapped in local optima. An alternative to NNI is (rooted) subtree prune and regraft, which is more commonly used in phylogenetic inference. An SPR operation prunes (i.e., detaches) a subtree of a tree T and regrafts (i.e., reattaches) to another branch of T. The size of 1-SPR neighborhood is O(n2), and contains the 1-NNI neighborhood as a subset. Naively, one can calculate the likelihood for each of the O(n2) trees in the 1-SPR neighborhood. However, this leads to O(n3m) running time for n cells and m SNV sites, because the time for finding the maximum posterior probability for a single tree is O(nm). This is too slow even for data of moderate size. We now present a novel algorithm that finds the best tree in the 1-SPR neighborhood in O(n2m) time.
High-level idea
We focus on computing the posterior probability P(T′) where T′ is obtained by a single SPR operation on the current tree T. This is illustrated in Figure 11. By Equation 2, we need to find the maximum over all the Q values in T′. The key observation is that we do not need to calculate all Q values of T′ from scratch. This is because T and T′ are very similar topologically. Almost all the nodes in T are also in T′. At these shared nodes, Q values in T′ either are the same as those in T or can be easily computed from the Q values in T. Careful consideration of the relations between the Q values in T and T′ leads to an efficient algorithm to find the largest value Q in T′ after some pre-processing is performed on T. We now give the details of this algorithm.
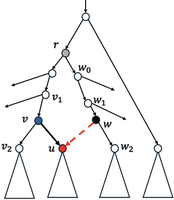
An illustration of (rooted) SPR local search. The subtree rooted at u (whose parent is v) is pruned and regrafted to the edge entering w2. Note: the lowest common ancestor of v and w2 in the tree before SPR is r.
We consider the SPR operation in Figure 11 that prunes a subtree Tu (rooted at the node u) of T and regrafts so that the parent of u is a new node w in T′, which breaks the edge (w1, w2) into two edges. We let v2 be the sibling of u and v1 be the parent of v in T. We let node r be the lowest common ancestor (LCA) of v and w2 in T. Recall that the posterior probability is the product of the maximum Qs(a) (over each node a of T) times a constant factor for each site s (Eq. 2). To simplify the notation, we omit the subscript s by writing Qs(a) as Q(a) throughout this paper with the understanding that Q(a) is for a specific site. We denote Q′(x) to be the updated Q(x) value for the node x on the new tree T′ after the SPR move shown in Figure 11.
We now show that we can find the maximum of Q′(x) values efficiently without calculating all Q′(x) values explicitly. Note that T′ has a new node w that is not in T, and v is in T but not in T′. All the other nodes are the same in T and T′. Recall that Q(a) is equal to the product of the ratios between the probability of the taxon t(b) being the mutant (allele 1) and that of being the wild type (allele 0) for each leaf b within the subtree Ta (Eq. 1). A path from the node u to the node v of a tree T is denoted as u → v, which consists of all nodes from u to v sequentially, and u and v are in the path unless otherwise stated. We have the following observation. For any node  , exactly one of the following four cases holds for Q′(x):
, exactly one of the following four cases holds for Q′(x):
-
x is a node in T that either (i) is not on the paths r → v and r → w1, or (ii) x = r. Then, Q′(x) = Q(x). This is because the SPR move does not change the set of taxa under node x, and thus Q′(x) is equal to the product of the same set of ratios as in Q(x).
-
x ≠ r and is on the path r → v1,
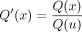 . This is again because Q(x) is the product of ratios of probabilities of genotypes within Tx being 1 versus 0; after the SPR move, the leaves under u are removed from
. This is again because Q(x) is the product of ratios of probabilities of genotypes within Tx being 1 versus 0; after the SPR move, the leaves under u are removed from 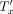 and thus are absent from the product in Q′(x).
and thus are absent from the product in Q′(x).
-
x ≠ r and is along the path r → w1,
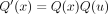 . This is because now the leaves under u are inserted in
. This is because now the leaves under u are inserted in 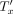 .
.
-
x = w (i.e., x is the only new node in T′ added by SPR). Then
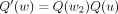 .
.
We now perform some preprocessing steps on T that allow the evaluation of multiple nodes together that fall into the same case as listed above. The preprocessing is performed only once for the current tree T before the iteration of local search starts from T.
Preprocessing of the original T
We have the following definitions for T. For each node a of T, 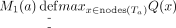 . That is, M1(a) is the maximum value of Q(x) over all nodes x within the subtree Ta (it is allowed x = a). For a leaf a0, M1(a0) = Q(a0). For an internal node a with two children a1 and a2,
. That is, M1(a) is the maximum value of Q(x) over all nodes x within the subtree Ta (it is allowed x = a). For a leaf a0, M1(a0) = Q(a0). For an internal node a with two children a1 and a2,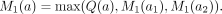 Note that the M1(a) values are for the nodes a in the original T before the SPR move. Recall that the Q(a) values are already computed. Therefore, all M1(a) values can be computed in O(n) time by performing a bottom-up traversal of T.
Note that the M1(a) values are for the nodes a in the original T before the SPR move. Recall that the Q(a) values are already computed. Therefore, all M1(a) values can be computed in O(n) time by performing a bottom-up traversal of T.
We denote the parent of node b in T as p(b). For each node a and a node b ≠ a where b ∈ nodes(Ta) (i.e., b is within the subtree Ta), define 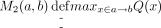 . That is, M2(a, b) is the maximum value of Q(x) over all nodes x that are on the path from a to b. M2(a, b) values can be computed in O(n2) time using the recurrence: M2(a, a) = Q(a); for b ≠ a:
. That is, M2(a, b) is the maximum value of Q(x) over all nodes x that are on the path from a to b. M2(a, b) values can be computed in O(n2) time using the recurrence: M2(a, a) = Q(a); for b ≠ a: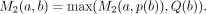 Finally, for each node a and a node b ≠ a where b ∈ nodes(Ta), we define
Finally, for each node a and a node b ≠ a where b ∈ nodes(Ta), we define
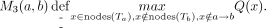
That is, M3(a, b) is the maximum value of Q(x) over all nodes x that are within Ta but not within Tb or along the path from a to b. As an example, in Figure 11, when calculating M3(r, v), the node u is excluded (because u ∈ Tv); similarly, the sibling of r is also excluded because it is not in Tr; on the other hand, w1 is included because w1 ∈ Tr,  , and
, and 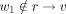 . All M3(a, b) values can be computed in O(n2) time. This is because there are O(n2) M3(a, b) values and each value takes O(1) time to compute. To see this, we first iterate over each node b; then walk the way up to the root from b; for each node a that is ancestral to b, let a1 be the child of a that is on the path a → b, and let a2 be the sibling of a1 but not on a → b. Then M3(a, a) = −∞. If a ≠ b,
. All M3(a, b) values can be computed in O(n2) time. This is because there are O(n2) M3(a, b) values and each value takes O(1) time to compute. To see this, we first iterate over each node b; then walk the way up to the root from b; for each node a that is ancestral to b, let a1 be the child of a that is on the path a → b, and let a2 be the sibling of a1 but not on a → b. Then M3(a, a) = −∞. If a ≠ b,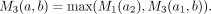
Constant-time calculation of maximum posterior probability for a single SPR move
We are now ready to show how to calculate the maximum Q′ value for T′ obtained by a single SPR move in O(1) time per SNV site. We define child(a, b) for a node a and a node b that is a descendant of a where child(a, b) is the immediate child of a that is also ancestral to b. For example, in Figure 11, child(v1, v2) = v. Now, consider a single SPR move that prunes a subtree rooted at u (u’s parent is v, v’s parent is v1, and u’s sibling is v2) to form a new node w by breaking an edge (w1, w2) as in Figure 11. With these definitions, we have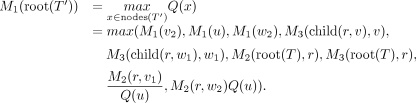 The maximum posterior probability P(T′) can be computed from M1(root(T′)) by Equation 2. Here, root(T′) is the root node of T′. Due to space limitations, we give the detailed algorithm for the SPR local search in the Supplemental Methods (Sec. S3). The correctness of the algorithm is given in the Supplemental Methods (Sec. S4).
The maximum posterior probability P(T′) can be computed from M1(root(T′)) by Equation 2. Here, root(T′) is the root node of T′. Due to space limitations, we give the detailed algorithm for the SPR local search in the Supplemental Methods (Sec. S3). The correctness of the algorithm is given in the Supplemental Methods (Sec. S4).
Time analysis
For a fixed SPR operation, the maximum Q′ values at each site s can be computed in O(1) time (Eq. 3) when the M1, M2, and M3 values are precomputed for this SPR operation. There are m sites. So, it takes O(m) time to compute the maximum posterior probability for a tree in the 1-SPR neighborhood whose size is O(n2). Precomputing M0, M2, and M3 values takes O(n2) time for each site. Therefore, finding the maximum posterior probability for a single SPR takes O(n2m) time in total.
Branch and bound speedup of SPR local search
Exhaustive search over all the O(n2) trees in the 1-SPR neighborhood can be slow when n is large. We now show that a branch and bound approach can, in practice, significantly speedup the SPR local search. We use Figure 11 for illustration. Suppose that we are to prune a subtree that is rooted at u. We need to find the best branch (w1, w2) to regraft for this subtree Tu. We let the LCA node of u and w2 be r. Let w0 be the child of r that is ancestral to w2; that is, (w1, w2) is within 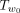 . Naively, examining all branches under
. Naively, examining all branches under 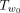 takes O(n) time because there are O(n) edges in
takes O(n) time because there are O(n) edges in 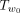 . The key idea is that an upper bound B of the maximum Q′ value for all trees that can be obtained by SPR moves that prune Tu to somewhere within
. The key idea is that an upper bound B of the maximum Q′ value for all trees that can be obtained by SPR moves that prune Tu to somewhere within 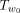 can lead to significant speed up: we can discard all SPRs that regraft Tu to be within the subtree
can lead to significant speed up: we can discard all SPRs that regraft Tu to be within the subtree 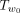 if B is no larger than the current maximum Q′ value we have found so far. Of course, to make the branch and bound approach work, this upper bound needs to be relatively
strong and also computable efficiently.
if B is no larger than the current maximum Q′ value we have found so far. Of course, to make the branch and bound approach work, this upper bound needs to be relatively
strong and also computable efficiently.
Due to space limitations, we provide the details of the branch and bound in the Supplemental Methods (Sec. S5).
Genotype calling
Once the optimal tree Topt is reconstructed, genotype calling becomes straightforward. We consider each SNV site s and find the branch b of Topt to place the single mutation for s that maximizes the Qs(v) value in Equation 2. The cells that are descendants of b are the mutants, and the rest are the wild types. We compute b by using the standard trace-back technique in dynamic programming.
Software availability
ScisTree2 source code is available on GitHub (https://github.com/yufengwudcs/ScisTree2) and as Supplemental Code. ScisTree2 is implemented in C++. We also provide a Python interface. The simulated data and scripts used to reproduce the results presented in this paper are available on Zenodo (https://zenodo.org/records/15620911).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
Research was partly supported by U.S. National Science Foundation grant IIS-1909425 (to Y.W.). We thank anonymous reviewers and Derek Aguiar for constructive comments on earlier versions of our manuscript.
Author contributions: Y.W. and T.G. conceived the study. H.Z., T.G., and Y.W. designed the methodologies. Y.W. and H.Z. implemented the methodologies. H.Z. and Y.Z. performed analyses on simulated data and the HGSOC data. H.Z., Y.Z., T.G., and Y.W. drafted and revised the manuscript. All authors reviewed and contributed to the writing of the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280542.125.
-
Freely available online through the Genome Research Open Access option.
- Received February 13, 2025.
- Accepted August 26, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








