
Fusion, fission, and scrambling of the bilaterian genome in Bryozoa
Abstract
Groups of orthologous genes are commonly found together on the same chromosome over vast evolutionary distances. This extensive physical gene linkage, known as macrosynteny, is seen between bilaterian phyla as divergent as Chordata, Echinodermata, Mollusca, and Nemertea. Here, we report a unique pattern of genome evolution in Bryozoa, an understudied phylum of colonial invertebrates. Using comparative genomics, we reconstruct the chromosomal evolutionary history of five bryozoans. Multiple ancient chromosome fusions followed by gene mixing led to the near-complete loss of bilaterian linkage groups in the ancestor of extant bryozoans. A second wave of rearrangements, including chromosome fission, then occurred independently in two bryozoan classes, further scrambling bryozoan genomes. We also discover at least five derived chromosomal fusion events shared between bryozoans and brachiopods, supporting the traditional but highly debated Lophophorata hypothesis and suggesting macrosynteny to be a potentially powerful source of phylogenetic information. Finally, we show that genome rearrangements led to the dispersion of genes from bryozoan Hox clusters onto multiple chromosomes. Our findings demonstrate that the canonical bilaterian genome structure has been lost across all studied representatives of an entire phylum, and reveal that linkage group fission can occur very frequently in specific lineages.
Advances in genome sequencing and assembly have in recent years made it possible to compare the organization of entire genomes across diverse branches of the animal kingdom. The emerging picture is one of a common set of metazoan genes, superimposed with extensive gene duplication, gene loss, and gene novelty in each lineage, plus a surprising degree of large-scale organizational similarity. Many groups of orthologous genes have been retained on the same chromosome for hundreds of millions of years and remain together in diverse and highly divergent lineages (Putnam et al. 2007, 2008; Simakov et al. 2013, 2022; Schultz et al. 2023; Lin et al. 2024). This conservation of chromosome-scale gene linkages, known as macrosynteny, is intriguing, as the selective pressures maintaining it and its significance for genome function remain largely unexplored.
The persistence of gene linkages enables the identification of orthologous chromosomes between distantly related phyla. Furthermore, it permits the reconstruction of ancestral linkage groups (ALGs): hypothesized sets of orthologous genes located on the same chromosome or chromosome arm in ancestral species (Simakov et al. 2022; Mackintosh et al. 2023a). Lineage-specific ALGs have been identified in several animal groups, including vertebrates (Sacerdot et al. 2018; Simakov et al. 2020) and Lepidoptera (butterflies and moths) (Wright et al. 2024), and the discovery of 24 ALGs shared between spiralians and deuterostomes (Simakov et al. 2022; Marlétaz et al. 2023) revealed the genome structure of the most recent bilaterian common ancestor. These highly conserved bilaterian blocks of orthologous genes are retained in species from phyla as divergent as chordates, echinoderms, molluscs, and annelids (Wang et al. 2017; Simakov et al. 2022; Marlétaz et al. 2023; Martín-Zamora et al. 2023; Lin et al. 2024), suggesting that there are substantial functional constraints to genome organization. Conservation of ALGs seems to be the norm at the phylum level, but several lineages at lower taxonomic levels have experienced genomic scrambling, including the breaking up or mixing of genes from different ALGs. These include planarians (Ivanković et al. 2024), tunicates (Plessy et al. 2024), and clitellate annelids (Lewin et al. 2024a; Schultz et al. 2024; Vargas-Chávez et al. 2024).
Despite this general conservation, fluctuations in chromosome number—often leading to ALG rearrangement—are a relatively frequent evolutionary process (White 1954; King 1995; Rice et al. 2015; Román-Palacios et al. 2021). In animals, most of these changes are attributed to inter-chromosomal rearrangements such as translocations and fusions, although they may also arise through whole genome duplication (WGD) (Orr 1990; Gregory and Mable 2005; Moriyama and Koshiba-Takeuchi 2018; Muffato et al. 2023). Following chromosome fusion, orthologous genes from different ALGs may be either maintained in separate chromosome sections or shuffled and interspersed along the chromosome in a process known as “fusion-with-mixing” (Simakov et al. 2022). Conversely, chromosomes can undergo fission, splitting into separate parts, but the division of ALGs through this process is rare (Simakov et al. 2022; Wright et al. 2024).
The interplay between the stability of orthologous gene sets and the dynamic nature of inter-chromosomal rearrangements shapes the genomic landscape. These rearrangements disrupt gene expression (Harewood and Fraser 2014), alter recombination rates (Yoshida et al. 2023), drive adaptation (Dunham et al. 2002; Coyle and Kroll 2008; Cheng et al. 2012), and contribute to macroevolutionary processes like speciation (Rieseberg 2001; de Vos et al. 2020; Mackintosh et al. 2023b; Berdan et al. 2024). This dynamic quality of chromosomal changes has made macrosynteny a powerful tool for phylogenetic studies (Steenwyk and King 2024). In particular, fusion-with-mixing events can be particularly powerful phylogenetic markers as the intermixing of genes from different ALGs is considered rare and irreversible. In light of this, macrosynteny was recently used to investigate early branching metazoan lineages (Schultz et al. 2023) and teleost evolution (Parey et al. 2023).
Even in the age of phylogenomics, when the amino acid sequences of thousands of deduced proteins are aligned and analyzed, there remains a small group of “Problematica”: taxa for which a robust phylogenetic placement remains out of reach (Jenner and Littlewood 2008; Jenner et al. 2009). Many of these taxa are rare or have few species. Yet one phylum—the Bryozoa—stands out among Problematica as highly abundant, species rich, and ecologically important (Gordon and Costello 2016; Lombardi et al. 2020), increasingly as alien invasive species (Loxton et al. 2017; Weaver et al. 2018; Pratt et al. 2022). Bryozoans are benthic, suspension-feeding, aquatic invertebrates that are important ecosystem engineers (Wood et al. 2012; Bock and Gordon 2013). They form colonies of individual zooids, generally with calcified skeletons (Zhang et al. 2021), and each zooid can be morphologically differentiated for specialized roles in feeding, brooding, or defense. Despite their unique biological traits, bryozoans and bryozoan genomes remain relatively understudied (Orr et al. 2021; Liao et al. 2023). Several chromosome-scale assemblies have recently been made available (Hoencamp et al. 2021; Wood et al. 2023; Bishop et al. 2023a,b, 2024), but these are without comparative analysis, and beyond an accepted placement within Spiralia, the phylum's phylogenetic position remains unresolved (Supplemental Fig. S1; Jenner and Littlewood 2008; Jenner et al. 2009; Khalturin et al. 2022; Liao et al. 2023).
Here, we present a comparative genomic analysis of five bryozoan genomes, focusing on genome rearrangements and chromosome evolution. Our study aims to (1) characterize the inter-chromosomal rearrangements that have occurred within the phylum Bryozoa, (2) determine how the 24 bilaterian ALGs are organized in bryozoans, (3) reconstruct the genome organization of the last common ancestor of extant bryozoans, and (4) use macrosynteny to elucidate the phylogenetic position of Bryozoa.
Results
Chromosome-scale genomes of five bryozoans
To reconstruct the evolutionary history of bryozoan chromosomes, we first assembled a data set of five chromosome-scale genomes. Four assemblies, Bugulina stolonifera (Wood et al. 2023), Cryptosula pallasiana (Bishop et al. 2023b), Membranipora membranacea (Bishop et al. 2023a), and Watersipora subatra (Bishop et al. 2024), are marine bryozoans in the class Gymnolaemata, which accounts for ∼90% of described living bryozoan species (Fig. 1A; Bock and Gordon 2013). The fifth, Cristatella mucedo (Hoencamp et al. 2021), is an outgroup belonging to the freshwater class Phylactolaemata (Taylor and Waeschenbach 2015). Four of the five assemblies were generated and made openly accessible by the Darwin Tree of Life (DToL) project (The Darwin Tree of Life Project Consortium et al. 2022), sequenced to chromosome-level using Pacific Biosciences (PacBio) HiFi long reads and high-throughput chromosome conformation capture (Hi-C) scaffolding (Wood et al. 2023; Bishop et al. 2023a,b, 2024).
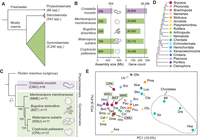
Comparative genomics of five chromosome-scale bryozoan genomes. (A) Relationships between the three classes of bryozoans. Class Phylactolaemata is the most early branching, containing just 86 species, all of which inhabit freshwater environments (Bock and Gordon 2013). Stenolaemata comprises 543 species, all of which are marine. Gymnolaemata is the most species-rich class, comprising 5240 species. (B) Genome size and gene model count of bryozoan genome assemblies used in this study. Dashed lines mark mean values. The clade containing W. subatra and C. pallasiana has larger genomes. (C) Tree of bryozoan species constructed with chromosome-scale genomes with the scallop Pecten maximus as the outgroup. P. maximus was selected as the outgroup owing to its conserved genome structure and slowly evolving sequences. The tree was constructed using the maximum likelihood method (LG + F + R6 model) with 1000 bootstrap replicates in IQ-TREE, utilizing 994 orthologous genes. The scale bar represents the number of substitutions per site. (D) Phylogenetic relationships among animal phyla, with dashed lines indicating areas of uncertainty in their placement. Phylogeny based on work of Telford et al. (2015) and Liao et al. (2023). (E) Principal component analysis (PCA) conducted using orthologous gene group counts as determined by OrthoFinder. Points are colored by their phylum, as indicated in panel D. PCs 1 and 2 explain 10.0% and 9.4% of the variance in the data set, respectively. Variance ratios for the top 30 principal components are available as Supplemental Figure S2. Species abbreviations: (Ava) Adineta vaga, (Aqu) Amphimedon queenslandica, (Aru) Asterias rubens, (Bfl) Branchiostoma floridae, (Bmi) Bolinopsis microptera, (Cel) Caenorhabditis elegans, (Cin) Ciona intestinalis, (Cte) Capitella teleta, (Dme) Drosophila melanogaster, (Dpu) Daphnia pulex, (Dre) Danio rerio, (Gfa) Galaxea fascicularis, (Hmi) Hofstenia miamia, (Hro) Helobdella robusta, (Hsa) Homo sapiens, (Lan) Lingula anatina, (Llo) Lineus longissimus, (Lva) Lytechinus variegatus, (Nve) Nematostella vectensis, (Obi) Octopus bimaculoides, (Ofu) Owenia fusiformis, (Pau) Phoronis australis, (Pma) Pecten maximus, (Pvu) Patella vulgata, (Sko) Saccoglossus kowalevskii, (Sma) Schistosoma mansoni, (Sme) Schmidtea mediterranea, (Sro) Symsagittifera roscoffensis, (Tad) Trichoplax adhaerens, (Tca) Tribolium castaneum, and (Xtr) Xenopus tropicalis.
We performed comprehensive gene prediction using the BRAKER3 pipeline (Gabriel et al. 2024), utilizing transcriptomic data as supporting evidence (Fig. 1B; Supplemental Tables S1–S3). Benchmarking Using Single-Copy Orthologs (BUSCO) (Simão et al. 2015; Manni et al. 2021) reveals a high degree of completeness in gene predictions as evidenced by completeness scores of 86.6% to 97.0% from the Metazoa Odb10 database (Supplemental Table S1), which consists of a set of genes that is near universal in metazoans (Manni et al. 2021). Given that accurate phylogenetic relationships are key to inferring chromosome rearrangement events, we built a maximum likelihood phylogenetic tree of the five bryozoan species using gene models obtained from these chromosome-scale genomes (Fig. 1C). This tree is consistent with recent phylogenetic analyses using mitochondrial and ribosomal data (Orr et al. 2022), placing the four gymnolaemates in a monophyletic group as sister to C. mucedo. Principal component analysis of orthologous gene counts finds bryozoans to have a gene set broadly similar to that of other lophotrochozoans (Fig. 1D,E; Supplemental Tables S4, S5).
Massive chromosome rearrangements within Bryozoa
Haploid chromosome number in our bryozoan data set ranges from eight in C. mucedo to 12 in C. pallasiana. To investigate how the chromosomes of each species are related and understand whether large genome restructuring events have occurred within the phylum Bryozoa, we used OrthoFinder (Emms and Kelly 2019) to identify 4242 single-copy orthologs and mapped the position of these orthologs within each genome. We then constructed synteny maps, which we displayed as ribbon plots (Fig. 2) and Oxford dot plots (Supplemental Fig. S3). Throughout our analysis of chromosome rearrangements, we apply the algebraic symbol conventions established by Simakov et al. (2022), wherever applicable. Specifically, we use a triple bar for one-to-one relationships, also known as syntenic equivalences (≡); a tensor for fusion with mixing (⊗); a dot product for end-to-end fusion (●); a down arrow for centric insertion (↘); and an addition symbol for fission (+) (Fig. 2A). However, although these descriptions are effective for taxa with relatively conserved genomes, we found that the highly derived nature of bryozoan genomes resulted in many chromosomes not fitting clearly into these predefined categories. Furthermore, the notation may become excessively complex and confusing when addressing chromosomes that have undergone numerous ALG fusions and have intricate evolutionary histories. We therefore also utilize narrative descriptions, especially for highly derived chromosomes.
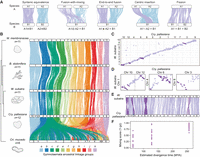
Chromosome rearrangement in gymnolaemate and phylactolaemate bryozoans. (A) Schematic representations of types of inter-chromosomal rearrangement events. White bars represent chromosomes, and colored lines connect orthologous genes; the same color line represents genes in the same ancestral linkage group (ALG). (B) Chromosome-scale gene linkage between five bryozoan species revealed by ribbon plots. Horizontal bars represent chromosomes, and vertical lines connect the genomic position of orthologous genes in each genome. Plot shows 4242 single-copy orthologous genes (average, 386 genes per linkage group). Chromosomes connected by many orthologs are inferred to be homologous (i.e., orthologous chromosomes). In gymnolaemate species, 11 ALGs a to k are highly conserved, although the chromosome containing ALG a underwent a fission event to form Chr 10 and Chr 12 in C. pallasiana. These ALGs are not conserved in the phylactolaemate species C. mucedo. Links between chromosomes sharing five or fewer genes between chromosomes are removed for clarity but can be seen in the Oxford dot plots. (C) Oxford dot plot revealing chromosome-scale gene linkage between the two most closely related species in our data set, C. pallasiana and W. subatra. Each axis represents the entire length of the genome of one species. Gray lines separate chromosomes. Each point represents a pair of orthologs placed by their ordinal position in each genome. Strings of orthologs perpendicular to the main diagonal axis reveal clear intra-chromosomal inversions (dark purple). (D) Single-chromosome Oxford dot plots for C. pallasiana and W. subatra chromosomes with clear intra-chromosomal inversions (dark purple). (E) Ribbon plot for C. pallasiana and W. subatra highlighting intra-chromosomal inversions (dark purple). (F) Microsynteny mixing score versus divergence time for gymnolaemate bryozoan chromosomes across species. Comparisons are grouped by estimated divergence time: <100 million years ago (MYA), C. pallasiana versus W. subatra; 100–150 MYA, C. pallasiana and W. subatra versus B. stolonifera; >250 MYA, C. pallasiana, W. subatra, and B. stolonifera versus M. membranacea. Each point represents a chromosome. The microsynteny mixing score measures how genes are shuffled within a chromosome by intra-chromosomal inversions (see Methods): A higher score indicates more mixing. Mixing scores increase with phylogenetic distance.
We used the chromosomal position of orthologous genes to identify orthologous chromosomes among the four gymnolaemate species. There is complete syntenic equivalence among three gymnolaemate bryozoans (B. stolonifera, M. membranacea, and W. subatra), which all have a haploid chromosome number of 11, suggesting there have been no instances of chromosome fusion or fission (Fig. 2B). Based on gene position in these species, we assigned 11 gymnolaemate ALGs, labeled ALGs a to k (Fig. 2B). The final gymnolaemate species, C. pallasiana, has undergone one fission event to diverge from this state, with the chromosome containing gymnolaemate ALG a (M. membranacea Chr 1 ≡ B. stolonifera Chr 1 ≡ W. subatra Chr 1) splitting to form two smaller chromosomes (C. pallasiana Chr 10 + Chr 12) (Fig. 2B). Given the reported rarity of chromosome fission events in animal evolution, we verified that this is not an assembly error by generating a Hi-C contact map, which confirmed that the fission is genuine (Supplemental Fig. S4). This aside, chromosome structure is highly conserved between these species. In contrast, a lack of conservation is observed when the phylactolaemate bryozoan C. mucedo is compared. None of the 11 gymnolaemate ALGs are conserved, and most are completely dispersed and scrambled across the entire C. mucedo genome (Fig. 2B). For instance, we identified four gymnolaemate ALGs on seven of eight C. mucedo chromosomes (gymnolaemate ALGs a, b, c, and g) (Supplemental Fig. S5). This represents an almost complete loss of chromosome relationships between the two groups. Overall, genome structure is highly conserved within gymnolaemate bryozoans, but large inter-chromosomal rearrangements have resulted in highly divergent genome structures between members of Gymnolaemata and Phylactolaemata.
Macrosynteny studies over long evolutionary distances often overlook the precise order of genes, known as microsynteny (Renwick 1971; Passarge et al. 1999). This is because intra-chromosomal rearrangements like inversions rapidly alter gene order, leading to weak microsyntenic signals, which are difficult to trace between distantly related species. Within our data set, there is minimal conservation of gene order between bryozoans with the exception of the two most closely related species, W. subatra and C. pallasiana (Supplemental Fig. S3), which are estimated to have diverged 60–90 million years ago (Orr et al. 2022). Conservation of microsynteny between these species permitted us to investigate the process by which intra-chromosomal rearrangements occur. By generating Oxford dot plots for whole genomes (Fig. 2C) and individual chromosomes (Fig. 2D), we identified six clear cases of large intra-chromosomal inversions between these two species (e.g., W. subatra Chr 1 vs. C. pallasiana Chr 12). We noticed many inversions with a complete lack of intermixing, suggesting that these inversions involved entire chromosome arms (e.g., W. subatra Chr 8 vs. C. pallasiana Chr 3) (Fig. 2D,E). We developed a microsynteny mixing score (see Methods) to quantify the extent of intra-chromosomal gene shuffling. Between the closely related species W. subatra and C. pallasiana, the mixing score varies greatly between chromosomes, suggesting genes are not shuffled at equal rates on different chromosomes (Fig. 2F). In addition, the mixing score increases with estimated divergence time in our data set (Fig. 2F; Supplemental Tables S6, S7). Overall, the mixing of genes within a chromosome increases with the divergence time between species and occurs at least in part owing to large inversions.
Near-complete disruption of bilaterian ALGs in Bryozoa
To facilitate synteny analysis using bilaterian ALGs, we created a new pipeline, SyntenyFinder (see Methods). SyntenyFinder integrates OrthoFinder and RIdeogram and is capable of conducting large-scale synteny comparisons. We first validated our synteny analysis methods using bilaterians with established chromosome relationships. We applied the bilaterian ALG chromosome assignment to the amphioxus Branchiostoma floridae and found it to be consistent with previous work (Simakov et al. 2022), identifying bilaterian ALG fusions on Chr 1 (A1 ⊗ A2), Chr 2 (C1 ⊗ J2), Chr 3 (C2 ● Q), and Chr 4 (O1 ● I). This result confirms the accuracy of our ortholog identification and macrosynteny methodologies.
We next investigated how bryozoan chromosomes relate to the conserved bilaterian genome organization. Using only the bryozoan data, it remains uncertain whether the Gymnolaemata, Phylactolaemata, or neither is similar to the ancestral state of bilaterians. To resolve this, in addition to the amphioxus B. floridae (Simakov et al. 2020), we compared the macrosynteny of bryozoans with two other bilaterians with conserved genome organization: the nemertean Lineus longissimus (Kwiatkowski et al. 2021) and the mollusc Pecten maximus (Kenny et al. 2020). Both L. longissimus and P. maximus belong to the Lophotrochozoa, the same major protostome clade as bryozoans, and the deuterostome B. floridae serves as an outgroup. We generated a data set of 995 single-copy orthologs and associated each ortholog with one of the 24 bilaterian ALGs previously described (Supplemental Data S1; Simakov et al. 2022). Synteny maps show that, despite their divergence at the base of the Bilateria, the genome organizations of B. floridae, P. maximus, and L. longissimus exhibit high levels of similarity, having largely preserved the bilaterian ALGs (Fig. 3; Supplemental Fig. S6; Supplemental Table S8). Our analysis revealed that P. maximus and L. longissimus share four fusion events that have also been recorded in annelids and brachiopods (J2 ⊗ L, O1 ⊗ R, O2 ⊗ K, and H ⊗ Q) (Martín-Zamora et al. 2023; Lewin et al. 2024b), suggesting that they are ancestral to lophotrochozoans. In addition, we detected several lineage-specific fusion events. Specifically, we identified a fusion on Chr 2 (B2 ⊗ M) in P. maximus and a fusion on Chr 2 (C1 ⊗ G) in L. longissimus. Overall, the deuterostome B. floridae and lophotrochozoans P. maximus and L. longissimus retain most bilaterian ALGs alone on single chromosomes, and each has a few lineage-specific fusion events. These changes are modest, considering that these species have been diverging for more than half a billion years since the last common ancestor of bilaterians.
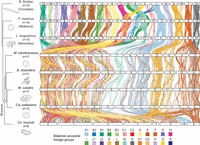
Extensive fusion-with-mixing of bilaterian ALGs in bryozoans. Chromosome-scale gene linkage between five bryozoan species and three outgroup bilaterians: a nemertean, a mollusc, and a chordate. Horizontal bars represent chromosomes. Vertical lines connect the genomic position of orthologous genes in each genome. Lines are colored by the 24 bilaterian ALGs (Simakov et al. 2022). These bilaterian ALGs are highly conserved in species ranging from the amphioxus B. floridae to the scallop P. maximus and the nemertean L. longissimus but are extensively scrambled in bryozoans. Species in the diagram are arranged to best illustrate patterns of genomic evolution, given that more information is gained from the nemertean–gymnolaemate comparison than from the nemertean–phylactolaemate comparison.
Like B. floridae, P. maximus, and L. longissimus, bryozoan genomes contain genes from all 24 bilaterian ALGs. However, in contrast to their highly conserved state in other lophotrochozoans and deuterostomes, these bilaterian ALGs have been extensively combined and mixed in the bryozoan lineage (Fig. 3; Supplemental Fig. S6; Supplemental Table S9). We found that genes from 18 out of the 24 bilaterian ALGs (75%) have been combined and mixed across four gymnolaemate chromosomes, indicating that gymnolaemate bryozoans underwent substantial irreversible fusion-with-mixing events followed by fragmentation of a single, ancestrally fused chromosome into four distinct ones. We also detect other bryozoan-specific fusions: ALGs A1 and C2 initially fused (A1 ⊗ C2) and then divided into two chromosomes (e.g., M. membranacea Chr 6 and Chr 8); ALG P joined with the lophotrochozoan fusion K ⊗ O2 (P ⊗ K ⊗ O2) and later separated into two chromosomes (e.g., M. membranacea Chr 9 and Chr 10); and ALGs F and N fused (F ⊗ N) and subsequently split into two chromosomes (e.g., M. membranacea Chr 5 and Chr 11). One chromosome contains only ALG M genes (e.g., M. membranacea Chr 7), although various genes from ALG M were translocated to other chromosomes. The identification of the same chromosome structure across M. membranacea, B. stolonifera, W. subatra, and C. pallasiana confirms that the observed genome scrambling is not caused by assembly errors but rather represents genuine evolutionary divergence. We also found that the C. mucedo genome arrangement, although unique from other bryozoans, similarly does not preserve the bilaterian ALGs on separate chromosomes. Massive rearrangements have independently taken place within the lineage leading to C. mucedo, resulting in the dispersion of bilaterian ALGs throughout its genome (Fig. 3). Overall, the genome organizations of the two classes of bryozoans studied here are distinct from each other and highly divergent from the bilaterian ancestral state.
The presence of several ALGs on two or four chromosomes in gymnolaemate bryozoans led us to question whether the splitting of ALGs was because of WGD or chromosome duplications rather than chromosome fission. We tested this using two complementary methods, each using Metaphire vulgaris, an annelid with a known WGD (Jin et al. 2020), as a positive control. We first tested for WGD using synonymous substitution per site (Ks) distributions for paralogs within each genome. The large peak in the distribution for M. vulgaris is caused by its WGD, and the absence of similar large peaks within the Ks distributions of gymnolaemate bryozoans does not support the hypothesis that a recent WGD occurred in this group (Supplemental Fig. S7). There may be very shallow peaks at higher Ks values (Ks = 3 to 4) in the gymnolaemate bryozoans, which could indicate ancient WGD in this lineage, but the signal is very weak (Supplemental Fig. S7).
Given this uncertainty, we then used MCScanX (Wang et al. 2012) to identify and align collinear blocks of genes within each genome. The presence of duplicated gene blocks on chromosomes with genes from the same ALG would suggest that they were formed by duplication rather than fission. Accordingly, strong pairwise relationships were identified between duplicated chromosomes in M. vulgaris. However, in gymnolaemate bryozoans, this analysis revealed very few such homologous blocks of genes and no pairwise chromosomal relationships in any species, which provides no evidence for WGD or whole-chromosome duplication events (Supplemental Fig. S8). Finally, we used a duplicate genes classifier (Wang et al. 2012) to determine the origin of duplicates found in the genomes. Although 23.5% of M. vulgaris genes are derived from WGD/segmental duplication events, only 0.4%–3.6% of genes in the four gymnolaemate species fall into this category (Supplemental Table S10). This again lends no support to the idea of a WGD in gymnolaemate bryozoans. In the phylactolaemate C. mucedo, we identified several sections of homology between C. mucedo chromosomes (e.g., Chr 1 with Chr 2, Chr 1 with Chr 5, and Chr 4 with Chr 6) (Supplemental Fig. S8) and several very small Ks peaks (Supplemental Fig. S7), suggesting possible segmental duplications in this lineage. Overall, we failed to find evidence that the presence of ALGs on two or four chromosomes in gymnolaemate bryozoans is the product of WGD or large-scale chromosomal duplications, although this cannot be conclusively ruled out.
The last common ancestor of extant bryozoans had a highly rearranged genome
Because the Gymnolaemata and Phylactolaemata have such distinct genome arrangements, we sought to uncover the genomic structure of their ancestor, the last common ancestor of extant bryozoan lineages. Did it maintain the general bilaterian organization or had rearrangements already occurred? We leveraged the conserved gene linkages found in modern genomes to reconstruct the ancestral state by searching for fusion and fission events that are shared by both gymnolaemate and phylactolaemate species. Because these two classes diverged at the base of the extant Bryozoa, inter-chromosomal rearrangements found in both sets of species can be inferred to have been present in their ancestor. This implicitly assumes that they did not arise multiple times independently, but the complex nature of the shared rearrangements makes that scenario highly improbable. This analysis revealed the likely presence of six linkage groups in the ancestor of living bryozoans, suggesting an ancestral karyotype of six chromosomes (designated here as α, β, γ, δ, ε, and ζ) (Fig. 4A,B). Notably, bryozoan ALGs α and β comprise 10 and eight bilaterian ALGs, respectively, indicating numerous events of sequential fusion-with-mixing (Fig. 4C; Supplemental Fig. S9; Supplemental Table S11). Thus, chromosomal rearrangements had fused and shuffled bilaterian ALGs extensively, even before the extant classes of bryozoans diverged.
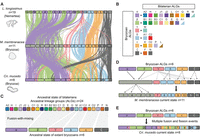
Reconstruction of ancestral genome architecture of extant bryozoans. (A) Chromosome-scale gene linkage between L. longissimus, M. membranacea, and C. mucedo. Links between orthologs are colored by the six bryozoan ALGs. Bryozoan ALGs are inferred from sets of genes that are colocated on the same chromosome (region) in both gymnolaemate and phylactolaemate bryozoans. L. longissimus is used as an outgroup because it largely preserves the 24 bryozoan ALGs on separate chromosomes but possesses the four fusion events that are shared by lophotrochozoans (J2 ⊗ L, O1 ⊗ R, O2 ⊗ K, and H ⊗ Q). (B) Composition of bryozoan ALGs by bilaterian ALGs, alongside the reconstructed bryozoan ancestral state (six chromosomes: α, β, γ, δ, ε, and ζ). Triangles indicate the inclusion of several genes from the bilaterian ALG, but the majority of genes from that ALG are elsewhere. (C) Reconstruction of the genome rearrangements that formed the bryozoan ancestral state. The top row shows bilaterian ALGs, and the bottom row shows bryozoan ALGs. (D) Reconstruction of the genome rearrangements that led from the bryozoan ancestral state to the extant gymnolaemate genomes. Solid black arrows indicate fusion-with-mixing. Solid gray arrows indicate dispersal around the genome. Dashed arrows indicate chromosome fission. (E) Reconstruction of the genome rearrangements that led from the bryozoan ancestral state to the extant phylactolaemate genomes.
Based on this ancestral state of extant bryozoans, we reconstructed the events leading to the observed genome structures in contemporary species (Fig. 4D). In the gymnolaemates, the chromosomes containing ALGs α and β underwent fusion-with-mixing (α ⊗ β) to create one enormous chromosome containing genes from 18 bilaterian ALGs. This chromosome then underwent sequential fission events to form four daughter chromosomes containing these 18 bilaterian ALGs (e.g., M. membranacea Chr 1 to Chr 4). Meanwhile, bryozoan ALGs δ, ε, and ζ each underwent a single fission event to form two separate chromosomes. ALG γ was maintained alone on one chromosome (Chr 7), but genes from this ALG also dispersed to all other chromosomes (Fig. 4D). The C. mucedo genome exhibits a structure more derived from the ancestral state, characterized by numerous fission and fusion events with limited mixing, resulting in chromosomes consisting of distinct segments derived from different bryozoan ALGs (Fig. 4E). Overall, the bryozoan common ancestor had a highly derived genome, and lineage-specific fusion and fission events have further modified the genomes of both gymnolaemate and phylactolaemate bryozoans.
Shared derived chromosomal rearrangements between bryozoans and brachiopods
Recent studies have proposed the application of macrosynteny analysis to phylogeny inference, arguing that the infrequency and irreversibility of fusion-with-mixing allows these events to serve as phylogenetic markers (Parey et al. 2023; Schultz et al. 2023; Steenwyk and King 2024). In light of the contentious debate surrounding the phylogenetic position of bryozoans, we sought to determine whether macrosynteny could offer insights in this regard. Two contrasting hypotheses exist: (1) the Lophophorata hypothesis, which proposes a close relationship to phoronids and brachiopods (Nesnidal et al. 2013; Luo et al. 2018; Laumer et al. 2019; Marlétaz et al. 2019), and (2) the Polyzoa hypothesis, which proposes closer affiliation to Entoprocta and Cycliophora (Supplemental Fig. S1; Hejnol et al. 2009; Kocot et al. 2017; Khalturin et al. 2022). Although no chromosome-scale genomes are available for the Cycliophora, Entoprocta, or Phoronida species, we recently sequenced a chromosome-level genome for the brachiopod Lingula anatina (Lewin et al. 2024b). We first validated the quality of this assembly using genome statistics and found it to be highly contiguous (Supplemental Tables S12, S13), and then we verified that the chromosome number was supported by cytological karyotype studies (n = 10) (Nishizawa et al. 2010). Next, we checked the assembly by comparing chromosome structure to two species with highly conserved genomes: P. maximus and L. longissimus (Supplemental Fig. S10). The recovery of many bilaterian ALGs on singular chromosomes further supports that the genome is correctly assembled (Supplemental Fig. S10).
We then conducted a macrosynteny comparison between the bryozoan M. membranacea and the brachiopod L. anatina to test for the existence of shared, derived, rare genomic changes in the form of chromosome fusion-with-mixing events. M. membranacea is taken as representative of Gymnolaemata because, other than the species-specific chromosome fission event in C. pallasiana, the genome structures of the four gymnolaemates are identical in terms of ALG composition so the same result is obtained whichever species is used. We identified nine fusion-with-mixing events shared between L. anatina and M. membranacea (Fig. 5A,B). Four of these events (J2 ⊗ L, O1 ⊗ R, O2 ⊗ K, and H ⊗ Q) are also shared with annelids, nemerteans, and molluscs (Fig. 3; Simakov et al. 2022; Martín-Zamora et al. 2023). The remaining five fusion-with-mixing events are exclusive to brachiopods and bryozoans, making them shared derived characteristics unique to these groups (Fig. 5C, marked by ⊗ occurring outside of parentheses). This suggests that bryozoans and brachiopods are closely related, having inherited these changes from a recent common ancestor, which supports the Lophophorata hypothesis (Fig. 5D, scenario type A). Fusion-with-mixing events are essentially irreversible in evolution, but convergent evolution cannot be ruled out (Steenwyk and King 2024). The changes could also be attributed to convergent evolution (Fig. 5E,F, type B) or a mixture of inheritance from a common ancestor and convergent evolution (Fig. 5G, type C), but inheritance from a recent common ancestor is the most parsimonious explanation, requiring the fewest fusion-with-mixing events.
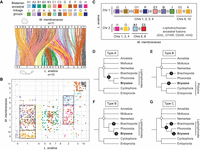
Shared derived chromosome features between bryozoans and brachiopods support the Lophophorata hypothesis. (A) Chromosome-scale gene linkage between the bryozoan M. membranacea and the brachiopod L. anatina. Horizontal bars represent chromosomes. Vertical lines connect the genomic position of orthologous genes in each genome. Lines are colored by bilaterian ALGs. (B) Oxford dot plot showing shared ALG fusion events between L. anatina and M. membranacea. Each point represents a pair of orthologs placed by their ordinal position in each genome, colored by their bilaterian ALG. Black boxes highlight shared fusion-with-mixing events, in which two ALGs appear together on the same chromosome in both species. (C) Representation of the nine ALG fusion-with-mixing events shared between L. anatina and M. membranacea. Colored squares represent ALGs; black lines represent chromosomes; and ⊗ marks ALG fusion-with-mixing events. ALGs J2, L, O1, R, C1, G, I, O2, and K are colocated on Chr 1 in L. anatina. ALGs J2, L, O1, R, C1, G, and I (Chrs 1, 2, 3, 4) and O2 and K (Chrs 9, 10) are also colocated in M. membranacea. Thus, J2 ⊗ L ⊗ O1 ⊗ R ⊗ C1 ⊗ G ⊗ I and O2 ⊗ K represent seven fusion-with-mixing events shared by both species. ALGs H, Q, A1, and C2 are colocated on Chr 2 in L. anatina. H and Q (Chrs 1, 2, 4) and A1 and C2 (Chrs 6, 8) are also colocated in M. membranacea. Accordingly, H ⊗ Q and A1 ⊗ C2 represent two additional fusion-with-mixing events shared by both species, for a total of nine. We note that several spiralian phyla share four of these events (“lophotrochozoan ancestral fusions”), represented by ⊗ within parentheses, although their timing remains uncertain. If these events are present in all spiralians, for instance, they may not be useful phylogenetic markers for the position of bryozoans. This leaves five confirmed derived fusion-with-mixing events (highlighted in bold) shared between bryozoans and brachiopods but not present in molluscs and annelids. These events suggest a close evolutionary relationship between bryozoans and brachiopods and are marked by ⊗ occurring outside of parentheses: (J2 ⊗ L) ⊗ (O1 ⊗ R) ⊗ C1 ⊗ G⊗I and A1 ⊗ C2. (D–G) Visual representation of four possible evolutionary scenarios that could give rise to the five derived fusion-with-mixing events shared between bryozoans and brachiopods (types A, B, and C). An open circle marks the base of the Lophotrochozoa. Black circles mark chromosome fusion-with-mixing events. (D) Scenario type A: Brachiopods, phoronids, and bryozoans are closely related, forming the Lophophorata. The five fusion-with-mixing events shared between bryozoans and L. anatina occurred in the ancestor of the Lophophorata. Total number of fusions required: five. (E) Scenario type B-1: Bryozoans, Cycliophora, and Entoprocta are closely related, forming the Polyzoa. Bryozoans are distantly related to brachiopods. The five fusion-with-mixing events shared between bryozoans and L. anatina occurred independently in bryozoans and brachiopods. Total number of fusions required: 10. (F) Scenario type B-2: Brachiopods, phoronids, and bryozoans are closely related, forming the Lophophorata. The five fusion-with-mixing events shared between bryozoans and L. anatina occurred independently in bryozoans and brachiopods. Total number of fusions required: 10. (G) Scenario type C: Brachiopods, phoronids, and bryozoans are closely related, forming the Lophophorata. Some of the five fusion-with-mixing events shared between bryozoans and L. anatina occurred in the ancestor of Lophophorata, whereas some occurred independently in bryozoans and brachiopods. Total number of fusions required: six to nine.
Under the assumption that fusion-with-mixing events are irreversible, the type A scenario would be contradicted if any bryozoan, phoronid, or brachiopod were discovered without the described fusion-with-mixing events. To test this, we examined whether the same fusion events are present in C. mucedo, the most divergent bryozoan in our data set. Despite C. mucedo having a highly divergent chromosome organization, the ALGs involved in all five derived fusion-with-mixing events shared by M. membranacea and L. anatina also occur together on the same chromosomes in C. mucedo (Supplemental Fig. S11). Altogether, we observed nine shared fusion events between bryozoans and brachiopods, with at least five being derived fusion-with-mixing events and thus considered potential phylogenetic markers. These events are notably absent in other lophotrochozoan phyla including molluscs, annelids, and nemerteans, which supports a close relationship between bryozoans and brachiopods and therefore the Lophophorata hypothesis. In the future, sequencing a phoronid genome to chromosome level will be a key test of this hypothesis, revealing whether the same fusions are found in this phylum.
Genome structure and Hox gene evolution
In bilaterians, Hox genes are essential for body patterning and cell type specification (Hubert and Wellik 2023), a role facilitated by their organization into tight clusters (Akam 1989; Duboule and Dollé 1989; Graham et al. 1989). We explored whether extensive genome scrambling has impacted Hox cluster organization in bryozoans. We annotated the Hox genes of the five bryozoan species (Supplemental Fig. S12; Supplemental Data S2) and identified extensive gene loss in the bryozoan Hox gene set, which lacks Hox1, Hox3, Scr, Antp, Lox2, and Post1 in all five species. The remaining Hox genes are distributed across two separate chromosomes in every assembly (Fig. 6A). In bilaterians such as P. maximus and L. longissimus, the Hox cluster is associated with the bilaterian ALG B2. This ALG is fragmented across four chromosomes (five in C. pallasiana) in each bryozoan genome, as a result of successive fusion and fission events (Fig. 6B). The main Hox cluster is located on orthologous chromosomes containing ALG B2 genes in each bryozoan genome assembly, and Hox genes separated from the cluster are also on a different orthologous chromosome that contains ALG B2 genes (Fig. 6C). This evidence suggests that the presence of Hox genes across two chromosomes in bryozoans is a unique case of partial cluster tandem duplication (Hox4, Lox5, Lox4, and Post2 genes) followed by separation owing to chromosome fission (Fig. 6D).
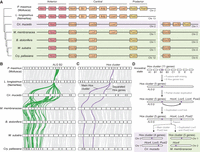
Evolution of the bryozoan Hox cluster. (A, left) Representation of the main Hox cluster of bryozoans and two outgroups: the nemertean L. longissimus and the mollusc P. maximus. Colored rectangles represent Hox genes. Horizontal black lines represent chromosomes. (Right) Separate Hox genes located on different chromosomes. (B) Positions of ALG B2 genes (green) in bryozoan genomes. (C) Positions of Hox genes (purple) in bryozoan genomes. (D) Representation of the evolutionary events that resulted in observed bryozoan Hox gene arrangements. In the bryozoan ancestral lineage, chromosomes containing bilaterian ALGs A1, B1, B2 (including the Hox cluster), B3, C1, D, E, and G underwent sequential fusion events to form bryozoan ALG β; the Hox cluster underwent a partial duplication, copying genes Hox4, Lox5, Lox4, and Post2; and the duplicated Lox4 gene was subsequently lost. In the lineage leading to C. mucedo (Phylactolaemata), the main cluster Post2 was lost, and the chromosome containing ALG β underwent fission, leaving the main cluster on Chr 2 and duplicated genes on Chr 1. In the lineage leading to M. membranacea (Gymnolaemata), the duplicated Lox5 and Post2 were lost, and the chromosome containing ALG β underwent fission, leaving the main cluster on Chr 1 and duplicated genes on Chr 2.
Given the critical dependence of Hox gene regulation on genomic organization, we hypothesized that physical separation might alter gene expression. To investigate this, we analyzed the expression of bryozoan Hox paralogs located on different chromosomes using published RNA sequencing (RNA-seq) data (Supplemental Fig. S13; Supplemental Table S14). In C. mucedo, both the Hox4b and Lox5b genes separated from the main cluster are expressed more highly than their main cluster counterparts Hox4a and Lox5a (paired t-tests P = 0.0009 and P = 0.0001, respectively) (Supplemental Fig. S13). Therefore, the physical separation of Hox cluster genes by genome rearrangements is associated with differences in expression levels.
Discussion
Our study reveals widespread rearrangements to bryozoan chromosomes that have extensively fragmented the 24 ALGs inherited by all bilaterians. This presents a stark contrast to reports of ALG conservation from other spiralians (Wang et al. 2017; Martín-Zamora et al. 2023), deuterostomes (Lin et al. 2024), and even nonbilaterians like cnidarians and ctenophores (Simakov et al. 2022; Schultz et al. 2023). We distinguish two distinct periods in which the bryozoan genome was drastically remodeled: the first generating the derived bryozoan ancestral state of just six ALGs from the 24 bilaterian ALGs, largely by chromosome fusion events, and the second independently modifying this state further in different bryozoan classes, including ALG fission events.
Three aspects of bryozoan genomes stand out as particularly remarkable. First is the magnitude of chromosome fusions present, with genes from 18 out of 24 bilaterian ALGs located together on the same chromosomes. Second, the occurrence of these massive fusion events across a phylum is exceptionally rare. Unlike dipterans, tunicates, and clitellate annelids, in which such extensive fusion events are specific to certain lineages within phyla (Zdobnov et al. 2005; Simakov et al. 2022; Lewin et al. 2024a; Plessy et al. 2024; Schultz et al. 2024; Vargas-Chávez et al. 2024), our data indicate that these rearrangements are present across the entire extant bryozoan phylum. This suggests that massive rearrangements had already occurred by the time of the last common ancestor of living lineages. However, we note that much of bryozoan diversity is represented only in the fossil record. Six of the seven orders of Stenolaemata are long extinct, and <40% of bryozoan genera have living representatives (Taylor and Waeschenbach 2015), and ∼70% of described bryozoan species are fossil (Gordon and Costello 2016). Third, many ALGs in bryozoans have been separated by ALG fissions. Major studies on animal chromosome evolution suggested that a general rule in metazoans is that ALGs are combined but not broken up (Simakov et al. 2022; Wright et al. 2024). In contrast, we identified numerous fissions occurring in the common ancestor of the four gymnolaemate species around 350 million years ago (Orr et al. 2022), with subsequent, lineage-specific fission in C. pallasiana <90 million years ago (Orr et al. 2022). This indicates a prolonged period during which fissions were evolutionarily tolerated, challenging the notion that they occur in brief, time-limited periods followed by evolutionary stasis (Simakov et al. 2022).
Why have bryozoans specifically undergone radical ALG fissions whereas many animal phyla retained the conserved ancestral state? Although our study does not offer a definitive answer, we can apply Mayr's classical framework (Mayr 1961) to outline the elements of a possible explanation. First, we must identify the mechanism that generated these rearrangements (the proximate cause). Second, we must understand why these rearrangements conferred a fitness advantage and were thus favored by selection (the ultimate cause). The high level of rearrangement in bryozoans could be caused by either (1) an increase in the rate of rearrangement events (the proximate cause), (2) an increase in the rate of fixation of rearrangement events (the ultimate cause), or (3) both. Loss of DNA repair machinery (Vargas-Chávez et al. 2024), transposable element invasions (Ahola et al. 2014; Höök et al. 2023), or environmental mutagenic agents (Schultz et al. 2024) have been proposed as potential proximate causes in other systems, although additional studies are needed to test whether these were contributors in bryozoans.
The ultimate cause of large-scale genome restructuring events is particularly intriguing from an evolutionary standpoint. One possibility is that rearrangements are selectively neutral or weakly deleterious but are fixed at an increased rate owing to genetic drift (Wright 1941; Mackintosh et al. 2023c). Notably, bryozoans are colonial and have an asexual stage in which they reproduce by budding (Schwaha 2020), and genetic drift and mutational load are elevated in asexual populations (Haag and Roze 2007). Alternatively, rearrangements may be adaptive and favored by positive selection, as has been suggested for loss of genome structure during the marine–freshwater transition in clitellate annelids (Sun et al. 2021; Lewin et al. 2024a; Schultz et al. 2024; Vargas-Chávez et al. 2024). The transition to obligate colonialism is a significant evolutionary innovation in bryozoans (Schwaha et al. 2020) that correlates with the timing of genome restructuring, but further work is required to understand whether this or other bryozoan adaptations such as biomineralization (Taylor et al. 2015) were facilitated by chromosomal rearrangement events.
A third possibility is that increased rate of fixation of rearrangements results from the relaxation in bryozoans of strong evolutionary pressures that have been maintained in other lineages. The role of conserved macrosyntenic gene linkages in genome functioning remains largely unknown, but their high-fidelity maintenance in diverse lineages for more than half a billion years (Simakov et al. 2022) implies at least some importance. One idea is that this is because of long-range interactions between distant elements on the same chromosome, which means that gene linkages are significant to gene expression (Tolhuis et al. 2002; Miele and Dekker 2008; Dean 2011). A change in the relative importance of mechanisms of gene regulation may therefore reduce the emphasis on long-range interactions and render conserved genome organization obsolete. Our work highlights the need for studies relating gene regulation to genome organization, which may be key to understanding the ultimate causes of the evolution of highly derived genomic architectures.
Having elucidated the organization of bryozoan genomes, we questioned whether macrosynteny could resolve their phylogenetic position. Substantial evidence has been presented supporting both their association with Phoronida and Brachiopoda (Lophophorata) (Nesnidal et al. 2013; Luo et al. 2018; Laumer et al. 2019; Marlétaz et al. 2019) and with Entoprocta and Cycliophora (Polyzoa) (Hejnol et al. 2009; Kocot et al. 2017; Khalturin et al. 2022). Our study identifies nine ALG fusion-with-mixing events shared between bryozoans and brachiopods. Five of these are confirmed as derived characters because they are not present in closely related lophotrochozoans like nemerteans, annelids, and molluscs, supporting the Lophophorata topology. If the other four fusions are not found in Entoprocta and Cycliophora and are therefore also derived characters, then the presence of at least nine shared derived fusion events in brachiopods and bryozoans would almost rule out convergence, becoming a conclusive piece of evidence in this ongoing debate. The future availability of chromosome-scale genomes from these groups is expected to provide clarity. Looking further, evidence from macrosynteny is likely to become increasingly prominent in discussions concerning phylogenetically challenging clades.
Overall, although macrosyntenic gene linkages are conserved across diverse bilaterian lineages, members of the phylum Bryozoa possess highly derived genomes with scrambling of these gene sets. Divergence has continued within the phylum, leading to distinct organizational structures in two bryozoan classes, and ALG fission, previously underappreciated in its importance, has occurred with high frequency. The addition of information on the third bryozoan class (the Stenolaemata, represented by present-day cyclostomes) when chromosome-level genomes become available will add considerably to this picture. Indeed, the genomes of two stenolaemates and more than 20 additional gymnolaemates will be sequenced in the coming years by the DToL Project (The Darwin Tree of Life Project Consortium et al. 2022). Furthermore, although we present comparative genomics data on Hox gene duplications, additional studies on the spatial expression patterns of bryozoan Hox genes are necessary to examine how genome rearrangements have affected their functions (Fuchs et al. 2011). We expect the bryozoan genomes annotated in this study to be crucial for the pursuit of such future research avenues.
Methods
Assembly acquisition and gene prediction
Chromosome-scale assemblies of the following are available from NCBI (https://www.ncbi.nlm.nih.gov/): B. stolonifera (accession no. GCA_935421135) (Wood et al. 2023), C. pallasiana (GCA_945261195) (Bishop et al. 2023b), M. membranacea (GCA_914767715) (Bishop et al. 2023a), and W. subatra (GCA_963576615) (Bishop et al. 2024), and were sequenced as part of the DToL project (The Darwin Tree of Life Project Consortium et al. 2022) and retrieved from the NCBI Datasets resource. A chromosome-scale assembly of C. mucedo (Hoencamp et al. 2021) was obtained from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE169088. None of the bryozoan assemblies used have publicly available gene models, so gene prediction was performed in-house. Repetitive elements were first annotated with RepeatModeler (v2.0.4) (Flynn et al. 2020) and masked using RepeatMasker (v4.1.5) in sensitive mode (Smit et al. 2015). Gene prediction and annotation were performed using the BRAKER3 pipeline (v3.0.3), incorporating hints from RNA-seq data as aligned reads in BAM format (Stanke et al. 2006, 2008; Li et al. 2009; Barnett et al. 2011; Lomsadze et al. 2014; Buchfink et al. 2015; Hoff et al. 2016, 2019; Gabriel et al. 2024).
BRAKER3 uses RNA-seq and genome data to train the gene prediction tools AUGUSTUS and GeneMark, producing highly supported gene transcripts. Raw RNA-seq reads for gene prediction (Supplemental Table S2) were sourced from the NCBI Sequence Read Archive (SRA) using the SRA Toolkit (v3.0.0) (Leinonen et al. 2011) with GNU parallel (https://play.google.com/store/books/details?id=sKdSDwAAQBAJ). Transcriptomic data for B. stolonifera, C. pallasiana, M. membranacea, and W. subatra were generated from adult colonies collected by the Marine Biological Association, Plymouth, United Kingdom, in association with the DToL project. For these four species, RNA extraction was performed on the same colony used for DNA sequencing to generate the reference assembly. For B. stolonifera and M. membranacea, this data set was supplemented with independent data sets from several developmental stages (Supplemental Table S2). RNA-seq data for C. mucedo were derived from mature adult colonies from a population different to that from which the genome assembly was produced (Supplemental Table S2). Quality control for RNA-seq data was performed with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were trimmed with Trimmomatic (v0.39) (Bolger et al. 2014) and aligned to the genome with STAR (v2.7.10b) (Dobin et al. 2013). BUSCO (v5.4.7) (Simão et al. 2015; Manni et al. 2021) was used to evaluate the quality of gene prediction. Hox genes were identified with reciprocal TBLASTN searches (Camacho et al. 2009), and orthology was confirmed using gene trees built in IQ-TREE (v2.2.2.3) (Minh et al. 2020). Hox annotations are consistent with previous work, when available (Saadi et al. 2023).
Phylogenetics
Single-copy orthologs were identified using OrthoFinder (v2.5.4) (Emms and Kelly 2019). Alignments for each set of orthologs were created with MAFFT (v7.520) (Katoh et al. 2002; Katoh and Standley 2013), trimmed with ClipKIT (v1.4.1) (Steenwyk et al. 2020), and concatenated with PhyKIT (v1.11.7) (Steenwyk et al. 2021). IQ-TREE (v2.2.2.3) (Minh et al. 2020) was used to construct maximum likelihood trees with automated substitution model selection using ModelFinder (Kalyaanamoorthy et al. 2017) and 1000 UFBoot2 ultra-fast bootstrap replicates (Hoang et al. 2018). Trees were rendered with iTOL (Letunic and Bork 2021). A tree of Hox protein sequences was generated using the same IQ-TREE method.
Macrosynteny analysis
We developed SyntenyFinder, a pipeline comprising a collection of Python and R scripts designed to facilitate macrosynteny analysis using chromosome-level genome assemblies. SyntenyFinder is a straightforward pipeline that performs three main steps. First, it uses OrthoFinder (v2.5.4) (Emms and Kelly 2019) to identify single-copy orthologs in a data set of protein sequence files. Next, it integrates the orthogroups identified by OrthoFinder with information from genome sequence FASTA files and annotation files to track the genomic position of orthologous genes across the data set. Finally, it utilizes a custom R script to generate dot plots and chromosome ideograms (using RIdeogram [v0.2.2]) (Hao et al. 2020) showing the positions of orthologous genes between two genomes. SyntenyFinder is made available on GitHub (https://github.com/symgenoevolab/SyntenyFinder) and can be run in two modes: either automatically downloading and running on annotated genomes from NCBI by their accessions or on local files parsed directly to the program. This second option requires three types of file as an input: (1) genome sequence FASTA file, (2) gene positions in the format of a GTF file, and (3) a protein sequence FASTA file.
For the purposes of this paper, two runs were performed: first, with only the five bryozoan species and, second, adding B. floridae (Chordata), P. maximus (Mollusca), L. longissimus (Nemertea), and L. anatina (Brachiopoda). In both cases, only orthologs that were single copy in all study species were used for downstream analysis. No gene loss or duplication was permitted. In the second case, to track the genomic position of bilaterian ALGs, we used an input data set of the genes used to define the 24 bilaterian ALGs (Simakov et al. 2022) and used OrthoFinder to find orthologs of these genes in bryozoan genomes. The restriction to genes with known ALG relationships reduces the noise within the data set. Macrosynteny plots for this paper were created in R (v4.3.0) (R Core Team 2023). The same set of genes is used for each comparison in both ideograms and dot plots of macrosynteny. In ideograms, links between chromosomes with fewer than five shared genes were removed for visual clarity. These represent single-gene or few-gene translocations. All genes, including translocated genes, are shown in dot plots. For plotting purposes and to ensure alignment with orthologous chromosomes, Chr 1, Chr 2, Chr 4, Chr 6, Chr 7, Chr 10, and Chr 11 in W. subatra and Chr 2, Chr 3, Chr 7, Chr 9, Chr 10, Chr 11, and Chr 12 in C. pallasiana were reversed from their orientations as presented in published data sets.
Examination of large-scale duplications
Two complementary methods were used to determine whether segmental duplication or WGD events have occurred in the bryozoan lineage. In each case, we use the annelid M. vulgaris as a positive control for a species with a known WGD (Jin et al. 2020; Lewin et al. 2024a; Vargas-Chávez et al. 2024). First, synonymous substitutions per site (Ks) distributions were calculated with wgd (v.2.0.26) (Chen et al. 2024). Briefly, paralogs were identified with “wgd dmd” using Diamond (v2.0.14.152) (Buchfink et al. 2015, 2021), and then, “wgd ksd” was run to calculate the Ks distribution using PAML (v4.9) (Yang 2007). The Ks between two paralogs is an estimate of their divergence time. The null expectation with no WGD is that the number of paralogs decreases exponentially as Ks increases. When a WGD occurs, an excess of duplicates are created simultaneously, creating a peak in the distribution (Tiley et al. 2018). Paralogs situated in collinear blocks in the genome, so-called “anchor duplicates,” are more likely to be produced by WGD than dispersed paralogs and are therefore more reliable for WGD detection (Chen et al. 2024). Anchor duplicates were identified using “wgd syn” to implement the collinearity search algorithm of i-ADHoRe (v3.0.01) (Proost et al. 2011). Ks distributions using only anchor duplicates were then plotted with “wgd syn” (Chen et al. 2024).
Second, MCScanX (Wang et al. 2012) was used to identify collinear blocks of duplicated genes within bryozoan genomes and to classify all duplicates within each genome as dispersed, proximal, tandem, or WGD/segmental. The presence of large homologous regions within a genome containing collinear blocks of duplicated genes is suggestive of WGD. The MCScanX output was plotted as a Circos plot (Krzywinski et al. 2009) using shinyCircos (v2.0) (Wang et al. 2023).
Validation of chromosome fission
Given the absence of a high-quality Hi-C contact map for C. pallasiana (Bishop et al. 2023b), the observed fission event of gymnolaemate ALG a in this species remained uncertain. To validate the fission event that led to the formation of C. pallasiana Chr 10 and Chr 12, as shown in Figure 2, we examined chromatin interactions by mapping Hi-C data to the chromosome-level assembly. Hi-C reads for C. pallasiana were downloaded from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number ERR9866429 using the SRA Toolkit. The SRA file was fetched with prefetch and converted to FASTQ format using fasterq-dump. The Hi-C reads were then aligned to the genome assembly (accession GCA_945261195.1) using BWA (v0.7.17-r1198) (Li 2013), following the Arima Hi-C mapping pipeline (A160156 v03). A Hi-C contact map was generated using YaHS (v1.2a.1) (Zhou et al. 2023) and JuicerTools (v1.9.9) and was visualized with JuiceBox (v1.11.08) (Durand et al. 2016).
Quantification of microsynteny mixing score
To assess the amount of intra-chromosomal gene shuffling, we first calculated Spearman's rank correlation coefficient (ρ).
This coefficient quantifies the strength and direction of association between the ranked order of orthologous genes along
orthologous chromosome pairs. The Spearman's coefficient is given by the following equation:
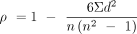 (1)
where ρ is the coefficient, di represents the difference in ranks between corresponding values of the two variables, and n is the total number of observations. Subsequently, we defined the synteny mixing score, an indicator of chromosomal rearrangement,
using the equation
(1)
where ρ is the coefficient, di represents the difference in ranks between corresponding values of the two variables, and n is the total number of observations. Subsequently, we defined the synteny mixing score, an indicator of chromosomal rearrangement,
using the equation
 (2)
where |ρ| denotes the absolute value of the Spearman's coefficient. This metric allows the synteny mixing score to vary from zero,
indicative of no intra-chromosomal rearrangement and thus a high degree of conserved microsynteny, to one, representative
of maximum rearrangement, signifying considerable disruption in the original gene order.
(2)
where |ρ| denotes the absolute value of the Spearman's coefficient. This metric allows the synteny mixing score to vary from zero,
indicative of no intra-chromosomal rearrangement and thus a high degree of conserved microsynteny, to one, representative
of maximum rearrangement, signifying considerable disruption in the original gene order.
Estimation of species divergence time
A phylogenetic tree, constructed as reported above from single-copy orthologs extracted from five bryozoan genomes and a mollusc outgroup (P. maximus), was used to estimate divergence times. We calibrated this tree with contemporary molecular and fossil data (Orr et al. 2022), focusing on the divergence between P. maximus and C. mucedo during the Cambrian Period (541 to 488 million years ago) (Erwin et al. 2011), employing the chronos function in the ape package (v5.7-1) (Paradis and Schliep 2019) in R.
Quantification of gene expression
Gene expression was quantified in a published C. mucedo RNA-seq data set obtained from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA594616. Raw reads were obtained from the NCBI SRA using the SRA toolkit (Leinonen et al. 2011) and GNU parallel (https://play.google.com/store/books/details?id=sKdSDwAAQBAJ). Hox gene expression was quantified with Salmon (v1.10.2) (Patro et al. 2017). Hox4a, Hox4b, Lox5a, and Lox5b gene expression levels were compared using t-tests with a Benjamini–Hochberg correction for multiple testing.
Data access
Gene models for all five bryozoan species have been deposited in Dryad (https://doi.org/10.5061/dryad.76hdr7t3f) and as Supplemental Data. Custom Python and R scripts used for macrosynteny analysis, calculating mixing scores, and estimating divergence times are available in our GitHub repository (https://github.com/symgenoevolab/SyntenyFinder) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was funded by a Royal Society Newton International fellowship (NIF\R1\201315) and an Academia Sinica Career Development award (AS-CDA-112-L06) to Y.-J.L. We acknowledge the efforts in sample collection by the Marine Biological Association (MBA)–Darwin Tree of Life collecting team: Patrick Adkins, Robert Mrowicki, Joanna Harley, and Helen Jenkins, as well as Nova Mieszkowska and Christine Wood of the MBA. We thank the members of the Symbiosis Genomics & Evolution Laboratory for their assistance and support.
Author contributions: T.D.L. and Y.-J.L. designed research. T.D.L., I.J.-Y.L., M.-E.C., and Y.-J.L. performed research. T.D.L., I.J.-Y.L., J.D.D.B., and Y.-J.L. contributed new reagents/analytic tools. T.D.L. and Y.-J.L. analyzed data. T.D.L., P.W.H.H., and Y.-J.L. wrote the paper. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279636.124.
-
Freely available online through the Genome Research Open Access option.
- Received May 29, 2024.
- Accepted October 31, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








