
The Chinese longsnout catfish genome provides novel insights into the feeding preference and corresponding metabolic strategy of carnivores
- Yulong Liu1,2,9,
- Gang Zhai1,9,
- Jingzhi Su3,9,
- Yulong Gong1,
- Bingyuan Yang4,
- Qisheng Lu1,2,
- Longwei Xi1,
- Yutong Zheng1,2,
- Jingyue Cao1,2,
- Haokun Liu1,
- Junyan Jin1,
- Zhimin Zhang1,
- Yunxia Yang1,
- Xiaoming Zhu1,
- Zhongwei Wang1,5,
- Gaorui Gong6,
- Jie Mei5,6,
- Zhan Yin1,
- Rodolphe E. Gozlan7,
- Shouqi Xie1,2,8 and
- Dong Han1,2,5
- 1State Key Laboratory of Freshwater Ecology and Biotechnology, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, Hubei 430072, China;
- 2College of Advanced Agricultural Sciences, University of Chinese Academy of Sciences, Beijing 100049, China;
- 3Wuhan DaBeiNong (DBN) Aquaculture Technology Company Limited, Wuhan, Hubei 430090, China;
- 4Department of Infectious Diseases and Public Health, Jockey Club College of Veterinary Medicine and Life Sciences, City University of Hong Kong, Kowloon, Hong Kong 999077, China;
- 5Hubei Hongshan Laboratory, Wuhan, Hubei 430070, China;
- 6College of Fisheries, Huazhong Agricultural University, Wuhan, Hubei 430070, China;
- 7ISEM, Université de Montpellier, CNRS, IRD, 34090 Montpellier, France;
- 8The Innovative Academy of Seed Design, Chinese Academy of Sciences, Beijing 100049, China
-
↵9 These authors contributed equally to this work.
Abstract
Fish show variation in feeding habits to adapt to complex environments. However, the genetic basis of feeding preference and the corresponding metabolic strategies that differentiate feeding habits remain elusive. Here, by comparing the whole genome of a typical carnivorous fish (Leiocassis longirostris Günther) with that of herbivorous fish, we identify 250 genes through both positive selection and rapid evolution, including taste receptor taste receptor type 1 member 3 (tas1r3) and trypsin. We demonstrate that tas1r3 is required for carnivore preference in tas1r3-deficient zebrafish and in a diet-shifted grass carp model. We confirm that trypsin correlates with the metabolic strategies of fish with distinct feeding habits. Furthermore, marked alterations in trypsin activity and metabolic profiles are accompanied by a transition of feeding preference in tas1r3-deficient zebrafish and diet-shifted grass carp. Our results reveal a conserved adaptation between feeding preference and corresponding metabolic strategies in fish, and provide novel insights into the adaptation of feeding habits over the evolution course.
Teleost fishes are the most successfully evolved of aquatic animals, displaying wide morphological, feeding, and metabolic diversity to adapt to complex living environments (Gui et al. 2022). Over millions of years of evolution, these vertebrates have developed distinct anatomical features related to feeding habits and food use, including the structure of the oropharyngeal and digestive organs, and have ultimately differentiated according to major feeding habits (Rangel et al. 2016; Bočina et al. 2017). These habits or niches include a carnivorous diet, a herbivorous diet, and an omnivorous diet (Hofer and Schiemer 1981; Burin et al. 2016; Kim et al. 2016). Recent studies have revealed the roles of olfactory and gustatory organs in fish feeding preferences, which contribute to dietary adaptation (Okada 2015; Kasumyan 2019; Levina et al. 2021), but little is known about the genetic basis or physiological response of metabolic strategies associated with fish feeding preferences. Therefore, understanding the genetic basis of feeding preferences and the corresponding metabolic strategies will provide important information on the evolution of vertebrate feeding habits and metabolism.
Deciphering the differentiation of feeding habits is one of the most important factors for understanding speciation (Grant and Grant 2006; Jiao et al. 2019). In cichlid fishes, long-term dietary selection for plastic traits can lead to allopatric speciation (Steinberg 2018). It has been reported that the oldest large herbivores evolved from small carnivorous terrestrial vertebrates (Reisz and Fröbisch 2014). Some teleost fish are carnivorous in the larval stage, and they switch from a carnivorous to a herbivorous diet as they mature (Wu et al. 2009; Wang et al. 2015; Liu et al. 2017; Gu et al. 2021). After digesting the yolk sac, they grow mainly by feeding on small planktonic animals (carnivores). As they mature, however, their feeding habits become more differentiated. To adapt to the environment in which they live and feed, fish become herbivorous, omnivorous, or carnivorous. Grass carp, a typical herbivorous fish, shifts from a carnivore feeding on plankton to a herbivore during the larval and juvenile stages (Wang et al. 2015), when the number of taste buds increases more than 10-fold (Wang et al. 1993). The dietary shift from carnivore to herbivore in grass carp provides an excellent model for understanding the genetic basis of dietary preferences between fish with different feeding habits by comparing its genome with that of other carnivorous fish.
Feeding, which involves a variety of biological processes such as food perception, absorption, and metabolism, is a complex quantitative trait consisting of food ingestion itself and foraging or appetitive behavior (Volkoff 2016). Feeding is regulated by central feeding centers in the brain, which process information from endocrine signals from both the brain and the periphery (Volkoff 2016). These signals include orexigenic hormones (e.g., neuropeptide Y-NPY; orexin; ghrelin) and anorexigenic hormones (e.g., cholecystokinin-CCK; cocaine and amphetamine-regulated transcript-CART) (Sobrino Crespo et al. 2014). Feeding is also affected by metabolic processes and neural signals that provide information on food ingestion and nutritional status (Ji et al. 2015; Lu et al. 2023). In addition, miRNA-based gene silencing has emerged as a key post-transcriptional mechanism that functions on a genome-wide scale to regulate feeding differentiation (Steinberg 2018). The complex regulation of feeding, the sensory system, the physiology of the digestive tract (e.g., trypsin), and the number of metabolic traits are closely linked to the adaptive plasticity of distinct feeding habits in fish (Volkoff et al. 2009; Hoskins and Volkoff 2012).
In fish, the sensory system includes the sense of taste, smell, etc. The sense of smell is important for the feeding behavior of fish (Kasumyan 2019; Kasumyan and Isaeva 2023), but the sense of taste is the key to the choice of food and therefore to the differentiation of feeding habits, because taste is the factor that determines the sensory characteristics of food (Yapici et al. 2016; Kasumyan 2019). The sense of taste is important in the differentiation of feeding preferences and feeding habits (Kasumyan 2019; Jiao et al. 2021). Taste is perceived via the taste receptor signaling pathway (including genes such as tas1rs) in the taste buds (Bruch et al. 1988; Bachmanov and Beauchamp 2007). In mammals, the taste receptors of the TAS1Rs have been reported to be highly sensitive to nutrients such as protein and amino acids (Nelson et al. 2002). In addition, the evolutionarily conserved microRNA (miR-263b) has been suggested to play a key role in the sensory system of Drosophila melanogaster (Klann et al. 2021). In fish, a few studies have been carried out on the taste receptor system in grass carp and mandarin fish, mainly focusing on the expression of TAS1Rs (Cai et al. 2018; Dou et al. 2018).
Feeding habits are also closely related to the adaptation of metabolic characteristics (Wang et al. 2020; Cheng et al. 2023). It has been suggested that specific proteolytic enzyme activity, protein metabolism, and dietary protein requirements are much lower in herbivorous species than in carnivorous species at the adult stage (Fish 1960; Hofer and Schiemer 1981). For grass carp, a decrease in protein requirement from 43.3% in the larval stage to 28% in the adult stage was observed (Dong et al. 2019; Yu et al. 2022). However, herbivorous fish are well known to have a greater capacity to use carbohydrates and higher amylase activity and expression than carnivores (Su et al. 2021). The amylase activity of grass carp was 54- to 82-fold higher than that of a carnivorous fish, the Chinese longsnout catfish (Leiocassis longirostris Günther), and grass carp showed a rapid increase and a faster clearance rate of plasma glucose after starch administration (Su et al. 2020). Although the adaptations in feeding habits and metabolic characteristics of carnivorous and herbivorous fish have been studied using differential gene expression, organ traits (Hofer and Schiemer 1981; Cai et al. 2018; Yuan et al. 2020; Ding et al. 2021), and morphology (He et al. 2013, 2020), the genetic basis of this adaptation during evolution has remained elusive.
To date, most Siluriformes fish for which genomes have been reported are omnivorous, including yellow catfish and channel catfish. Compared to herbivores, omnivores, and other Siluriformes fish, L. longirostris Günther is an excellent carnivorous fish model with a low efficiency in the utilization of food carbohydrate and a high demand for food protein content (≥45%). In the past decades, a vast majority of studies on nutrition, feeding behavior, immune response, and genetics of L. longirostris have been done (Zhu et al. 2004; Tan et al. 2007; Su et al. 2020; Dai et al. 2022). Therefore, we sequenced and assembled the whole genome of L. longirostris. We obtained candidate genes using whole-genome sequencing of Chinese longsnout catfish, as well as comparative genomics of carnivorous and herbivorous fish through a joint analysis of positive selection and rapid evolution. We then used functional studies to confirm the conserved role of candidate genes in the feeding preference and metabolic strategy in fish with distinct feeding habits. Our results uncovered a genetic basis for understanding the evolution of carnivorous fish and their adaptive traits in terms of feeding habits and metabolism.
Results
Chromosome-level genome assembly and evaluation of a typical carnivorous fish species
To identify differences in the genetic basis of fish feeding habits, we used third-generation Nanopores combined with Hi-C-assisted assembly technology to characterize the basic features of the L. longirostris (Fig. 1A) genome (Belton et al. 2012). Nanopore sequencing yielded 113.66 Gb of reads at a 151.55× coverage depth. In addition, 122.48 Gb of reads were obtained from an Illumina sequencing library for genome assembly. After Hi-C assisted assembly and removal of low-quality reads, we obtained a genome of 693.16 Mbp at the chromosome level, with an anchoring ratio of 99.32% (Fig. 1B,C; Supplemental Figs. S1–S3; Supplemental Tables S1–S4). The contig N50 was 8.98 Mbp, and the scaffold N50 was 27.70 Mbp. Genomic sequences were scaffolded into 26 chromosomes (Table 1); the Benchmarking Universal Single-Copy Orthologs (BUSCO; version: vertebrata_odb9) assessment was 96.5% (Supplemental Table S5); and the Core Eukaryotic Genes Mapping Approach (CEGMA) was 94.35% (Supplemental Table S6). Short fragment reads were aligned to the assembled genome using BWA software to assess complementation and consistency. The comparison ratio of 99.40% and the coverage ratio of 98.95% suggest that our assembled genome exhibits satisfactory uniformity (Supplemental Table S7). The heterozygous single-nucleotide polymorphism (SNP) ratio and the homozygous SNP ratio were 0.138745% and 0.001405%, respectively, indicating that the assembly has a high ratio of exact base pairs (Supplemental Table S8). Using the database of Swiss-Prot, Non-Redundant Protein Database (Nr), Kyoto Encyclopedia of Genes and Genomes (KEGG), InterPro, Gene Ontology (GO), and Protein Families (Pfam), the number of predicted genes was 24,216, of which 23,078 were annotated genes (Supplemental Fig. S4; Supplemental Table S9). The genome of L. longirostris had a syntenic relationship with the genome of Ictalurus punctatus and Pelteobagrus fulvidraco (Supplemental Fig. S5). We found that all chromosomes in P. fulvidraco have strong genomic collinearity with L. longirostris, suggesting that the relationship between them is very similar.
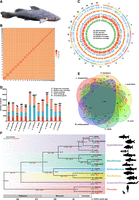
Chromosome-level genome assembly of the Chinese longsnout catfish (Leiocassis longirostris Günther) and the comparative genomics analysis with 10 species from mammals to teleosts. (A) Representative photo of Chinese longsnout catfish. (B) Genome-wide chromosomal contact map of Chinese longsnout catfish based on chromatin interaction data sets analyzed with Hi-C. Among chromosomes, a heat map from yellow to red intensity was used to represent the Hi-C links interaction frequency distribution (yellow to red represents low to high intensity). (C) Visualization of the whole-genome alignment is shown using the Circos plot (Krzywinski et al. 2009). Outer to the inner circles: chromosome length (each tick mark represents 1/5 Mb), gene density, GC percentage distribution (in nonoverlapping 100 kb windows), repeat percentage, Pacific Biosciences (PacBio) coverage, and Illumina coverage. (D) Distribution of genes of different types in different species. Single-copy genes and multiple-copy genes are the gene families found in the 13 species. Single-copy genes correspond to the number of genes in a gene family in which only one species has genes. Other genes refer to the number of genes in gene families that are not species specific. The number of nonclustered genes in each species. (E) Venn diagram showing the overlap of orthologous genes in Chinese longsnout catfish and five other fishes, including yellow catfish (Pelteobagrus fulvidraco), channel catfish (Ictalurus punctatus), zebrafish (Danio rerio), grass carp (Ctenopharyngodon idellus), and blunt snout bream (Megalobrama amblycephala). (F) Phylogenetic tree and time tree showing the expansion and contraction of gene families in 13 species ranging from mammals to teleosts, namely, grass carp (C. idellus), blunt snout bream (M. amblycephala), common carp (Cyprinus carpio), zebrafish (D. rerio), channel catfish (I. punctatus), yellow catfish (P. fulvidraco), Chinese longsnout catfish (L. longirostris Günther), rainbow trout (Oncorhynchus mykiss), giant grouper (Epinephelus lanceolatus), largemouth bass (Micropterus salmoides), sterlet (Acipenser ruthenus), Homo sapiens, and mouse (Mus musculus). The blue numbers indicate the times of divergence between the different species; the green numbers indicate the number of gene families that have expanded over the course of evolution; and the red numbers indicate the numbers of gene families that have contracted.
Characteristics of the Chinese longsnout catfish (Leiocassis longirostris Günther) whole-genome sequence
We analyzed the distribution of genes in L. longirostris and 12 other species ranging from mammals to teleosts (Supplemental Table S10). In L. longirostris, we observed similar numbers of single genes, single-copy genes, multiple-copy genes, and other genes as in yellow catfish and channel catfish (Fig. 1D). L. longirostris shared 14,983 gene families in common with channel catfish and 13,764 gene families in common with yellow catfish (Fig. 1E).
A phylogenomic tree for the 13 species mentioned above was established. Compared with the fishes of the Cyprinidae family, the evolutionary position of L. longirostris was closer to other fishes of the Siluriformes order, including the yellow catfish and the channel catfish. As a species in the family Cyprinidae, the grass carp is most closely related to the blunt snout bream. A chronological tree of L. longirostris and the 12 species mentioned above was constructed. The divergence between the Chinese longsnout catfish and yellow catfish dates to around 20.6 million years ago. Compared with its most recent ancestor, the L. longirostris showed 353 genes expansion events and 839 genes contraction events (P < 0.05) (Fig. 1F). The expansion of genes related to protein digestion and uptake, calcium ion transport, and protein dimerization activity was observed in L. longirostris, as well as the contraction of genes related to carbohydrate digestion and uptake and diabetes (Supplemental Tables S11–S14).
Joint analysis of positive selection and rapid evolution provides insights into the feeding preference and corresponding metabolic strategy
For positive selection analysis, we defined L. longirostris as the foreground branch, and blunt snout bream, grass carp, channel catfish, yellow catfish, common carp, and zebrafish as the background branch. A total of 379 L. longirostris genes were positively selected (false-discovery rate [FDR] < 0.05) (Fig. 2A). The positively selected genes of tas1r3, trypsin, p2y purinoceptor 1 (p2ry1) (Kinnamon and Finger 2013), tyrosine 3-monooxygenase (th), myosin light chain kinase (mylk), and cytosol aminopeptidase (lap3) are involved in taste transduction, pancreatic secretion, and protein metabolism pathways in L. longirostris (Supplemental Table S15).
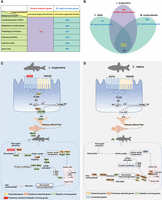
A representative diagram of the joint analysis of rapid evolution and positive selection in L. longirostris Günther. (A) Details of the analysis concerning positive selection and rapid evolution of Chinese longsnout catfish (L. longirostris Günther) compared with grass carp (C. idellus), blunt snout bream (M. amblycephala), yellow catfish (P. fulvidraco), channel catfish (I. punctatus), common carp (C. carpio), and zebrafish (D. rerio). For the positive selection analysis, Chinese longsnout catfish (L. longirostris) was defined as the foreground branch and other fish species as the background branch. The number of genes identified by positive selection is shown in red. For the rapid evolution analysis, the number of genes identified from L. longirostris compared with other fish is shown in blue. (B) List of genes (250 genes) in L. longirostris from the joint analysis of positive selection (379 genes, compared with other six fish) and rapid evolution analysis (1007 genes compared with grass carp; 1003 genes compared with blunt snout bream). (C,D) Diagram of expanded, contracted, positively selected, and rapidly evolving genes related with the feeding preference and corresponding metabolic pathways in Chinese longsnout catfish and grass carp. The genes involved in these pathways are labeled with different colors. The genes represented in orange are positively selected, and those represented in green have evolved rapidly. Genes in red are both positively selected and rapidly evolving. Genes in yellow are expanding, and genes in light blue are contracting.
Compared with grass carp, blunt snout bream, yellow catfish, channel catfish, common carp, and zebrafish, Chinese longsnout catfish exhibited 1007, 1003, 1517, 1470, 1045, and 987 genes, respectively, that evolved rapidly (Fig. 2A). Among the rapidly evolved genes compared between Chinese longsnout catfish and grass carp, htr1, calhm1, cck, npy2r, cela2, slc7a9, vars2, cars2, mars, hoga1, auh, and got1 of taste transduction, pancreatic secretion, and protein metabolism pathway were enriched (Supplemental Table S16). Rapidly evolved gene families of Chinese longsnout catfish compared with grass carp were enriched in GO terms, including amino acid metabolic process (GO:0006520, biological process), calcium ion binding (GO:0005509, molecular function), and somatostatin receptor activity (GO:0004994, molecular function) (Supplemental Table S17). Taking the intersection of genes included in the GO terms as indicating rapid evolution in the Chinese longsnout catfish relative to grass carp and blunt snout bream, as well as positively selected genes in Chinese longsnout catfish relative to blunt snout bream, grass carp, channel catfish, yellow catfish, common carp, and zebrafish, the tas1r3, trypsin and other 248 genes were identified (Fig. 2B; Supplemental Table S18), again suggesting that positively selected and rapidly evolving genes may contribute to taste transduction, amino acid metabolism, and protein synthesis in Chinese longsnout catfish.
In addition to the tas1r3 and trypsin genes, which were both positively selected and rapidly evolved, the expanded, positively selected, or rapidly evolved genes were also analyzed in Chinese longsnout catfish. Expanded genes pde1 and prdm7; positively selected genes p2ry1, lap3, th, and mylk; and rapidly evolved genes htr1, calhm1, cck, npy2r, cela2, slc7a9, vars2, cars2, mars, hoga1, auh, and got1 were mapped in pathways related to taste transduction, amino acid metabolism, and protein synthesis in Chinese longsnout catfish (Fig. 2C). In addition, in grass carp, genes that were contracted, expanded, positively selected, or rapidly evolved were analyzed. In the GO enrichment, significant GO terms related to feeding habits and metabolic strategy were observed; especially, the single contracted gene (odc1), the expanded genes (dak and pfkfb2), the positively selected genes (treh, slc2a9, ppp1r3g, pgk, chrebp, mdh1, and cs), and the rapidly evolved genes (lpf1 and hk) were mapped into pathways involved in carbohydrate metabolism in grass carp, among which the carbohydrate metabolic process GO term (GO:0005975) was the most abundantly enriched (Fig. 2D; Supplemental Table S19). These results showed that fish with different metabolisms could be identified at the genomic level by their adaptive traits for feeding habits, as revealed by the expanded genes and positively selected genes obtained from the comparative genomic analysis that were illustrated (not compared) (Fig. 2C,D).
Validation of the genetic adaptation of feeding preference
Grass carp, which undergo a natural transition from a carnivorous to a herbivorous diet, are an excellent animal model for the study of feeding habits' transition. To determine whether TAS1R3 plays a role in fish feeding preference, we examined the tas1r3 transcript levels before and after the transition in feeding habits in grass carp (Fig. 3A). It has been suggested that the oropharynx, brain, and intestine are the main tissues for the expression level of the gene tas1r3 in grass carp (Cai et al. 2018). Here, we observed that transcript levels of tas1r3 were significantly downregulated in the oropharyngeal, brain, and intestinal tissues of grass carp following the feeding habit transition (FHT) from carnivore of chironomid larvae to herbivore of Hydrilla verticillata (Fig. 3B). We observed an upregulation of tas1r3 in adult L. longirostris compared with the juvenile stage (Supplemental Fig. S6). Compared to those before FHT, the expression of key genes in the taste transduction pathway (pde1a, plcb2, and p2ry1) was significantly downregulated in the oropharyngeal tissues of grass carp after FHT from carnivore to herbivore (Supplemental Fig. S7A). In fish, feeding is regulated by central effectors in the brain that process information from endocrine signals (e.g., CCK and NPY) from both the brain and peripheral tissues. The food intake–related genes, cck and npy2r, in the brain were significantly downregulated in grass carp after the shift in feeding habits (Supplemental Fig. S7B). To elucidate the function of tas1r3 in fish feeding preference, tas1r3-deficient zebrafish were generated using CRISPR-Cas9 technology. Two knockout lines with 1 and 11 bp deletions were obtained, resulting in premature protein translation (Fig. 3C). PCR products with genomic DNA and cDNA were used as templates for mutation validation (Fig. 3D). The relative expression levels of tas1r3 in the oropharyngeal, brain, and intestinal tissues were extremely downregulated in tas1r3-deficient zebrafish compared with those in the wild-type control fish (Fig. 3E). This is probably because the mutant mRNA of the target gene containing a premature stop codon could be degraded by the nonsense mRNA decay mechanism (Popp and Maquat 2016).
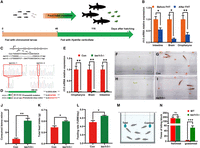
tas1r3 is the determinant element in fish feeding preference. (A) Schematic of the sampling timeline in grass carp before and after the feeding habit transition (FHT) from carnivores of chironomid larvae to herbivores of Hydrilla verticillata. (B) Expression of tas1r3 in the intestinal (n = 6), brain (n = 4), and oropharyngeal (n = 8) tissues of grass carp before or after the FHT from carnivory to herbivory. Grass carp before and after the FHT were sampled when they reached a body length of 2.82 ± 0.18 cm and 6.21 ± 0.63 cm at 46 and 116 days post hatching, respectively. (C) Generation of two independent knockout lines of zebrafish, tas1r3-1 and tas1r3-2, contained deletions of 1 bp and 11 bp, respectively. (D) Predicted protein sequence encoded by the wild-type control or the two tas1r3 mutated zebrafish. (E) Relative expression of tas1r3 in the oropharynx, brain, and intestinal tissues of the wild-type control or tas1r3-deficient zebrafish (n = 3). (F–I) Assay of food intake in zebrafish fed duckweed. (F,H) Images of wild-type control and tas1r3-deficient zebrafish before feeding duckweed, respectively. (G,I) Images of wild-type control and tas1r3-deficient zebrafish after feeding duckweed for 12 h, respectively. (J) Amount of duckweed consumed by wild-type control or tas1r3-deficient zebrafish in 12 h (n = 10). (K) Total food intake of soybean meal diet by wild-type control or tas1r3-deficient zebrafish (n = 3). (L) Feeding rate of soybean meal diet consumed by wild-type control or tas1r3-deficient zebrafish (calculated on a body weight basis per day; n = 3). (M) Experimental equipment for analyzing the biting times of fishmeal and grassmeal blocks for zebrafish. (N) Biting times of fishmeal and grassmeal blocks in control and tas1r3-deficient zebrafish (n = 8). (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (ns) not significant.
We found that when fed duckweed after 24 h of starvation, tas1r3-deficient fish showed a significantly higher intake of duckweed than wild-type control fish (Fig. 3F–J). Subsequently, a 7 day feeding trial was carried out using zebrafish fed a soybean meal (SM) diet. It was observed that tas1r3-deficient fish had a significantly higher feed intake than did wild-type control fish after being fed the plant-based diet (Fig. 3K). A significant increase in feeding rate was also observed in tas1r3-deficient fish compared with wild-type control fish (Fig. 3L). Our further experiments suggested that the appetite of the tas1r3-deficient fish fed on a normal (Artemia salina) diet is not affected compared with the wild-type control fish (Supplemental Fig. S8). In addition, biting times of fishmeal and grassmeal blocks in wild-type control and tas1r3-deficient fish were analyzed using experimental equipment that allowed the zebrafish to self-select different block types (Fig. 3M). The biting times of the grassmeal blocks of tas1r3-deficient zebrafish were significantly increased compared with those of the control groups (Fig. 3N; Supplemental Videos S1, S2).
To investigate the underlying mechanism for the downregulation of tas1r3 during the diet shift from carnivory to herbivory, we performed microRNA sequencing analysis and filtered 50 differentially expressed miRNAs in diet-shifted grass carp, including 31 upregulated miRNAs and 19 downregulated miRNAs (Fig. 4A). We found that the 3′ UTR of tas1r3 contained the putative miR-216b binding site (Fig. 4B). Using stem-loop RT-PCR, miR-216b expression was significantly upregulated after the change in feeding habit (Fig. 4C). A luciferase assay confirmed that miR-216b significantly reduced the luciferase activities of pGL3-tas1r3-3′ UTR (Fig. 4D). These functional studies revealed that TAS1R3 is regulated by miRNAs, that it is linked to the adaptive traits of taste transduction, and that it may contribute to the feeding habits of carnivorous fish.
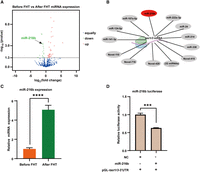
miR-216b is responsible for regulating the expression of tas1r3. (A) Volcano map of intestine miRNAs expression levels in grass carp before or after the FHT from carnivore to herbivore. (B) The miRNAs putative binding to tas1r3 3′ UTR. Gray means no significantly difference; red means significantly differentially expressed miRNAs of grass carp before and after the FHT. (C) The expression analysis of microRNA expression in intestine of grass carp before and after the FHT (n = 10). (D) Relative dual-luciferase activity analysis was performed to detect the effects of microRNA on tas1r3 3′ UTR activity (n = 3). (***) P < 0.001, (****) P < 0.0001.
Feeding preference affects metabolic strategy in fish
First, we examined the transcript level of hepatic trypsin in tas1r3-deficient zebrafish. Inactivation of tas1r3 decreased the level of trypsin transcription (Fig. 5A). We also found that in diet-shift grass carp, the transcript level and enzymatic activity of Trypsin coincided with a reduction in the expression of tas1r3 following the diet switch from carnivore to herbivore (Fig. 5B,C). Furthermore, the metabolic strategy of grass carp was altered after the change in feeding preference, with evidence of decreased plasma total amino acids and increased enzymatic activity of α-amylase, live glucokinase (GCK), and pyruvate kinase (PK) (Fig. 5D–G), suggesting an association of metabolic strategy corresponding to feeding preference. To validate this association in fish feeding habits, we performed a hepatic transcriptome analysis and compared metabolic profiles before and after the transition in feeding preferences of grass carp. The results showed that the hepatic differentially expressed genes (DEGs) belonging to the glucose metabolism and amino acid metabolism pathways were significantly enriched, suggesting that adaptation of the feeding habit is closely linked to metabolic preference. Based on the heat map, most DEGs in the glucose metabolism pathway were upregulated, and DEGs in the amino acid metabolism pathway were downregulated after the transition in feeding preferences of grass carp (Fig. 5H). The transcriptome results were verified by RT-PCR (Fig. 5I–J). Furthermore, tas1r3-deficient zebrafish showed a metabolic strategy similar to that of herbivorous fish, as the transcription levels of hepatic genes related to amino acid metabolism were consistent with the reduction in trypsin expression (Fig. 5K).
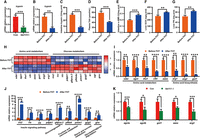
Feeding preference affects metabolic traits in fish. (A) Relative expression of hepatic trypsin in wild-type control or tas1r3-deficient zebrafish (n = 8). (B) Relative expression of hepatic trypsin in grass carp before and after the FHT from carnivores of chironomid larvae to herbivores of H. verticillate (n = 6). (C–G) Intestinal trypsin activity (n = 6), plasma TAA (n = 6), intestine α-AMS (n = 6), and hepatic GCK (n = 4), and PK activities (n = 4) of grass carp before and after the FHT from carnivory to herbivory. (H) Heat map of hepatic differentially expressed genes (DEGs) associated with amino acid and glucose metabolism in grass carp before or after the FHT from carnivory to herbivory. (I,J) qPCR validation of RNA-seq results for amino acid and glucose metabolism in grass carp before or after the FHT from carnivory to herbivory (n ≥ 4). (K) Relative expression of hepatic genes related to amino acid metabolism in wild-type control or tas1r3-deficient zebrafish (n ≥ 6). (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001.
Validation of the relationship between feeding preferences and corresponding metabolic strategies in fish with distinct feeding habits
Compared with herbivorous species (grass carp Ctenopharyngodon idellus and blunt snout bream Megalobrama amblycephala), carnivorous fish with high dietary protein requirements and a low capacity to utilize dietary carbohydrates (Chinese longsnout catfish L. longirostris and largemouth bass Micropterus salmoides) showed higher trypsin activities (Fig. 6A) and higher plasma amino acid levels (Fig. 6B). Furthermore, gut trypsin activities were consistent with feeding activity, as higher levels of animal protein in the feed can significantly increase trypsin activity regardless of how the fish are fed (Fig. 6C). Consistent with trypsin activity, there was an increase in plasma TAA and liver GOT and GPT in grass carp fed a fishmeal diet (Fig. 6D–F), and the expression levels of corresponding genes related to protein and amino acid metabolism were increased (Fig. 6G). However, when ingesting the plantmeal diet, the relative expression of genes related to glucose metabolism increased significantly (Fig. 6H). Similarly, in L. longirostris, the transcription of genes linked to amino acid metabolism and protein digestion was more active in the fishmeal group, whereas plantmeal induced the expression of genes linked to glucose metabolism (Fig. 6I,J).
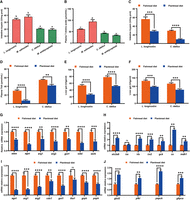
The relationship between feeding preferences and corresponding metabolic strategies in fish with distinct feeding habits. (A,B) Intestinal trypsin activities and plasma TAA contents in two typical carnivorous fish, Chinese longsnout catfish (L. longirostris Günther) and largemouth bass (M. salmoides), and two typical herbivorous fish, grass carp (C. idellus) and blunt snout bream (M. amblycephala) (n ≥ 7). Values marked with different superscripts above the bars indicate significant differences (P < 0.05). (C–F) Intestinal trypsin activities, plasma TAA contents, and GOT and GPT activities of the liver of L. longirostris and C. idellus fed a fishmeal or plantmeal diet, respectively (n = 6). (G,H) Relative expression of hepatic genes related with amino acid metabolism and glucose metabolism in grass carp (C. idellus) fed a fishmeal or plantmeal diet, respectively (n = 12). (I,J) Transcriptional level of hepatic genes related with amino acid and glucose metabolism in L. longirostris fed a fishmeal or plantmeal diet, respectively (n = 12). (ns) not significant, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001.
Discussion
With the development of sequencing technology, whole-genome sequencing analysis has become a powerful tool to reveal the evolution of animal traits. In the present study, we sequenced and assembled the genome of Chinese longsnout catfish, L. longirostris Günther. To our knowledge, this is the second chromosome-level genome with high-quality genome annotations of this typical carnivorous fish. Compared with the genome assembled by He et al. (2021), the current genome of Chinese longsnout catfish is 693.16 Mbp in size (previously 685.53 Mbp), with a contig N50 8.98 Mbp (previously 4.29 Mbp) and a chromosomal anchoring rate of 99.32% (previously 97.44%). These data suggest that the genome sequenced in this study is of higher quality, and the high-quality genome serves as an invaluable resource for functional genomics studies and provides crucial information for comparative genomics in the study of the evolution of special traits.
The feeding habits of fish species are diverse, and they may represent critical nodes in the evolution of vertebrate feeding habits (Chakrabarti et al. 1995; Cai et al. 2018; He et al. 2020). It has been proposed that the most primitive and oldest large herbivores evolved from small carnivorous vertebrates (Reisz and Fröbisch 2014). Therefore, insight into the transition of feeding habits from carnivores to herbivores is essential for understanding the evolution of feeding habits. The evolution of carnivory has previously been analyzed using comparative genomics of carnivorous, omnivorous, and herbivorous mammals previously (Kim et al. 2016). The carnivore genomes showed evidence of shared evolutionary adaptations in genes associated with diet and a clear contraction in gene families involved in starch and sucrose metabolism (Kim et al. 2016). However, given that mammalian speciation is derived from ancient fish, it may be useful to step back and explore the feeding preferences and corresponding metabolic strategy of carnivorous fish to better understand the basal and natural characteristics of genomic adaptations.
The genes identified by comparative genomics were broadly enriched in taste/appetite regulation between carnivorous and herbivorous fish. Feeding habits are assumed to be linked to taste perception (Li et al. 2005; Shi and Zhang 2006; Feng and Zhao 2013). Previous studies have shown that TAS1R1 pseudogenization is linked to the herbivorous feeding habits in other vertebrate species, such as the giant panda, which mainly feeds on bamboo, indicating that these evolutionary adaptations are indeed closely related to the feeding preference and food habits (Li et al. 2010; Hu et al. 2017). In the present study, the taste transduction of tas1r3, identified through the joint analysis of positive selection and rapid evolution, and its downstream genes (expanded gene pde1, positively selected gene p2ry1, and GO rapidly evolved genes calhm and htr1) were screened out for specific carnivore-related genomic features. Here, we further investigated the function of the tas1r3 gene and highlighted its role in feeding preference using the zebrafish model. Compared with the whole genome with the allotetraploid common carp (50 chromosomes), which has been duplicated, resulting in doubled chromosomes, the number of chromosomes in zebrafish (25 chromosomes), grass carp (24 chromosomes), Chinese longsnout catfish (26 chromosomes, this study), and yellow catfish (26 chromosomes) is almost comparable. More specifically, a single copy of tas1r3 has been reported in these fish, including zebrafish and grass carp (Cai et al. 2018), and a duplicated tas1r3 is present in the genome of common carp (located on Chromosomes A11 and B11, respectively, as annotated in the NCBI database).
For the tas1r3 in zebrafish, two independent knockout lines were established. The mutation of the tas1r3 has been validated both in the DNA and RNA levels of tas1r3-deficient zebrafish. The reduced expression of tas1r3 itself is observed in tas1r3-deficient fish. We know that the knockout of the specific gene should be examined using the antibody against the target protein by western blot. Unfortunately, before and during the submission and revision of this paper, we tried several times to generate antibodies against Tas1r3, but that did not work well in examining the Tas1r3 in zebrafish. Despite this, the feeding preference phenotype exhibited by tas1r3-deficient fish is specific, as assessed by the zebrafish in a double-blind feeding experiment. Collectively, we think that these feeding behavior experiments of tas1r3-deficient zebrafish provided direct evidence that Tas1r3 is essential for carnivores. Meanwhile, currently, we also could not completely rule out the possibility that the CRISPR-Cas9-mediated knockout procedure might also affect other genes rather than the target genes. We agree that the effects on sweet taste caused by tas1r3 depletion could not be excluded yet, as Tas1r3 has been reported to be involved in sweetness by interacting with Tas1r2 to form a heterodimer (Nuemket et al. 2017). However, we validated that tas1r3 plays an important role in regulating feeding habits, a phenotype exhibited by Chinese longsnout catfish. This is not in contrast to its function as a heterodimer, which does not affect the conclusion of this study. The adult L. longirostris showed an upregulation of tas1r3 compared with the juvenile stage. Although the functional study of tas1r3 using the genetic manipulation in L. longirostris is difficult to achieve owing to the long period of sexual maturation (usually takes 4–5 years), the positive correlation between upregulated tas1r3 and FHT was observed. In fact, many genes are known to vary between different stages of fish, and such changes have been helpful in deciphering their roles, as they correspond to developmental cascades, such as reproduction (He et al. 2014), metabolism (Li et al. 2024), or feeding preference (in this study). On the other hand, the feeding habits of grass carp after FHT from carnivore to herbivore during the larval and juvenile stages are accompanied by the tas1r3 downregulation, which was regulated by miR-216b. PLCB2, coexpressed with taste receptors in rat taste bud cells, forms a taste transduction pathway consisting of Gi2 and PLCB2 that occurs in a subset of taste cells (Asano-Miyoshi et al. 2000; Miyoshi et al. 2001). The significantly downregulated plcb2 in the oropharyngeal tissues of grass carp after FHT from carnivore to herbivore confirms that the Tas1rs-Plcb2 signals are important for taste transduction in fish.
Feeding habits are closely linked to metabolic characteristics that are associated with the digestive physiology and metabolic strategies (Jones et al. 1997; Polakof et al. 2012; Hecker et al. 2019). Digestive enzymes, including trypsin and amylase, play crucial roles in food digestion and adaptation to feeding habits (Yi et al. 2013). The genes of trypsin and amylase evolving rapidly or being positively selected have been reported to be consistent with feeding habits (Chen and Zhao 2019). In the present study, trypsin was shown to be both positively selected and rapidly evolving in the carnivore (L. longirostris), accompanied by many genes clustered in protein or amino acid metabolism according to the comparative genomic analysis. In contrast to carnivores, these positively selected, and rapidly evolved genes in the herbivore (Ctenopharyngodon idella) were largely involved in carbohydrate metabolism. Dietary shifts are known to alter the nutrient properties and thus the associated metabolic adaptations in animals (Zhou et al. 2014; Jiao et al. 2019; Huang et al. 2021). These results demonstrate that different metabolic characteristics are responding to feeding habit traits at the genomic level. It should be noted there have been many studies on the relationship between the host gut microbiota and dietary habits (Liu et al. 2016; Xu et al. 2022). The microbiome of fish species vary greatly depending on their living environment. Although not studied here, we believe that future research on the microbiome of fish using standard rearing trails and systemic analysis will provide additional insights into the biology of feeding behavior.
The further investigations of the metabolic profiles in grass carp suffering from FHT, the comparison between L. longirostris and M. salmoides versus C. idellus and M. amblycephala, and the feeding trials in L. longirostris and C. idellus all indicated that the metabolic strategies were preferred to protein metabolism in carnivores and glucose metabolism in herbivores. We speculate that carnivores and herbivores have undergone adaptive evolution in metabolic processes to respond to differentiated feeding habits. This may also be supported by the contraction of gene families for starch and sucrose metabolism in mammalian carnivore genomes (Kim et al. 2016). We therefore attribute it to the fact that carnivores ingest animal source foods containing a high content of protein and amino acids, whereas herbivores consume plant source foods with a high content of carbohydrates (Wilson 2003; Polakof et al. 2012; Teles et al. 2020). These data revealed that the metabolic strategies are strongly related to the feeding preferences of fish with different feeding habits, as well as a previously unrecognized link between fish feeding preference and metabolic strategy.
Compared with that of carnivores, the diet of herbivores is fibrous and low in lipid, which has more plant-derived galactolipids than animal material (Willmott et al. 2005). Galactolipids are the major lipids found in the membranes of plants, algae, and photosynthetic microorganisms (Sahaka et al. 2020). In herbivores such as fish, rabbits, horses, and ruminants, galactolipids may be the main source of dietary fatty acids owing to feeding habits and digestive physiology (De Caro et al. 2008). In herbivorous fish, unlike other animals, the pancreatic carboxyl ester hydrolase (CEH) is the main galactolipase involved in the digestion of galactolipids (Sahaka et al. 2020). It has been reported that the expression of genes involved in the digestion of galactolipids including bile salt-stimulated lipase/CEH, secretory phospholipase A2 (PLA2), and pancreatic elastase were significantly upregulated in grass carp after FHT from feeding with chironomid larvae Chironomus tentans to feeding with duckweed Lemna minor (He et al. 2015). This is in agreement with the present results on galactolipid digestion in the grass carp model before and after FHT, as well as bile metabolism (CYP8B1) (Supplemental Fig. S9). Therefore, these adaptations could be used to release and absorb the fatty acids from the galactolipids in food, which is important for the lipid metabolism of herbivores.
We recognized that although a number of complementary models were used in the present study, they may not adequately represent the general genetic basis of feeding habits in fish, as there are more than 32,000 species of fish, and distinct feeding habits have been classified (Nelson et al. 2016). Given the complexity of fish feeding habits, our results have at least revealed a potential genetic basis for the differentiation of feeding habits and the corresponding metabolic adaptations in fish. Further studies are needed to analyze the regulatory network of the Tas1r3 pathway regarding the interactions between trypsin activity and critical components of feeding habits.
In summary, we identified the function of tas1r3 in the feeding preference of carnivorous fish and that of trypsin in the associated metabolic strategy in fish with distinct feeding habits. The present study has revealed a previously unrecognized link between feeding preference and the corresponding metabolic strategy. We also demonstrated that altered metabolic strategies accompanied the transition of feeding preferences in several experimental models, which is important for coping with both the living environment and developmental stages. Our results provide genetic insight into the evolution of carnivores and the corresponding adaptive traits in terms of feeding preference and metabolism. Understanding the molecular mechanism of feeding habit differentiation could serve as a starting point for future functional studies of molecularly guided breeding, feeding enhancement through genetic manipulation, and diet-related metabolic syndromes.
Methods
Ethics
All fish experiments were conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals and were approved by the Institute of Hydrobiology, Chinese Academy of Sciences (approval number: IHB/LL/2021006).
Genome sequencing
The Hi-C library was prepared according to the standard protocol described by Belton et al. (2012) with certain modifications. The Hi-C library was constructed using muscle of the catfish as inputs. The muscle tissues were cross-linked with a 4% formaldehyde solution to fix the conformation of DNA. Cross-linked DNA was digested with a four-cutter restriction enzyme MboI (400 units) at 37°C on a rocking platform. The next steps involved labeling the DNA ends with biotin-14-dATP and ligating the cross-linked fragments to the blunt end. Proximal chromatin DNA was religated with the ligation enzyme. Nuclear complexes were reverse-cross-linked by incubation with Proteinase K at 65°C. DNA was purified by phenol–chloroform extraction. Biotin was removed from the ends of unligated fragments using T4 DNA polymerase. The ends of fragments sheared by sonication (200–600 bp) were repaired by a mixture of T4 DNA polymerase, T4 polynucleotide kinase, and Klenow DNA polymerase. Biotin-labeled Hi-C samples were specifically enriched using streptavidin C1 magnetic beads. After addition of A-tails to fragment ends and ligation with Illumina paired-end (PE) sequencing adapters, Hi-C sequencing libraries were amplified by PCR and sequenced on an Illumina PE150 platform. The methods for genomic DNA extraction are described in the Supplemental Methods.
Genome and chromosome assembly using Hi-C data
Using the ONT platform, we generated 113.66 Gb reads. All nanopore data were used for the catfish (L. longirostris) genome assembly. First, the Wtdbg2 (v2.5) package was used for preliminary genomic contigs assembly. Second, Racon (v1.3.1) was used to correct the genomic contigs with the nanopore data. Third, double-terminal sequencing was performed using the Illumina HiSeq platform, and we obtained 122.48 G of sequence data. The Pilon (v1.22) package was used to correct genomic contigs with the Illumina data.
Based on the Hi-C data obtained using the Illumina PE150, the assembled contig/scaffold sequence was mounted to the chromosome level by using LAchesis (v201701) software.
Assessment of the assembled genome
BUSCO (version: vertebrata_odb9) (Manni et al. 2021; http://busco.ezlab.org/) and CEGMA (Parra et al. 2007; http://korflab.ucdavis.edu/datasets/cegma/) were used to assess the integrity of the genome sequence. Small fragment libraries were compared to the assembled genome using BWA software (http://bio-bwa.sourceforge.net/) to assess genome integrity and sequence consistency.
Annotation of the assembled genome
Homology alignment and de novo prediction were used to identify repeat sequences in the whole genome. Homology prediction was performed using RepeatMasker (https://www.repeatmasker.org) software and in-house scripts (RepeatProteinMask) with default parameters to identify repeat sequences, as in the Repbase database (http://www.girinst.org/repbase). For ab initio prediction, a database of de novo repetitive elements was first constructed by LTR_FINDER (http://tlife.fudan.edu.cn/ltr_finder/), RepeatScout (Price et al. 2005; https://github.com/mmcco/RepeatScout), and RepeatModeler (https://www.repeatmasker.org/RepeatModeler/) with default parameters. RepeatMasker was used for ab initio prediction. In addition, tandem repeats were extracted by TRF (http://tandem.bu.edu/trf/trf.html). AUGUSTUS (http://bioinf.uni-greifswald.de/augustus/), GlimmerHMM (https://ccb.jhu.edu/software/glimmerhmm/), and SNAP (http://homepage.mac.com/iankorf/) were used for structural annotation of the genome incorporating ab initio prediction.
Gene functions were assigned according to the best match by aligning gene sequences in databases such as SwissProt (https://www.uniprot.org), Nr (https://www.ncbi.nlm.nih.gov/protein), Pfam (http://pfam.xfam.org/), KEGG (https://www.genome.jp/kegg/), and InterPro (https://www.ebi.ac.uk/interpro/). The methods for the transcriptome and the complete transcriptome for annotation of the assembled genome are described in the Supplemental Methods.
Comparative genomic bioinformatic analysis
After obtaining the genome sequence map of the catfish (L. longirostris), we identified common and different genes by comparison with the genomes of other species and inferred the evolutionary relationships between species and identified genes or gene families related to biological traits.
Cluster analysis of gene families
Gene families have clear similarities in structure and function, as they encode similar proteins. The same gene families can be closely grouped together to form a gene cluster, but more often, they are dispersed at different locations on the same chromosome or exist on different chromosomes, with different patterns of expression regulation. OrthoMCL (https://orthomcl.org/orthomcl/app) is a commonly used gene family identification process that employs the following steps (Li et al. 2003). In step 1, gene sets from individual species are filtered. First, where there are multiple transcripts of a gene, only the transcript with the longest coding region is retained. Second, genes that encode for proteins of fewer than 50 amino acids and that have stop codons are eliminated. In step 2, the similarity between the protein sequences of all species is obtained by BLASTP all-versus-all, and the default e-value is e–5. In step 3, OrthoMCL software is used to cluster the above results, with an expansion coefficient of 1.5.
Phylogenetic tree analysis
Multiple sequence comparisons were performed for all single-copy gene families using MUSCLE software (http://www.drive5.com/muscle/). The above results were then combined to form a superalignment matrix. Finally, RAxML (version 8.1.19) with the parameters “-m GTRGAMMA –f a –x 271828 –N 100 –p 12345” (Stamatakis 2014) was used to construct a phylogenetic tree for L. longirostris using maximum likelihood (ML TREE). The individual gene trees for each orthorlog using RAxML were built and were further summarized by ASTRAL (v.4.7.12) (Stamatakis 2014). To estimate the divergence time between different species, the MCMCTree program (Yang 2007) was used to implement the Bayesian relaxed-molecular clock (BRMC) method. The time correction points were taken from the TimeTree website (http://www.timetree.org/).
Gene family expansion and contraction
Gene family expansion and contraction refer to a significant difference in the number of genes in gene families between one or more species, and paralogous genes were used for expansion and contraction analysis. Depending on the results of the gene family clustering, the CAFÉ (v4.2) (Hahn et al. 2005; De Bie et al. 2006; https://sourceforge.net/projects/cafehahnlab/) can be used to filter the genes present in the abnormal gene families of each species in order to analyze gene family expansion and contraction with the following parameters: -p 0.05, -t 4, -r 10,000. The results of phylogenetic relationship and divergence time were used as input in this analysis.
Positive selection analysis
Positive selection, which refers to a single-copy gene family that has nonsynonymous mutations at the codon of DNA sequence, was analyzed by comparing genomes between species. The analysis of positive selection calculates the values of Ka and Ks, as well as the Ka/Ks ratio of the terminal branches and uses a likelihood ratio test in order to detect the presence of positive selection. Ka/Ks is the ratio of the nonsynonymous mutation rate to the synonymous mutation rate. If Ka/Ks > 1, we consider that there is a positive selection effect; if Ka/Ks = 1, we consider that there is neutral selection. A value of Ka/Ks < 1 indicates purifying selection. Ks = number of synonymous SNP mutations/number of synonymous mutation sites; Ka = number of nonsynonymous SNP mutations/number of nonsynonymous mutation sites.
The MUSCLE software was used to perform a multisequence alignment on the protein sequences of the orthologous genes of the species subjected to positive selection analysis, and the results of protein sequence alignment were then used as a template to produce the corresponding CDS alignment results. For each gene family, the branch-site model with a M0 model of the CodeML tool in the PAML software package was used to determine whether the gene family was positively selected between the different species. The LRT was performed to test whether the branch-site model containing positively selected codons (Ka/Ks > 1) fits better than the null mode, which includes the neutral or negative selection (Ka/Ks ≤ 1). In the present study, to identify the positively selected genes dependent on the feeding preference, the Chinese longsnout catfish (L. longirostris; the only carnivorous fish in these species analyzed) was selected as the foreground branch for positive selection analysis, and omnivorous fish of channel catfish (I. punctatus), yellow catfish (P. fulvidraco), common carp (Cyprinus carpio), zebrafish (Danio rerio), and herbivorous fish of blunt snout bream (M. amblycephala) and grass carp (C. idellus) were chosen as the background branch.
Rapid evolution analysis
Rapid evolution refers to the rate of substitution of the gene. To identify genes undergoing rapid evolution, the orthologous genes were compared and analyzed between Chinese longsnout catfish (L. longirostris) and channel catfish (I. punctatus) or yellow catfish (P. fulvidraco) or common carp (C. carpio) or zebrafish (D. rerio) or grass carp (C. idellus) or blunt snout bream (M. amblycephala). Ka/Ks values were calculated by using the branch model in the CodeML tool in the PAML software, and GO enrichment analysis was performed. Next, the Ka/Ks > 200 data were filtered out to weed out anomalies and to reduce the false-positive results (Ma et al. 2018; Peichel et al. 2020). GO terms with fewer than five genes were also filtered out. A Wilcoxon rank-sum test was then performed on the Ka/Ks values of the genes in each selected GO term. The P-value was corrected by the FDR. The GO terms with P < 0.05 were considered to be rapidly evolving.
Feeding behaviors of tas1r3–/– zebrafish
Zebrafish were fed with duckweed (L. minor) according to the method of Cai et al. (2021). Prior to the experiment, three wild-type control and three tas1r3–/– zebrafish were randomly selected and individually distributed in tanks (15 cm × 9 cm × 8 cm) (Supplemental Methods). After 24 h of acclimation, the zebrafish were fed 15 pieces of duckweed per tank. At 12 h after feeding, the remaining duckweed was quantified. The same experiment was repeated 10 times. Fishmeal and grassmeal were selected for the behavioral feeding trial. Briefly, eight wild-type control and eight tas1r3–/– zebrafish were randomly selected and individually allocated to tanks (25 cm × 20 cm × 15 cm). After acclimation, test blocks of fishmeal/grassmeal or control blocks (blank) were placed in the corners of the tanks. The zebrafish were allowed to choose between the different blocks. The formulation of the blocks was as follows: 1 g carboxymethylcellulose sodium was mixed with 0.2 g of fishmeal or grassmeal; then, 2 mL of water was added to the mixture. The mixture was processed into uniform-size blocks and dried at 60°C. After placing the control and test blocks, the videos of the preference behavioral testing were continuously recorded by a camera for 5 min, and the first biting and the times of biting the two types of blocks were recorded. The tests were repeated eight times using the same methods in the short-term behavior test. The double-blind test of the genotyping and feeding experiments was conducted as follows: The genotyping was performed with DNA extracted from the caudal fin, and the feeding experiments were performed by two groups of students. Students who conducted the feeding experiments were not informed of the genotyping. The methods for tas1r3 knockout in zebrafish are described in the Supplemental Methods.
Food consumption of tas1r3–/– zebrafish
A plantmeal-based diet was prepared using SM as the main protein source (Supplemental Table S20). The experimental diet was processed through an extruder (Modern Yanggong Machinery S&T Development) and made into 0.5 mm diameter pellets. Six wild-type control and six tas1r3–/– zebrafish were randomly selected and weighed, and fish with similar sizes were individually distributed into tanks (25 cm × 20 cm × 15 cm). There are three replicates for each genotype. After acclimation, the zebrafish were fed to apparent satiation twice daily at 08:30 and 16:30 for 7 days. Daily food intake was recorded, and the residual diets were collected and measured.
FHT of grass carp and sample collection
Grass carp, as a typical herbivorous fish, transitions from a carnivorous diet of plankton to a herbivorous diet of aquatic plants during the larval and juvenile stages. A FHT trial in grass carp was carried out according to the method of Wang et al. (2015). Briefly, grass carp larvae were reared in tanks and fed with chironomid larvae (C. tentans). At 46 days post hatch (body length = 2.82 ± 0.18 cm), nine fish were randomly selected for sampling before the FHT. After sampling, the remaining grass carp were fed H. verticillata. At 116 days post hatch (body length = 6.21 ± 0.6 cm), nine grass carp were randomly selected for sampling after the FHT. All sampled fish were anesthetized with MS-222 (200 mg/L), and plasma, oropharyngeal, brain, liver, and whole intestine tissues were collected from grass carp, frozen in liquid nitrogen, and stored at −80°C for further analysis. RNA samples from the liver and intestine were sent to the Novogene Technology Company to establish RNA and miRNA libraries and were sequenced on Illumina platforms. The methods for transcriptome, real-time qPCR, and biochemical analyses are described in the Supplemental Methods. The primers used in this study are listed in Supplemental Table S21.
Comparative feeding trial with two carnivorous and two herbivorous fish species
Two typical carnivorous fish and two typical herbivorous fish were selected to examine the genetic adaptations for metabolic preference. Grass carp and blunt snout bream (M. amblycephala) were fed with H. verticillata twice daily during the experiment, which are typical herbivores with no stomach, intestines almost twice the length of the body, food passing through the intestines in 8–18 h, intestinal microbiome contributing to carbohydrate turnover, and fermentation. Catfish (L. longirostris) and largemouth bass (M. salmoides) were fed with zebrafish twice daily during the experiment, which are typical carnivorous fish with stomach, shorter intestines with more microvilli, and high protease activity. After 7-day feeding, nine individuals of each species were randomly selected for sampling. After being anesthetized with MS-222 (200 mg/L), plasma, liver, and intestinal tissues were collected. All samples were frozen in liquid nitrogen and stored at −80°C until analysis.
Fishmeal and plantmeal diet feeding experiment with carnivorous and herbivorous fish
To better understand the role of trypsin in the metabolic strategy of fish with distinct feeding habits, we conducted a feeding trial using fishmeal and plantmeal diets in grass carp and Chinese longsnout catfish. To simulate the natural diets of herbivorous and carnivorous fish, we used fishmeal as the main source to prepare a fishmeal diet, and soybean as the main source to prepare a plantmeal diet. Seventy-two grass carp with similar size were divided into six tanks, and 72 Chinese longsnout catfish were divided into six tanks (12 fish per tank). Fish in three tanks were fed a plantmeal diet, and fish in the other three tanks were fed a fishmeal diet. The fish were fed to apparent satiation twice daily at 8:30 and 16:30 for 7 days. After the feeding experiment, three fish from each tank were randomly selected for sampling. After being anesthetized with MS-222 (200 mg/L), the plasma, liver, and intestinal tissues were collected and stored at −80°C for analysis.
Small RNA library sequencing and bioinformatics analysis
Raw data were obtained by Illumina sequencing and were processed by removing adapter-containing reads, poly-N-containing reads, and low-quality reads. After quality control, sequencing libraries were generated using the NEB next multiplex small RNA library prep set for Illumina (NEB). After purification and size selection, libraries with insertions between 18 and 40 bp were ready for Illumina sequencing with SE50. The small RNA tags were mapped to the reference sequence by Bowtie without mismatch to analyze their expression and distribution on the reference (Langmead et al. 2009). The program miRBase20.0 was used as a reference, and the modified mirdeep2 and srna-tools-cli software were used to obtain the potential miRNA and to draw the secondary structures. To eliminate tags from protein-coding genes, repeat sequences, rRNA, tRNA, snRNA, and snoRNA, small RNA tags were mapped to RepeatMasker, the Rfam database, or these types of data from the specified species itself. To ensure that each unique small RNA mapped to a single annotation, we followed the priority rule: known miRNA > rRNA > tRNA > snRNA > snoRNA > repeat > gene > NAT-siRNA > gene > novel miRNA > ta-siRNA. The proportion of total rRNA was used as a marker of sample quality. miRNA target gene prediction was performed by psRobot_tar in miRanda. miRNA expression levels were estimated by transcripts per million (TPM) according to the following criteria: normalized expression = mapped readcount/total reads × 1,000,000. Differential expression analysis of two conditions/groups was performed using the DESeq R package (1.8.3) (Zhou et al. 2010). The P-values were adjusted using the Benjamini and Hochberg (1995) method. A corrected P-value of 0.05 was set as the default threshold for significantly differential expression. The methods for miRNA target gene validation are described in the Supplemental Methods.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 8.0.1 (GraphPad Software). All data were obtained from at least three independent experiments. Statistical significance was determined using two-tailed unpaired Student's t-tests between two groups or one-way ANOVA between multiple groups. A P-value < 0.05 was considered statistically significant.
Data access
All sequencing data generated in this study have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA967986, PRJNA1130114, and PRJNA1130120. The relevant source data involved in this study have been uploaded to the Science Data Bank at https://www.scidb.cn (CSTR ID: 31253.11.sciencedb.16106). The relevant data at the Science Data Bank can be found in Supplemental Data and Supplemental Materials.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Key R&D Program of China (2023YFD2400605 and 2022YFD2400900), the National Natural Science Foundation, China (U21A20266, 31672670, 31972771, 32061133009), the Youth Innovation Promotion Association of CAS (2013223 to D.H. and 20200336 to G.Z.), and the State Key Laboratory of Freshwater Ecology and Biotechnology (2022FBZ03).
Author contributions: S.-Q.X. and D.H. conceived the study and supervised the project. Y.-L.L., G.Z., J.-Z.S., Y.-L.G., and B.-Y.Y. performed genome data analyses. Y.-L.L., G.Z., and J.-Z.S. performed the laboratory work. Q.-S.L., L.-W.X., Y.-T.Z., and J.-Y.C. validated the results. H.-K.L., J.-Y.J., Z.-M.Z., Y.-X.Y., X.-M.Z., Z.-W.W., G.-R.G., J.M., Z.Y., and R.E.G. helped with analyzing the results. Y.-L.L., G.Z., and D.H. prepared the figures and wrote the manuscript, with input from all the authors. All authors reviewed and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278476.123.
-
Freely available online through the Genome Research Open Access option.
- Received September 6, 2023.
- Accepted July 15, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








