
Analysis of a cell-free DNA–based cancer screening cohort links fragmentomic profiles, nuclease levels, and plasma DNA concentrations
- Yasine Malki1,2,3,5,
- Guannan Kang1,2,3,5,
- W.K. Jacky Lam1,2,3,4,5,
- Qing Zhou1,2,3,
- Suk Hang Cheng1,2,3,
- Peter P.H. Cheung2,3,
- Jinyue Bai1,2,3,
- Ming Lok Chan2,3,
- Chui Ting Lee2,3,
- Wenlei Peng1,2,3,
- Yiqiong Zhang2,3,
- Wanxia Gai1,2,3,
- Winsome W.S. Wong1,2,3,
- Mary-Jane L. Ma1,2,3,
- Wenshuo Li1,2,3,
- Xinzhou Xu2,3,
- Zhuoran Gao2,3,
- Irene O.L. Tse2,3,
- Huimin Shang1,2,3,
- L.Y. Lois Choy1,2,3,4,
- Peiyong Jiang1,2,3,4,
- K.C. Allen Chan1,2,3,4 and
- Y.M. Dennis Lo1,2,3,4
- 1Centre for Novostics, Hong Kong Science Park, Pak Shek Kok, Hong Kong SAR, China;
- 2Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China;
- 3Department of Chemical Pathology, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China;
- 4State Key Laboratory of Translational Oncology, Sir Y.K. Pao Centre for Cancer, The Chinese University of Hong Kong, Prince of Wales Hospital, Shatin, New Territories, Hong Kong SAR, China
-
↵5 These authors contributed equally to this work.
Abstract
The concentration of circulating cell-free DNA (cfDNA) in plasma is an important determinant of the robustness of liquid biopsies. However, biological mechanisms that lead to inter-individual differences in cfDNA concentrations remain unexplored. The concentration of plasma cfDNA is governed by an interplay between its release and clearance. We hypothesized that cfDNA clearance by nucleases might be one mechanism that contributes toward inter-individual variations in cfDNA concentrations. We performed fragmentomic analysis of the plasma cfDNA from 862 healthy individuals, with a cfDNA concentration range of 1.61–41.01 ng/mL. We observed an increase in large DNA fragments (231–600 bp), a decreased frequencies of shorter DNA fragments (20–160 bp), and an increased frequency of G-end motifs with increasing cfDNA concentrations. End motif deconvolution analysis revealed a decreased contribution of DNASE1L3 and DFFB in subjects with higher cfDNA concentration. The five subjects with the highest plasma DNA concentration (top 0.58%) had aberrantly decreased levels of DNASE1L3 protein in plasma. The cfDNA concentration could be inferred from the fragmentomic profile through machine learning and was well correlated to the measured cfDNA concentration. Such an approach could infer the fractional DNA concentration from particular tissue types, such as the fetal and tumor fraction. This work shows that individuals with different cfDNA concentrations are associated with characteristic fragmentomic patterns of the cfDNA pool and that nuclease-mediated clearance of DNA is a key parameter that affects cfDNA concentration. Understanding these mechanisms has facilitated the enhanced measurement of cfDNA species of clinical interest, including circulating fetal and tumor DNA.
Much research effort has been made in obtaining diagnostic information from cell-free DNA (cfDNA) to investigate various physiological and pathological states in the context of pregnancy, oncology, and organ transplantation (Lo et al. 2021). There has also been growing research interest in understanding the biological life cycle of cfDNA molecules in circulation. Several key mechanisms of cfDNA release have been proposed, including the different modes of cell death, the release of neutrophil extracellular traps, and active cellular secretion (Grabuschnig et al. 2020; Heitzer et al. 2020; Han and Lo 2021). The rapid clearance of cfDNA has been demonstrated by studying the kinetics of fetal cfDNA in pregnant women after delivery (Lo et al. 1999; Yu et al. 2013). The measurement of cfDNA concentration in plasma at a given time reflects upon an interplay between DNA release from cells and clearance from circulation.
Many pathophysiological conditions are characterized by an altered equilibrium of plasma cfDNA concentration. An elevation of cfDNA concentration has been observed in patients with different cancers (Mattox et al. 2023), systemic lupus erythematosus (Tug et al. 2014), and infectious diseases (Han et al. 2020a; Cheng et al. 2021), compared with healthy controls. Performing physical exercise, such as a 40 min run, could lead to a mean increase of 18-fold in the total cfDNA concentration (Fridlich et al. 2023). The concentration of cfDNA serves as an important parameter in liquid biopsy, as it affects the sensitivity of disease diagnosis and the reproducibility of quantitative measurements. An enhanced understanding of the production and clearance of cfDNA may give rise to novel diagnostic approaches with greater sensitivity for early disease detection. Such a postulation has been recently supported by a study in which priming agents were developed to inhibit the clearance processes of cfDNA (Martin-Alonso et al. 2024). Administration of these priming agents prior to blood collection increased the recovery of circulating tumor DNA by more than 10-fold, as demonstrated in a preclinical model of mice with lung cancer (Martin-Alonso et al. 2024).
It is currently understood that part of the clearance process of cfDNA involves enzymatic degradation by the action of DNA nucleases. It has been revealed that fragmentomic features, such as fragment sizes and end motifs, are linked to the activity of these nucleases (Serpas et al. 2019; Chan et al. 2020; Han et al. 2020b; Han and Lo 2021; Lo et al. 2021; Chen et al. 2022; Zhou et al. 2023). For instance, deoxyribonuclease 1L3 (DNASE1L3) preferentially cleaves DNA in a manner that results in a preponderance of C-ends (Serpas et al. 2019; Chan et al. 2020; Han et al. 2020b), whereas deoxyribonuclease 1 (DNASE1) preferentially cleaves DNA into T-ends (Chen et al. 2022). Our work has revealed that DNASE1L3 plays a critical role in the fragmentation process of cfDNA in plasma, as evidenced by the aberrant plasma DNA fragmentation patterns in a Dnase1l3-deficient mouse model and human subjects (Serpas et al. 2019; Chan et al. 2020), as well as the presence of DNASE1L3 protein in plasma (Chen et al. 2022). We also identified DNA fragmentation factor subunit beta (DFFB) as a major player in the fragmentation of newly released DNA from dying cells (Han et al. 2020b). We hypothesized that the process of nuclease-mediated fragmentation is linked to the total concentration of cfDNA.
Previous reports have shown considerable variation in the plasma cfDNA concentration of healthy individuals (Alborelli et al. 2019; Meddeb et al. 2019; Ørntoft et al. 2021; Zhu et al. 2023). We reason that characteristic fragmentomic patterns in the cfDNA pool between subjects with different cfDNA concentrations may hold clues to mechanisms that regulate the levels of cfDNA in plasma. In the present study, we have analyzed the distribution of plasma cfDNA concentrations in 862 individuals. These subjects are part of a cohort of individuals who had undergone screening for the early detection of nasopharyngeal carcinoma (NPC) in Hong Kong (Chan et al. 2017, 2023). The NPC screening was performed through the detection of Epstein–Barr virus (EBV) DNA in the circulating cfDNA pool. This cohort was used to investigate whether the overall concentration of circulating DNA in plasma is associated with changes in fragmentomic features, which might provide hints toward nuclease-mediated fragmentation, or other factors that contribute to inter-individual differences in cfDNA concentrations.
Results
Subject cohort characteristics and cfDNA concentration distribution
The aim of the study was to evaluate fragmentomic characteristics of cfDNA in individuals with different concentrations of plasma cfDNA. We therefore retrieved cfDNA concentration values from individuals in a cancer screening cohort and analyzed the size profiles, end motif patterns, and nuclease contributions from sequencing data of plasma cfDNA (Fig. 1). The individuals studied were participants who had undergone screening for NPC (Chan et al. 2017, 2023). These subjects underwent plasma EBV DNA testing by real-time polymerase chain reaction (PCR) for NPC screening. The screening trial was conducted between the years 2017–2020. All subjects were ethnically Chinese men.
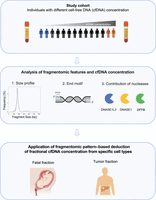
Overview of the study design. The plasma cfDNA concentration of all 862 individuals from the study cohort was measured. The fragmentomic features of the cfDNA were analyzed, including the size profile and end motif distributions. The contribution of nucleases was explored using the cleavage profile end motif patterns, and protein quantification of DNASE1L3 in plasma. A machine learning model was trained using fragmentomic features to predict cfDNA concentration. We further explored whether a similar model could be applied to study fractional DNA concentration in predicting the fetal and tumor fraction of plasma cfDNA in pregnant subjects and patients with HCC, respectively.
Individuals who were tested as EBV-positive were subjected to targeted sequencing of plasma EBV DNA as a reflex test to enhance the specificity of NPC detection (Lam et al. 2018). We retrieved and analyzed the targeted sequencing data of 862 EBV-positive subjects to explore the relationship between fragmentomic features and cfDNA concentrations. To minimize the potential effects of target capture on the fragmentomic analysis, only “off-target” reads were used to study fragmentomic features of cfDNA (see Methods). The median age of the 862 subjects was 54 years (interquartile range of 49–57 years).
The distribution of cfDNA concentration is shown in Figure 2. A large variation was observed in the cfDNA concentration among subjects, with values ranging from 1.61–41.01 ng/mL, and therefore a 25.5-fold difference between subjects with the highest and lowest plasma cfDNA concentration. The median cfDNA concentration was 7.39 ng/mL. The skewness of the plasma DNA concentration was 2.043 (P < 0.001), and the kurtosis of the distribution was 6.974 (P < 0.001). The high positive value of skewness (beyond the range of +1 and −1) indicates a positively skewed distribution of plasma DNA concentrations. The kurtosis value from our cohort represents a leptokurtic distribution (above three), generally indicating a large proportion of individuals on both extremes of the distribution spectrum. The values of plasma cfDNA concentration deviates from a normal distribution (Shapiro–Wilk test: P < 0.001). We have demonstrated that plasma cfDNA concentrations can be fitted into a gamma distribution (Supplemental Fig. S1A). The significant skewness of plasma cfDNA concentration, particularly in subjects with higher cfDNA concentrations, may indicate potential biological or physiological factors that may play a role in modulating cfDNA concentration, resulting in large deviations of cfDNA concentrations from the median in some individuals.
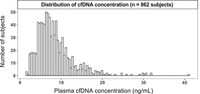
Frequency distribution histogram plot showing the distribution of plasma cfDNA concentration from 862 individuals of the study cohort.
To further validate the findings and conclusions, we sequenced and analyzed an additional 497 individuals from the same clinical study. These individuals had no detectable EBV-DNA in plasma, referred to as “EBV-negative cohort.” The subject characteristics of both cohorts are summarized in Supplemental Table 1. The cfDNA concentration ranged from 1.62–25.25 ng/mL, with a median of 7.56 ng/mL, consistently exhibiting a positively skewed distribution (Supplemental Fig. S2). The skewness of the cfDNA concentration from this cohort was 1.461 (P < 0.001); kurtosis was 3.537 (P < 0.001); and the Shapiro–Wilk test resulted in P < 0.001. The distribution of cfDNA concentrations also fit a gamma distribution function (Supplemental Fig. S1B).
Quantification of the plasma cfDNA concentration was performed on all subjects of the cohort using fluorometric measurements (see Methods). Fluorometric methods measure the total DNA concentration, including DNA from both nuclear and mitochondrial origins. The readings from fluorometric measurements (n = 25) were well correlated with results obtained from the droplet digital PCR assay, which targeted a single-copy gene valosin-containing protein (VCP) (Supplemental Fig. S3). This highlights the reliability of using fluorometric methods to reflect the total cfDNA of nuclear origin.
Fragmentomic study 1: size profile analysis
One goal of this study was to examine how fragmentomic features, namely, the size distribution and end motif patterns of cfDNA, varied among individuals with different concentrations of cfDNA in plasma. Figure 3, A and B, shows the overall size distribution profile of plasma cfDNA from the highest 10% of individuals (red) and lowest 10% of individuals (blue), as well as the median distribution (black) in terms of cfDNA concentration. Plasma DNA of all subjects exhibited a modal peak size of ∼166 bp, consistent with reports investigating the mononucleosome units of cfDNA. The logarithmic plot shows the presence of the di- and trinucleosome peaks, with sizes of ∼350 bp and 520 bp.
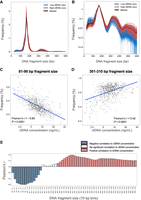
Size profile of plasma DNA fragments in subjects of different cfDNA concentrations. The size profiles of the selected lowest and highest 10% of subjects, and the median distribution of the cohort, are shown in linear scale (A) and logarithmic scale (B). Correlation between the frequency of DNA fragments within each 10 bp window bin and cfDNA concentration was assessed for all subjects, for the 81–90 bp fragment size range (C), 301–310 bp fragment size range (D), and across all 10 bp bins from 20 to 600 bp (E). Blue and red labels indicate 10 bp bins with a statistically significant negative or positive correlation to cfDNA concentration, respectively.
In comparison to subjects with low cfDNA concentrations, subjects with high cfDNA concentrations appeared to have increased frequencies of DNA fragments >250 bp, with an enhancement in the di- and trinucleosomal peaks. The increased frequency for fragments >250 bp in subjects with high cfDNA concentration resembled previous findings in Dnase1l3-deficient mice, which exhibited an elevated frequency of di- and trinucleosomal DNA in plasma (Serpas et al. 2019). These results suggested that subjects with high cfDNA concentrations might exhibit an impaired DNASE1L3-mediated fragmentation process. Short DNA fragments within the size range of 20–120 bp were decreased in subjects with high cfDNA concentrations. These shorter fragments might represent intermediate products (i.e., subnucleosomal DNA) derived from mononucleosomal DNA during the degradation process. Size distribution plots of different cfDNA concentration ranges (<5, 5–10, 10–15, ≥15 ng/mL) also show this gradient pattern of decreased frequency of DNA fragments within the 20–120 bp range and of increased frequency of DNA fragments >250 bp (Supplemental Fig. S4).
We next studied the size profile frequencies in all 862 subjects. The frequencies of cfDNA fragments for each 10 bp bin size were measured and correlated to cfDNA concentrations. As examples, the frequency of cfDNA fragments between 81 and 90 bp was negatively correlated with cfDNA concentration (Pearson's r = −0.65, P < 0.0001) (Fig. 3C), whereas the frequency of cfDNA between 301 and 310 bp was positively correlated (Pearson's r = 0.42, P < 0.0001) (Fig. 3D). As shown in Figure 3E, the cfDNA frequencies in bins <160 bp were found to be negatively correlated to cfDNA concentrations, whereas bins >230 bp size were positively correlated to cfDNA concentrations. The proportional quantification of DNA molecules within certain size ranges may serve as a parameter to approximate the degradation rate of cfDNA in plasma. The process of apoptosis releases DNA with a wide range of sizes (Ungerer et al. 2022; Zhu et al. 2023; Davidson et al. 2024), whereas subsequent cell-extrinsic cleavage occurs via nucleases in blood, namely, DNASE1L3 (Serpas et al. 2019). Factors that affect the degradation of cfDNA would alter the proportion of DNA molecules within certain size ranges. The overall larger size profiles in individuals with high cfDNA concentrations might suggest decreased activity of extracellular DNA clearance in blood.
Fragmentomic study 2: end motif analysis
We have shown that the concentration of cfDNA was associated with changes in the size distribution of the cfDNA pool. We next investigated whether the distribution of 5′ end motifs varied with cfDNA concentration (for end motif analysis, see Methods). To this end, we systematically studied the correlations of 256 end motifs (4-mer) to the plasma cfDNA concentrations. A total of 79 end motifs were found to be significantly correlated with cfDNA concentrations, with 34 motifs being negatively correlated and 45 being positively correlated after adjustment for multiple comparisons using Bonferroni's correction (Supplemental Tables 2, 3). Motifs with a negative correlation were mostly C-end motifs (such as CACT, CATC, CACC), with several T-end and A-end motifs, whereas the positively correlated motifs were nearly all G-end motifs (such as GCAA, GAAC, GGCA).
We performed heatmap analysis to visualize the pattern of the significantly correlated motifs to cfDNA concentration, as shown in Figure 4A. Each row corresponds to a particular 4-mer motif, with the composition of the first base indicated, and columns represent plasma DNA from each subject. For better visualization, column-wise normalization (z-score) was applied to the motif frequencies. A gradual change in motif frequencies was observed across the whole spectrum of cfDNA concentrations, with 4-mer end motifs starting with a 5′ C nucleotide appearing to gradually decrease as the cfDNA concentration increased. However, the 4-mer end motifs starting with a 5′ G nucleotide showed a rising gradation. The most pronounced difference in end motifs frequencies was observed in cfDNA concentration ranges of <5 ng/mL and ≥15 ng/mL (Supplemental Fig. S5). The observed trends in fragmentomic features were also present in the highest and lowest 10 subjects of the EBV-negative cohort (Supplemental Fig. S6). By evaluating the highest and lowest 10% of individuals from the main study cohort, the selected 79 significantly correlated motifs also showed a consistent trend when subclassified into Alu regions, CpG islands, and gene body regions (Supplemental Fig. S7), suggesting that these changes in end motif profiles appear to be present across different genomic regions. Furthermore, eliminating the highest and lowest 10% of the cohort yielded similar results regarding size distribution and end motif patterns (Supplemental Fig. S8), indicating that the observed patterns were not exclusively influenced by the extreme values. Of note, fragmentomic features derived from the “off-targeted” reads in the targeted sequencing data set were in good agreement with features in those paired samples based on genome-wide paired-end sequencing (Supplemental Figs. S9, S10). These results demonstrated the validity of using “off-target” reads in our fragmentomic analysis.
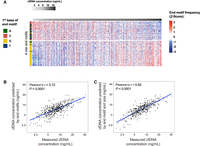
End motifs with significant correlation to cfDNA concentration. The frequencies of all 256 end motifs (4-mer) were correlated to the cfDNA concentration. The resulting analysis revealed 34 negatively correlated end motifs to cfDNA concentration and 45 positively correlated end motifs. (A) Heatmap analysis with rows indicating a particular 4-mer motif, in which the first base of the motif highlighted by a specific color in the left-most column (A, C, G, and T are colored by green, red, yellow, and blue, respectively). Each column indicates a plasma DNA sample from one subject. The frequency z-score, calculated for each end motif, is shown by the color scale. A support vector regression (SVR) model was trained with fragmentomic features to predict cfDNA concentration, using end motif frequencies (B) and both end motif and size profiles (C).
Motivated by the gradation patterns observed in the heatmap, we explored whether we could use regression to model the relationship between cfDNA concentration and 4-mer end motifs. We adopted a support vector regression (SVR) model, with a random selection of 50% of subjects used as the training set and the remaining 50% as the testing set. Indeed, we observed a correlation between the cfDNA concentration predicted by the SVR model and the cfDNA concentration measured by fluorometric quantification in the validation set (Pearson's r = 0.72, P < 0.0001) (Fig. 4B). By utilizing the size profile in addition to end motif frequencies, the correlation could be further improved, with a Pearson's r of 0.82 (P < 0.0001) (Fig. 4C). In addition, we used the 10 subjects with the lowest cfDNA concentrations and the 10 subjects with the highest cfDNA concentrations of the EBV-negative cohort as an additional test set for the trained model, which also showed a strong positive correlation between predicted and measured cfDNA concentration (Pearson's r = 0.94, P < 0.0001) (Supplemental Fig. S11). Hence, these results further validate that the overall cfDNA concentration was associated with characteristic fragmentomic patterns in cfDNA pool.
Deconvolutional analysis of end motifs
Our group has previously reported on six distinct cleavage patterns via a nonnegative matrix factorization (NMF) algorithm, termed “founder” end motif profiles (F-profiles), which were linked to different biological processes (Zhou et al. 2023). F-profiles I, II, and III represent the contribution of DNASE1L3, DNASE1, and DFFB, respectively. F-profile IV is a cleavage profile with a high C-end preference (e.g., CG-end preference), whereas F-profile V exhibits a strong G-end preference. Profile VI represents nonspecific cleavage patterns, speculated to originate from chemical factors such as oxidative stress. We investigated whether the F-profile contributions varied between subjects of different cfDNA concentrations.
We stratified the DNA fragments into three size ranges for the end motif based deconvolutional analysis, which were 20–160 bp, 161–230 bp, and 231–600 bp, selected based on the correlation to cfDNA concentration in Figure 3E. We reasoned that each size range might result from different stages of the fragmentation process of DNA in plasma. We visualized the differences in F-profile contributions with different cfDNA concentrations using a heatmap analysis, with z-score normalization applied to each F-profile. Of the three size ranges, the 231–600 bp size range showed the most distinct variation in F-profile contributions with cfDNA concentration (Fig. 5). The contribution of F-profile I (DNASE1L3), II (DNASE1), and III (DFFB) exhibited a gradation pattern with increased cfDNA concentration. F-profile V showed a contrasting trend with cfDNA concentration, consistent with the higher frequency of G-end motifs associated with increased cfDNA concentration. Overall, the larger plasma DNA fragments with sizes around the di- and trinucleosome peaks had a higher frequency of G-end motifs, as well as decreased end motif frequencies from DNASE1L3, DNASE1, and DFFB. No particular gradation pattern was observed amongst all six F-profiles in the 21–160 bp size range (Supplemental Fig. S12). The gradation patterns in F-profiles I, III, and V were also observed in the 161–230 bp size range (Supplemental Fig. S12).
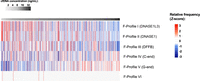
Deconvolutional analysis of end motifs to deduce the contribution of the six “founder” end motif profiles (F-profile) of plasma DNA fragments within the 231–600 bp size range, among subjects of different cfDNA concentrations. Heatmap analysis was performed, with each row showing the contributions (expressed as z-scores) of each F-profile across subjects with different cfDNA concentrations.
A multiple linear regression analysis was performed on all six F-profiles across three size ranges to determine which profiles were significantly associated with cfDNA concentration (Supplemental Table 4). The analysis revealed that F-profiles I (DNASE1L3), III (DFFB), and V (G-ends) of molecules within the 231–600 bp range, and F-profile V within the 161–230 bp range, were significantly associated with the cfDNA concentration. These results provide evidence that DNASE1L3 and DFFB are factors that may regulate the concentration of circulating cfDNA in plasma.
Concentration of DNASE1L3 protein in plasma
Fragmentomic analysis suggested that the activity of DNA nucleases might be at least in part responsible for regulating the concentration of cfDNA in plasma. Previous work has revealed that DNASE1L3 is secreted by macrophages and dendritic cells into plasma circulation (Shiokawa and Tanuma 1998; Sisirak et al. 2016) and plays an important role in the cell-extrinsic fragmentation of cfDNA (Sisirak et al. 2016; Chan et al. 2020). We therefore developed an assay to quantify the concentration of DNASE1L3 in plasma to determine whether subjects with different cfDNA concentrations were associated with varied levels of DNASE1L3. We focused primarily on the subjects with the highest and lowest cfDNA concentrations to evaluate whether such a difference in DNASE1L3 protein levels was observed.
As a result, the highest five subjects showed a significantly decreased level of DNASE1L3 protein in plasma compared with the lowest five subjects (52.5% reduction in DNASE1L3 levels; P = 0.0079, Wilcoxon test) (Supplemental Figs. S13, S14). However, the significance of this trend was diminished when extended to the highest and lowest 10 subjects (14.5% reduction in DNASE1L3 levels; P = 0.2475, Wilcoxon test) (Supplemental Figs. S13, S14). It is possible that only subjects with particularly elevated levels of cfDNA in plasma, such as those subjects exceeding 30 ng/mL, were associated with decreased levels of DNASE1L3 in plasma. Nevertheless, this analysis suggested that the levels of DNASE1L3 in plasma might partially account for the variation in cfDNA concentrations among different individuals.
Tissue-of-origin analysis of cfDNA in individuals with different cfDNA concentrations
As we observed that the activity of nucleases was linked to cfDNA concentration, we questioned whether the variation in cfDNA concentration might be associated with the release of cfDNA from a particular cellular source. We employed the fragmentomics-based methylation analysis (FRAGMA) to deduce the methylation status of cfDNA (Zhou et al. 2022). We selected cell type–specific hyper- and hypomethylated CpG sites for six cell types (liver, neutrophils, B cells, T cells, erythroblasts, and megakaryocytes), as they represented major cellular sources contributing to the cfDNA pool (Loyfer et al. 2023). The deduced tissue contribution was assessed based on methylation status at these CpG sites (see Methods) and was correlated to the cfDNA concentration (Supplemental Fig. S15). The results showed that the deduced contribution of each cell type was not significantly correlated to cfDNA concentration, with a weak correlation coefficient (Pearson's r < 0.1 for all cell types). Hence, the production of cfDNA from various cell types might not be an important factor contributing to the variation of cfDNA concentration.
Follow-up collection of subjects with extreme cfDNA concentrations
To investigate whether the cfDNA concentration might persist in subjects with the highest and lowest cfDNA concentrations, we contacted a total of 40 subjects (the highest and lowest 10 from both cohorts) for a follow-up blood collection. Samples from 26 individuals were obtained, as the remaining individuals either developed cancers, were lost to follow-up, or refused participation. The median interval time between blood collection from our existing cohorts for NPC screening and blood recollection was 76 months (interquartile range of 64–77 months).
The concentrations of cfDNA were well correlated between the two collection points for each subject (Pearson's r = 0.75, P < 0.0001) (Supplemental Fig. S16A). The difference in cfDNA concentration between the high and low cfDNA groups was statistically significant (P < 0.0001) (Supplemental Fig. S16B). A comparison of the fragmentomic profiles between paired matched samples from the two collection time points showed a significant correlation in the proportion of DNA fragments within 81–90 bp and 301–310 bp (Supplemental Fig. S17A,B) and in the pattern of end motif frequencies from the selected 79 end motifs from Figure 4A (Supplemental Fig. S17C,D). Overall, this suggests that cfDNA concentration might be mediated by certain intrinsic physiological factors, leading to persistent extremes spanning a median collection interval of ∼6 years.
Relationship between clinical laboratory parameters and cfDNA concentration
We further explored the possible correlation of other biochemical parameters in blood with the concentration of plasma cfDNA. Laboratory tests were performed on these individuals from the follow-up collection (n = 26 individuals), which included blood cell counts (red blood cells, neutrophils, lymphocytes, monocytes, eosinophils, and platelets), the concentration of plasma proteins (total plasma proteins, albumin, globulins, alkaline phosphatase [ALP], alanine aminotransferase [ALT], C-reactive proteins [CRP]), and nonprotein components (sodium, potassium, bilirubin, urea, creatinine). Data for each parameter were correlated with the concentration of plasma cfDNA, with a false-discovery rate (FDR) adjustment for multiple comparisons (Supplemental Table 5). Multiple linear regression was also performed to study the effects of plasma protein components on cfDNA concentration (Supplemental Table 6).
The correlation analyses revealed that the levels of ALT (Pearson's r = 0.676, P = 0.0024), CRP (Pearson's r = 0.595, P = 0.0168), and total plasma proteins (Pearson's r = 0.549, P = 0.0296) were significantly correlated to plasma cfDNA concentrations (Supplemental Table 5; Supplemental Fig. S18). The multiple linear regression analysis revealed that only ALT (P = 0.0028) and CRP (P = 0.0095) were significant independent variables affecting plasma cfDNA concentrations (Supplemental Table 6). The levels of ALT and CRP from these subjects were within the reference intervals (ALT < 53 IU/L; CRP < 9.9 mg/L) (Supplemental Fig. S18). These results demonstrate that plasma cfDNA levels were also associated with physiological factors such as liver function and inflammation.
Deduction of fractional DNA concentrations from different cell types using fragmentation patterns
As the total cfDNA concentration of individuals could be predicted using fragment sizes and end motifs, we investigated whether such fragmentomic features could be applied to subsets of cfDNA species. We used circulating tumor DNA in HCC patients and circulating fetal DNA in the plasma of pregnant women as examples, in which fragmentomic features were used to predict tumoral and fetal DNA fraction, respectively (Fig. 1). Our previous work has demonstrated that fetal and tumor-derived DNA have a shorter modal size of 143 bp (Yu et al. 2014; Jiang et al. 2015; Jiang and Lo 2016). This may be attributed to differences in chromatin packing and methylation density (Sun et al. 2018; Pastor and Kwon 2022). Characteristic changes in the end motif profiles have also been observed in cancer and pregnancy, for example, decreased frequencies of DNASE1L3 representative motifs such as CCCA in patients with HCC (Jiang et al. 2020). As such, an alteration in fetal or tumoral DNA fraction would lead to changes in fragmentomic features of the cfDNA pool (Yu et al. 2014, 2017; Jiang et al. 2015, 2020). We reasoned that the integrative analysis of the whole spectrum of fragment sizes and end motifs would enable more accurate prediction of fetal or tumoral DNA fraction.
The size profile and end motif profiles of cfDNA from 30 pregnant women (including first, second, and third trimester cases, all from the “first cohort” in Supplemental Table 7) were used to train an SVR model. We adopted the leave-one-out procedure (see Methods) and compared the predicted fetal fraction (%) by fragmentomic features to the fetal fraction measured using informative single-nucleotide polymorphisms (SNPs). The predicted fetal fraction was in good agreement with the SNP-based fetal DNA fraction (Pearson's r = 0.81, P < 0.0001) (Fig. 6A). The magnitude of correlation exceeded a previous approach that employed the motif diversity score (Spearman r = −0.46, P = 0.01) (Jiang et al. 2020). A similar analysis was performed to predict tumor fraction from 20 patients with HCC, including individuals in early, intermediate, and advanced stages (“first cohort” in Supplemental Table 8). The fragmentomic-based predicted tumoral DNA fraction was well correlated to the tumor fraction measured by copy number aberration (Pearson's r = 0.84, P < 0.0001) (Fig. 6B). These results also lead to an enhanced correlation of tumor fraction compared with the motif diversity score (Pearson's r = 0.65, P = 0.0019) (Jiang et al. 2020). We further validated our model of fetal and tumor fraction prediction by using a second cohort of pregnancy (n = 30) (Supplemental Table 7) and HCC cases (n = 20) (Supplemental Table 8), with an SVR model trained with the first cohort (50% training, 50% testing). Our results supported the validity and robustness of fetal fraction prediction (Pearson's r = 0.81, P < 0.0001) (Supplemental Fig. S19A) and of tumor fraction prediction (Pearson's r = 0.75, P < 0.0001) (Supplemental Fig. S19B) using fragmentomic features. The use of LASSO regression on the fetal and tumor prediction allowed for the identification of features that are most informative in the model (Supplemental Tables 9, 10). The most informative features for fetal fraction prediction were the end motifs ACGT, CCGA, and CCGT, whereas that of tumor fraction prediction was the end motif ACGA. The underlying biology causing the observed results for certain end motifs warrants further investigation. Taken together, the use of fragmentomic patterns not only offered a means to predict the absolute total concentration of cfDNA but also allowed the deduction of fractional DNA contributions from particular cell types.
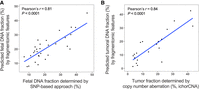
Application of fragmentomic pattern–based deduction of the fractional DNA concentration from specific cell types. Fragmentomic features, including size profile and end motif distribution, were used to train SVR models in the prediction of the fractional DNA percentage, which was correlated to the proportional tissue DNA contribution. (A) Correlation between the fetal DNA fraction predicted by the fragmentomic features and SNP-based methods, using 30 pregnant subjects. (B) Correlation between the tumoral fraction predicted by fragmentomic features and copy number aberration (ichorCNA).
Discussion
In this study, we explored the long-standing biological puzzle of the high inter-individual variability of plasma cfDNA concentrations. We showed that the total cfDNA concentration was linked to changes in the fragmentomic profiles (i.e., the size profile and end motif distribution) of the cfDNA pool. Several lines of evidence also pointed toward the involvement of nucleases, such as DNASE1L3 and DFFB, in the modulation of cfDNA concentration. The use of a machine learning model (i.e., the SVR model) allowed for the cfDNA concentration to be inferred from fragmentomic features. A similar model could also be employed to predict the fetal and tumoral DNA fractions in pregnant women and patients with HCC, respectively. The capability to accurately estimate the fractional contributions of specific cell types to the plasma DNA pool holds significant clinical relevance, for example, possibly facilitating the advancement in noninvasive prenatal testing, as well as cancer detection and monitoring.
The end motif analysis revealed that 79 end motifs (4-mers) were significantly correlated to cfDNA concentration. Motifs with a negative correlation to cfDNA concentration were mostly C-end and T-end motifs, whereas those with a positive correlation were nearly all G-ends. Similarly, the deconvolution analysis of the end motifs revealed the increased contribution of Profile V in subjects with higher cfDNA concentrations, which is a cleavage profile characterized by a series of G-end motifs (Zhou et al. 2023). The results raise the possibility that other nucleases or biological processes might have a preference for the generation of these G-end motifs. It is possible that some intermediate processes, or effects of DNA fragmentation during apoptosis, might have increased the frequencies of these G-end motifs, such as GCAA and GAAC. Further exploration of other nucleases, such as the other members of the DNase family, endonuclease G, as well as potential non-enzyme-mediated fragmentation processes (Zhou et al. 2023), might shed mechanistic insights into the fragmentation pattern of cfDNA and the regulation of cfDNA concentration in plasma.
The importance of DNASE1L3 in plasma cfDNA fragmentation has previously been demonstrated in Dnase1l3 knockout mouse models (Serpas et al. 2019), DNASE1L3-deficient patients (Chan et al. 2020), and patients with hepatocellular carcinoma (Jiang et al. 2020). Our current work revealed the link between DNASE1L3-related fragmentation signatures and cfDNA concentration. Subjects with high cfDNA concentrations exhibited fragmentomic features that resembled a Dnase1l3-deficient mouse model and human subjects, such as the enhanced di- and trinucleosomal patterns in size profile and the decreased C-end motifs (Serpas et al. 2019; Chan et al. 2020). The contribution of DNASE1L3 cleavage profile (Profile I) showed a decreasing frequency with cfDNA concentration and was most noticeable in the 231–600 bp size range. It is possible that an attenuated DNASE1L3 activity might hinder the degradation of longer cfDNA molecules into shorter fragments.
The reduced activity of DNASE1L3 was at least partially evidenced by the decreased protein levels of DNASE1L3 in plasma from subjects with the highest cfDNA concentrations (comparing the five highest- and lowest-ranked subjects). However, this trend was diminished when expanding further to the highest and lowest 10 subjects. We speculate that only individuals with extremely elevated cfDNA concentrations (i.e., ∼30 ng/mL or above) might be associated with a deficiency in DNASE1L3 concentrations in plasma. Such individuals represented the top 0.58% (top five of 862 individuals) in terms of plasma cfDNA concentrations from our cohort. It is worth noting that the contribution of DFFB also shows a similar gradation pattern with cfDNA concentration, with a decreased contribution in subjects with higher cfDNA concentrations. DFFB plays a major role in DNA fragmentation during apoptosis, and our previous work has demonstrated that newly released DNA exhibited strong A-end and G-end preference that was associated with DFFB activity (Han et al. 2020b). Further work is required to assess the activity of these nucleases and how it affects the process of DNA clearance.
The characteristic fragmentomic patterns observed allowed us to train a machine learning model to directly infer the cfDNA concentration. We demonstrated that such a model that integrates the size profile and end motif distribution could have useful clinical applications, such as the prediction of fetal and tumoral DNA fractions. This approach is shown to be more strongly correlated to the fetal or tumoral DNA fractions compared with a previous method of using end motif diversity scores (Jiang et al. 2020). Such a machine learning method provides a unique advantage as it incorporates both the size and end motif differences associated with a particular physiological condition or disease. These findings open up many further possibilities for analyzing the cfDNA concentration shed from various tissues using cfDNA fragmentomics. However, there is still room to enhance the current model to enable the differentiation between different pathophysiological states. For example, both pregnancy and cancer might exhibit certain overlapping fragmentation patterns, such as the shortening of the cfDNA size distribution (Jiang and Lo 2016; Lo et al. 2021). Similiarities in fragmentomic features would make it challenging for the model to differentiate between various pathophysiological statuses. Further work may involve the analysis of larger data sets of sequencing data during the model training, encompassing a variety of physiological states, such as pregnancy, cancer, and patients with other conditions like autoimmune diseases.
Of note, it is important to acknowledge several additional considerations for the current work. As our main subject cohort (n = 862) originated from a previous clinical study for NPC screening (Chan et al. 2023), information such as body weight, time of day, and the time delay between food intake was not reported. Detailed clinical laboratory tests were not routinely performed on paired blood collections from the main study cohort. Potential effects of physiological status and lifestyle factors have not been explored in this study. It is worth noting that the current protocol of the screening study is implemented large-scale in clinical settings, with previous reports of high negative predictive value (>99%) and a false-positive rate of <0.7% (Lam et al. 2018; Chan et al. 2022, 2023). We believe potential preanalytical factors may not be a substantial factor affecting our plasma processing and extraction, given such diagnostic potential and clinical utility. We have confirmed that fragmentomic patterns that vary with cfDNA concentration were also observed in a second cohort of EBV-negative individuals (Supplemental Fig. S6). However, future studies are warranted to explore the impact of preanalytical factors on plasma cfDNA concentration and fragmentomic profiles. It is also likely that different protocols for sample processing, methods of extraction, and DNA measurement could affect the measurement of cfDNA concentration. Further investigations into the effects of different experimental techniques would facilitate comparisons across different cohorts. Our present work evaluates fragmentomic profiles of plasma DNA using short-read sequencing from Illumina platforms, which has a readout limit of ∼600 bp. It would be beneficial to further expand our fragmentomic analysis to longer cfDNA molecules (>1000 bp) in our analysis of cfDNA concentration, using long-read sequencing platforms such as single-molecule, real-time sequencing (by Pacific Biosciences) and nanopore sequencing (e.g., by Oxford Nanopore Technologies). In addition, different extraction methods would enable the analysis of various subpopulations of DNA in plasma. These include the bead-based approaches to isolate ultrashort single-stranded DNA (Cheng et al. 2022; Hudecova et al. 2022), which would be of great biological interest in the context of this study.
This study elucidated several potentially biologically important underpinnings for the variability of cfDNA concentration in physiologically normal individuals. The understanding of the biology governing cfDNA concentration paves the way to personalize cfDNA presentation patterns in liquid biopsies and facilitates technological advances that increase the sensitivity of detection in studying diseases. This was recently demonstrated by Martin-Alonso et al. (2024), in which priming agents were developed to target the clearance process of cfDNA, therefore enabling increased recovery of tumor-derived cfDNA. Furthermore, it would be of great research interest to explore the biology of cfDNA concentration and associated fragmentomic features in various physiological conditions, such as in the context of pregnancy, cancer, and organ transplantation.
Methods
Study design and subject recruitment
Samples used in this study were obtained from prospectively collected plasma from previously published studies, aiming to detect the presence of EBV DNA in plasma for the early detection of NPC (Chan et al. 2017, 2023). The study was conducted in Hong Kong during 2017–2020, recruiting participants from organized public health education sessions. The exclusion criteria for the screening were individuals with cancer, with autoimmune diseases, or with symptoms of NPC, as well as the use of systemic glucocorticoids or immunosuppressive therapy. Blood collection from the NPC clinical study was conducted in a controlled setting, following the established protocols that ensured subjects were in a resting state prior to the blood collection. In this study, the cfDNA concentration and sequencing data from 862 subjects with detectable EBV DNA at baseline screening were used (EBV-positive cohort). These subjects did not develop NPC or other types of cancer identified within 1 year of sample collection. A selection of 497 subjects with no detectable EBV DNA (EBV-negative cohort) was used as a second cohort to validate findings from the main study cohort. The 10 subjects with the highest cfDNA concentrations and the 10 subjects with the lowest cfDNA concentrations from both cohorts were called back for a follow-up collection for validation, with 26 individuals returning for blood collection and analysis in this study.
Sample collection and processing
Blood collection from the screening study was done using Roche cell-free DNA collection tubes (07832389001). Samples were stored at 4°C for no longer than 6 h before processing. The blood was first centrifuged at 1600g for 10 min at 4°C. The plasma portion was further subjected to centrifugation at 16,000g for 10 min at 4°C to pellet out residual cells and debris. Plasma was stored in aliquots at −80°C until required for experimental work. Subsequent DNA extractions for all samples were carried out after a single freeze–thaw cycle, with samples frozen during plasma processing and thawed only for DNA extraction. No samples underwent multiple freeze–thaw cycles. We excluded hemolyzed plasma from further sequencing and analysis in this study cohort.
DNA extraction and measurement of cfDNA concentration
Plasma cfDNA was extracted as previously described (Chan et al. 2022, 2023). Briefly, 2 mL of plasma of each sample was extracted using the MagMAX cell-fee DNA isolation kit in the KingFisher flex system (Thermo Fisher Scientific) according to the manufacturer's instructions. The cfDNA concentration of each extracted sample was measured using a Qubit 4 fluorometer instrument (Invitrogen).
Measurement of cfDNA concentration by digital droplet PCR assay
The digital droplet PCR (ddPCR) assay was performed as previously described (Gai et al. 2023) for a selection of 25 samples from EBV-negative cohort as validation. We have adopted this method to target the VCP, using one set of primer and probe. Briefly, ddPCR reactions were performed by the QX ONE droplet digital PCR system (Bio-Rad). The DNA samples used were eluted with the same volume during DNA extraction. The reaction was prepared in 20 µL, with equal volumes of DNA added per reaction. Components added to the reaction include 2× ddPCR supermix for probes (Bio-Rad), final concentration of 900 μmol/L of each primer, and 250 nmol/L of the DNA probe. The ddPCR thermal profile consisted of an initial incubation for 30 min at 37°C followed by the denaturation step for 10 min at 95°C . It was further followed by 45 cycles of amplification, with each cycle including a denaturation 30 sec at 94°C for and annealing for 1 min at 57°C. A final incubation for 10 min at 98°C was carried out. Data and calculations were Poisson corrected. To measure the cfDNA concentration, the number of DNA templates for the VCP gene was quantified from cfDNA samples. The primer sequence used for this assay was
-
VCP forward primer, 5′-GGGAGGTCTGTGGACCCTATC-3′;
-
VCP reverse primer, 5′-GGGAGGTCTGTGGACCCTATC-3′; and
-
probe sequence, 5-AMCTTCCCCAACCATCAGMGB-3′.
Target-capture enrichment and analysis
Routine clinical screening for EBV DNA for NPC detection involves target capture enrichment of viral DNA molecules from the total pool of plasma DNA, as it improves the positive predictive value of NPC detection (Lam et al. 2018). Enrichment in the screening study was performed using a hybridization-based capture with probes that cover the entire EBV genome and selected human autosomal DNA regions. Data from the target capture of all subjects (862 subjects) were retrieved for analysis. Only off-target DNA fragments were analyzed in this study to avoid the effects of target capture on fragmentomic analysis. Off-target reads were defined as DNA fragments with no full or partial alignment to the human autosomal target sites. The robustness of using “off-target” reads for fragmentomic analysis has been validated in comparison to the paired genome-wide sequencing data (Supplemental Figs. S8, S9).
Genome-wide sequencing DNA library preparation
For the selected subjects with the highest and lowest concentrations of cfDNA in the EBV-positive and EBV-negative cohort, genome-wide sequencing was performed without target capture. Extracted DNA from 2 mL of plasma was performed using TruSeq DNA nano library prep kit (Illumina), with purification steps done using MinElute reaction cleanup (Qiagen) according to the manufacturer's instructions. Adaptor-ligated DNA was amplified using eight cycles of PCR. Quality control of the prepared libraries was done by Qubit and Agilent 4200 TapeStation (Agilent).
Sequencing and alignment
Target-captured DNA libraries were sequenced on the NextSeq 500 System (Illumina) with 75 bp × 2 (150 cycles) paired-end sequencing. Sequenced DNA was aligned to the human genome GRCh37 (hg19). Fragmentomic analysis was performed only using the paired-end reads uniquely aligned to the human autosomal chromosomes, which was consistent with our previous work (Jiang et al. 2020). Reanalysis of the existing data using the GRCh38 (UCSC hg38) human reference genome would not significantly alter the results, as the major difference between the two versions of the human reference genomes is the sequence representation for highly repetitive regions and centromeres. Short sequencing reads obtained from those regions would have multiple alignments and would therefore not be utilized in our downstream analysis.
Analysis of 5′-end motifs
Our analysis of end motif profiles was performed as previously described (Serpas et al. 2019; Chan et al. 2020). We analyzed the first 4 nt sequence (termed the 4-mer end motif) at each 5′ fragment end of plasma DNA molecules. The frequency of each of the 256 possible 4-mer end motifs (44 combinations) was calculated and normalized by the total number of ends. In our analysis, we correlated the frequency of each end motif to the plasma cfDNA concentration (n = 862 subjects). The P-value of each correlation was adjusted using Bonferroni's correction for multiple comparisons (Supplemental Tables 2, 3). Our analysis is limited to the 5′ end motif, as the identity of the original 3′ end motifs will be modified during the end repair step, as reported in our previous work (Jiang et al. 2020).
Analysis of the contribution of F-profiles
The data matrix and deduced percentage contributions of the six F-profiles were performed as previously described (Zhou et al. 2023). Briefly, the percentage contribution of each F-profile in a cfDNA sample was deduced using nonnegative least squares–based deconvolution analysis of the previously established data matrix. We performed the F-profile analysis using DNA fragments stratified into different size ranges: 20–160 bp, 161–230 bp, and 231–600 bp. Multiple linear regression was performed on all the significantly correlated F-profiles in each size range.
Deduction of the total cfDNA concentration or fractional cfDNA concentration using fragmentomic features
The SVR model was built by the “e1071” package in R 4.1.2. The 862 subjects from the target sequencing data set were randomly grouped into training set (n = 431 subjects) and testing set (n = 431 subjects) without overlap. The training data set was used to build a SVR model to predict the cfDNA concentration based on fragmentomic features. Frequencies of 4-mer motifs (256 motifs) and frequencies of cfDNA at different sizes (20∼600 bp; 581 sizes) were used as features to build the model. The prediction target was the cfDNA concentration after logarithmic transformation. The default radial kernel was used in the SVR model, and the gamma and cost values were tuned using the function “tune.svm.” After the model was built, the testing data set was used to evaluate the predictive accuracy, with 20 subjects from EBV-negative cohort were used as a second testing data set.
The SVR models were built using the leave-one-out strategy to predict fetal fraction in pregnant subjects (n = 30) and tumor fraction in patients with HCC (n = 20). The same fragmentomic profiles (i.e., end motif and size) were used as training features, with the fetal fractions and tumor fractions as target values for building the SVR model. The samples were from previously published sequencing data (Jiang et al. 2020). Raw sequencing data for HCC and pregnancy cases were obtained from a previous study (Jiang et al. 2020), from the European Genome-phenome Archive (EGA; https://ega-archive.org) under accession number EGAS00001003409. The predicted fetal and tumor fraction based on fragmentomic features were correlated to the fractions quantified by FetalQuant and ichorCNA, respectively. To demonstrate the robustness of the SVR models, we included another 20 HCC patients and 30 pregnant subjects as validation (Supplemental Fig. S19). To identify the most informative fragmentomic features in predicting fetal fraction and tumor fraction, we performed LASSO regressions using the first data set of 30 pregnant women and 20 patients with HCC (Supplemental Tables 9, 10). The LASSO regression coefficients were used to indicate the importance of each feature.
Quantitative analysis of DNASE1L3 levels in plasma
DNASE1L3 levels in plasma between selected samples were evaluated using the Jess automated western blotting system (ProteinSimple). Fifty-microliter aliquots of plasma were diluted 200-fold using 1 × phosphate-buffered saline (PBS) and mixed with 2.5× fluorescent master mix containing DTT. The samples were boiled for 5 min to 95°C. The samples were loaded onto the 12 × 230 kDa separation module containing 25 capillary cartridges with the following settings: separation voltage 375 volts, 25 min; “primary antibody time” and “secondary antibody time” as 30 min; “detection profile–chemi”; and DNASE1L3 was immuno-detected using polyclonal anti-DNASE1L3 antibodies (Thermo Fisher Scientific PA5-107113) and secondary anti-rabbit antibodies (anti-rabbit detection module, DM001 protein simple). Chemiluminescent signals were quantified by band intensity area using Compass for SW software. Samples were run in two replicates, with the correlation between the two replicates shown in Supplemental Figure S14.
Tissue-of-origin analysis by FRAGMA
FRAGMA-based tissue deconvolution analysis is based on the deduction of the methylation status of cytosine–phosphate–guanine
(CpG) sites, by the preferential cleavage of methylation sites compared with unmethylated sites, using the CGN/NCG motif ratios
as previously described (Zhou et al. 2022). Hypermethylated and hypomethylated CpG sites are defined as CpG sites with a methylation index of >70% and <30%, respectively.
Unique hypermethylated CpG sites for each cell type (liver, neutrophils, B cells, T cells, erythroblasts, and megakaryocytes)
were identified by bisulfite sequencing tissue references for each cell type. The percentage contribution of each tissue was
deduced by the difference in CGN/NCG motif ratio between hyper- and hypomethylated CpG sites, normalized by the CGN/NCG motif
ratio from the reference tissue, using the following equation:
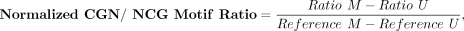 where “ratio” denotes the raw CGN/NCG motif ratio, and “M” and “U” represent methylation and unmethylated sites, respectively.
This equation has been described in a patent application (US2023/0374601–“fragmentation for measuring methylation and disease”)
prior to this work.
where “ratio” denotes the raw CGN/NCG motif ratio, and “M” and “U” represent methylation and unmethylated sites, respectively.
This equation has been described in a patent application (US2023/0374601–“fragmentation for measuring methylation and disease”)
prior to this work.
Data collection of clinical lab test parameters
Whole blood, serum, and plasma samples from individuals of the follow-up blood collection were subjected to a detailed clinical laboratory analysis in the Prince of Wales Hospital, from the Immunology, Hematology and Chemical Pathology Laboratory, run by the Hospital Authority (Hong Kong). Data from these parameters outlined in Supplemental Table 5 were provided and used in this study.
Statistical analysis
Sequencing analyses were performed by bioinformatic programs written in the Perl and R languages. The P-values for all comparisons are stated in the figures and results section. The Pearson correlation coefficient, r, was used to assess correlations. Statistical comparisons between two groups were performed using the Wilcoxon rank-sum test with two-tailed comparison. Statistical tests and plots were performed using R (R Core Team 2024) and GraphPad Prism 9. A value of P < 0.05 was considered statistically significant.
Data access
All raw and processed sequencing data generated in this study have been submitted to the European Genome-Phenome Archive (EGA; https://ega-archive.org/), under accession number EGAS50000000692.
Competing interest statement
K.C.A.C. and Y.M.D.L. hold equities in DRA, Take2, and Insighta. P.J. and W.K.J.L. hold equities in Illumina. P.J. is a consultant to KingMed Future. W.P. is a consultant to Take2. K.C.A.C. and P.J. are directors of DRA, Take2, KingMed Future, and Insighta. W.K.J.L. is a director of DRA. Y.M., G.K., W.K.J.L., P.J., K.C.A.C., and Y.M.D.L. have filed a patent application based on the data in this study. Patent incomes are received from Grail, Illumina, DRA, Take2, Insighta, and Xcelom.
Acknowledgments
This work was supported by the Innovation and Technology Commission of the Hong Kong SAR Government (InnoHK initiative) and the Li Ka Shing Foundation. We thank the Biomedical Technology Support Centre of the Hong Kong Science and Technology Parks for instrument use and technical support of the Jess automated Western Blotting (Protein Simple) device. We thank Chris Kum and Xingqian Li for their technical assistance.
Author contributions: Y.M., G.K., W.K.J.L., Q.Z., P.J., K.C.A.C., and Y.M.D.L. designed research. Y.M., G.K., W.K.J.L., Q.Z., S.H.C., P.P.H.C., J.B., M.L.C., C.T.L., Y.Z., W.G., W.W.S.W., M.-J.L.M., X.X., Z.G., and H.S. performed research. W.K.J.L., I.O.L.T., L.Y.L.C., and K.C.A.C. took part in clinical recruitment. Y.M., G.K., W.K.J.L., Q.Z., J.B., W.P., W.W.S.W., W.L., P.J., K.C.A.C., and Y.M.D.L. analyzed data and performed statistical analysis. Y.M., G.K., W.K.J.L., P.J., K.C.A.C., and Y.M.D.L. wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279667.124.
-
Freely available online through the Genome Research Open Access option.
- Received June 5, 2024.
- Accepted November 25, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








