
Streamlined spatial and environmental expression signatures characterize the minimalist duckweed Wolffia australiana
- Tom Denyer1,
- Pin-Jou Wu1,
- Kelly Colt2,
- Bradley W. Abramson2,3,
- Zhili Pang4,
- Pavel Solansky1,
- Allen Mamerto2,
- Tatsuya Nobori2,5,6,
- Joseph R. Ecker2,5,6,
- Eric Lam4,
- Todd P. Michael2 and
- Marja C.P. Timmermans1
- 1Center for Plant Molecular Biology, University of Tübingen, Tübingen 72076, Germany;
- 2Plant Molecular and Cellular Biology Laboratory, The Salk Institute for Biological Studies, La Jolla, California 92037, USA;
- 3Applied Sciences and Life Sciences Laboratory, Noblis, Reston, Virginia 20191, USA;
- 4Department of Plant Biology, Rutgers, The State University of New Jersey, New Brunswick, New Jersey 08901, USA;
- 5Howard Hughes Medical Institute, The Salk Institute for Biological Studies, La Jolla, California 92037, USA;
- 6Genomic Analysis Laboratory, The Salk Institute for Biological Studies, La Jolla, California 92037, USA
Abstract
Single-cell genomics permits a new resolution in the examination of molecular and cellular dynamics, allowing global, parallel assessments of cell types and cellular behaviors through development and in response to environmental circumstances, such as interaction with water and the light–dark cycle of the Earth. Here, we leverage the smallest, and possibly most structurally reduced, plant, the semiaquatic Wolffia australiana, to understand dynamics of cell expression in these contexts at the whole-plant level. We examined single-cell-resolution RNA-sequencing data and found Wolffia cells divide into four principal clusters representing the above- and below-water-situated parenchyma and epidermis. Although these tissues share transcriptomic similarity with model plants, they display distinct adaptations that Wolffia has made for the aquatic environment. Within this broad classification, discrete subspecializations are evident, with select cells showing unique transcriptomic signatures associated with developmental maturation and specialized physiologies. Assessing this simplified biological system temporally at two key time-of-day (TOD) transitions, we identify additional TOD-responsive genes previously overlooked in whole-plant transcriptomic approaches and demonstrate that the core circadian clock machinery and its downstream responses can vary in cell-specific manners, even in this simplified system. Distinctions between cell types and their responses to submergence and/or TOD are driven by expression changes of unexpectedly few genes, characterizing Wolffia as a highly streamlined organism with the majority of genes dedicated to fundamental cellular processes. Wolffia provides a unique opportunity to apply reductionist biology to elucidate signaling functions at the organismal level, for which this work provides a powerful resource.
Multicellular organisms comprise specialized tissues accommodating diverse cell types. This variety is required to achieve the array of functions necessary for an organism to develop and thrive in dynamic environments, and comes about through precise coordination of complex gene regulatory networks that integrate responses to internal and external cues. Plants, in particular, have evolved intricate mechanisms to accommodate diverse biotic and abiotic environmental inputs, leading to changes in their metabolism, physiology, and development at cellular to organismal levels. Understanding commonalities and variations in these responses on a molecular level is vital to begin decoding the fundamental drivers of tissue distinction and how responses to various inputs may be coordinated at the organismal level to achieve a desired phenotype. Advances in single-cell-resolution RNA sequencing (scRNA-seq) have opened the possibilities for systems approaches to query cellular specialization during development and their specific responses to environmental cues, allowing for transcriptomic profiling at high spatiotemporal resolution (Seyfferth et al. 2021; Denyer and Timmermans 2022). In plants, scRNA-seq studies published to date have largely focused on aspects of Arabidopsis development at the organ or tissue level (e.g., Denyer et al. 2019; Kim et al. 2021; Zhang et al. 2021; Shahan et al. 2022). However, although many of these studies have examined in depth, for example, cell lineage progressions within a developing tissue, no studies have yet profiled at the whole-plant level to sufficient depth to enable observations at the system level. This is principally owing to the high level of structural complexity and large cell numbers in most plant models.
A seminal study published in 2017 did profile the transcriptome for every individual cell within the model nematode Caenorhabditis elegans (Cao et al. 2017). A key characteristic that enabled this feat is the simplicity of body plan and low number of cells and cell types within this model animal. A plant paralog to such a model can be found in the Wolffia genus of the Lemnaceae family. These plants, ranging in size from <1 mm to several millimeters, have a highly reduced architecture (Lam and Michael 2022). Wolffia plants have no roots or vasculature system but exhibit clear developmental distinctions between top and bottom portions relative to the air/water interphase. For example, the upper epidermal surface of Wolffia fronds is characterized by the presence of stomata and pigment cells, which can contain phenolic compounds that turn brown in response to UV damage (Li et al. 2023). Similarly, the above-water approximately three to eight parenchymal cell layers comprise highly chlorophyllous and relatively compact cells, whereas the underwater parenchymal tissue, which forms the bulk of the frond, contains highly vacuolated cells with significantly lower chlorophyll levels (Fig. 1A). Further benefitting Wolffia as a model is its compact genome with >90% of conserved core eudicot gene functions represented by small gene families (Michael et al. 2021; Lam and Michael 2022), which can facilitate an understanding of the fundamental principles of plant responses to its environmental circumstances.
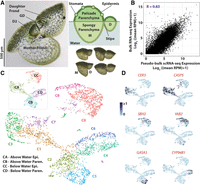
A scRNA-seq atlas of Wolffia australiana cells reveals two core cell types optimized for life in air versus water. (A) W. australiana comprises a mother (M) frond from which successive daughter (D) fronds bud off. Within the plant, a granddaughter (GD) frond is often also seen. A developmental progression of a daughter frond (anticlockwise from top left) is shown on the bottom right. The plant can generally be divided into epidermal and parenchymal cells, with stomata dispersed across the above-water epidermis and the parenchyma divided into photosynthetic palisade parenchyma at the top of the plant and spongy parenchyma in the hull (top right). A conical cavity is seen near the center of the frond below the surface of the water. (B) Pearson correlation analysis shows that gene expression values in merged single-cell and bulk-tissue RNA-seq of whole Wolffia plants are highly correlated. (C) UMAP visualization of the dusk scRNA-seq atlas reveals nine distinct cell clusters, organized around four superclusters (inset) corresponding to above-water epidermis (CA), below-water epidermis (CC), above-water parenchyma (CB), and below-water parenchyma (CD). (D) UMAP projections of normalized expression profiles of select DEGs within the atlas showing cluster and supercluster specificity. Names of Arabidopsis orthologs are given (see Supplemental Data Set S10).
Most, if not all, green organisms partition their biological activities to a specific time-of-day (TOD), and they do this both developmentally and ecologically to ensure synchronization with the daily light–dark cycles on earth (Michael et al. 2003; Sanchez and Kay 2016; Steed et al. 2021; Oravec and Greenham 2022). TOD regulation of specific biological activities is, in part, controlled internally by the circadian clock, which is highly conserved from single-cell algae to higher plants (Michael 2022a; Laosuntisuk et al. 2023). In general, the internal circadian clock, in conjunction with daily changes in light and temperature, controls up to 90% of the transcriptome, focusing biological activities to a specific TOD. For instance, the expression of genes associated with photosynthesis and phytohormones peaks in the morning, with nitrogen and carbon assimilation in the late afternoon, with cold acclimation and defense at dusk, with cell cycle in the early evening, and with lignin and protein biosynthesis in the middle of the night (Supplemental Fig. S1; Supplemental Text; Bläsing et al. 2005; Covington and Harmer 2007; Filichkin et al. 2011; Ferrari et al. 2019). In Wolffia, only ∼13% of genes are TOD regulated under the diurnal conditions of light/dark cycles (Supplemental Text; Michael et al. 2021). The lower percentage of TOD-regulated genes in Wolffia may reflect its compact genome. Alternatively, it is becoming increasingly clear that TOD expression can vary by cell type (Endo 2016; Swift et al. 2022) and that bulk RNA-seq analysis may miss such cell type–specific timing information.
Results
Wolffia cells can be broadly divided into four distinct subpopulations
We transcriptionally profiled Wolffia at single-cell resolution to describe, in an unbiased manner, cell types and states within this budding frond system. We first optimized a protoplast isolation method, considering the distinct cell wall composition of Wolffia and the presence of a cuticle as an adaptation to life on water (Borisjuk et al. 2018). Viable protoplasts from clonally propagating plants (accession wa8730) were then prepared and processed through the 10x Genomics scRNA-seq cell capture and library production pipeline. Plants were gown under intermediate day length (12 h light and 12 h dark), and cells profiled at both dawn (lights on) and dusk (lights off), to capture cell type–dependent TOD-responsive gene expression. The dusk data set comprises 4327 cells isolated across two replicates, which were further filtered to 3151 cells of high quality (mean, 1333 genes/cell detected; mean, 3907 UMIs/cell). This number approaches 1× coverage of all cells within this organism and is likely to capture a broad spectrum of cell types and developmental transitions. Indeed, a total of 12,825 unique genes were identified in the data set, corresponding to 92% of all annotated genes in the Wolffia genome (Michael et al. 2021; Ernst et al. 2023). In addition, replicate libraries are highly congruent (Pearson correlation coefficient R > 0.99) (Supplemental Fig. S2A,B), and pseudobulked scRNA-seq data correlate well (R > 0.83) with a bulk RNA-seq data set of pooled Wolffia plants (Fig. 1B; Michael et al. 2021), demonstrating that despite inevitable technical limitations in capturing, for example, rare cells or large cells, the atlas provides a rich data set of high quality.
The dusk atlas resolves as nine cell clusters (C0 to C8), quite distinctly presented as four major lobes in a UMAP projection, referred to henceforth as “superclusters” CA to CD (Fig. 1C; Supplemental Table S1; Supplemental Interactive 3D UMAP S1). To understand the transcriptomic basis for this arrangement, we used differential gene expression (DEG) analysis to first identify genes defining each of the four superclusters (log2 fold change >1 and adjusted P-value < 0.05) (Supplemental Data Set S1). Just greater than 500 differentially expressed genes (DEGs) were identified across all superclusters. This number is unexpectedly low compared with expression variation revealed by scRNA-seq analysis across tissues in, for example, the root, shoot, or leaf of other model species (e.g., Denyer et al. 2019; Kim et al. 2021; Zhang et al. 2021). This distinction is even more apparent when considering DEGs expressed in >10% of cells within a given supercluster (PCT1) and in <10% of remaining cells (PCT2), criteria commonly used to identify tissue-specific marker genes. Under these conditions, we identified a total of 113 supercluster marker genes (Supplemental Data Set S1). The lack of specification is likely in part explained by the limited structural complexity of the plant with no root or vasculature, for example, but also points to a very streamlined system with the majority of genes dedicated to basic cellular processes. The detection of 92% of all annotated genes in the single-cell transcriptome data sets corroborates this idea.
Wolffia, similar to other duckweeds, has a compact genome with fewer protein-coding genes than most other plant species (Harkess et al. 2021; Michael et al. 2021; Ernst et al. 2023). However, like most nonmodel genomes, functional annotation of the predicted genes is sparse compared with model species such as Arabidopsis, maize, or rice. Therefore, orthologs for supercluster DEGs were identified from well-characterized model species, focusing primarily on Arabidopsis, in order to enable Gene Ontology (GO) term enrichment analysis and to discern distinguishing functions for cells within each supercluster (Supplemental Data Sets S1, S10). Accordingly, cells in CA are marked by expression of genes predicted to function in wax biosynthesis and cuticle development, pointing to an epidermal identity (Supplemental Data Set S1). For example, genes encoding VERY-LONG-CHAIN ALDEHYDE DECARBONYLASE 3 (CER3) and CER8, required for cuticle biosynthesis (Lü et al. 2009), are strongly expressed in cells of this cluster (Fig. 1D), as is WRINKLED 4 (WRI4), encoding a transcription factor (TF) that promotes oil and wax biosynthesis (Park et al. 2016; Supplemental Fig. S2C). In addition, the ortholog of CASPARIAN STRIP MEMBRANE DOMAIN PROTEIN 5 (CASP5) in Arabidopsis, involved in the formation of lignified apoplastic barriers (Roppolo et al. 2011), is specifically expressed in cells of this supercluster (Fig. 1D). Further, select basic helix–loop–helix (bHLH) and MYB TF genes linked to drought-stress responses are uniquely expressed in cells of the CA supercluster. These include MYB41 and MYB49, which are part of a regulatory circuit activated in response to desiccation, and MYB60, which has been linked to stomatal closure under drought stress (Wang et al. 2021; Supplemental Fig. S2C).
Cells in supercluster CC also host several DEGs with predicted functions in lipid biosynthesis and metabolism, but these relate particularly to the sphingolipid class. Examples of this include the ortholog to SPHINGOID BASE HYDROXYLASE 2 (SBH2) (Fig. 1D), as well as Wa8730a009g003220 and Wa8730a014g002170, whose orthologs have been linked to sphingolipid biosynthesis in other species (Supplemental Fig. S2C; Supplemental Data Set S10). Additional DEGs for this supercluster encode the aquaporin PIP1B and the iron transporter IRT1. Indeed, in addition to sphingolipids, supercluster CC DEGs are also enriched for transport-linked GO terms (Supplemental Fig. S2C; Supplemental Data Set S2). Curiously, two markers are orthologous to YABBY2, which encodes a TF that promotes growth and abaxial cell fate in Arabidopsis lateral organs (Fig. 1D; Siegfried et al. 1999). Given the description of DEGs with roles in processes such as lipid biosynthesis, it is likely that CC, like CA, comprises epidermal cells, albeit of a different fundamental nature. That epidermal cells are well distinguished from other major cell types is seen commonly in scRNA-seq studies of plant cells (e.g., Kim et al. 2021; Zhang et al. 2021). In root atlases, an additional strong distinction is seen between hair and nonhair epidermal cells (e.g., Denyer et al. 2019; Shahan et al. 2022). The division seen here is likely of an altogether different nature, and the half-submergence of Wolffia may point to different functions needed for those epidermal cells that interact with the two distinct environments of above or below the air/water interphase.
A majority of DEGs for supercluster CB are associated with light responses and photosynthesis functions and include many encoding light-harvesting chlorophyll (LHC) binding proteins (Fig. 1D; Supplemental Data Set S1; Supplemental Fig. S2C). As such, CB appears to comprise parenchyma cells of the photosynthetic heart of the plant. This supercluster is further characterized by the enriched expression of several orthologs in the GIBBERELLIC ACID–STIMULATED Arabidopsis (GASA) gene family, which encodes highly conserved cysteine-rich peptides involved in redox sensing and hormonal responses (Bouteraa et al. 2023). Curiously, of all DEGs for this cluster, none are orthologous to Arabidopsis TFs. This is in stark contrast to the other three superclusters. In addition, TFs are prominent among tissue-specific or cell type–specific genes in studies of other model plants (e.g., Denyer et al. 2019; Knauer et al. 2019; Marand et al. 2021). It is conceivable that the strong photosynthetic signature of these parenchymal cells may be driven primarily by environmental cues integrating into existing gene regulatory networks.
Supercluster CD is close to CB in the cluster cloud (Fig. 1C). However, based on the 180 DEGs distinguishing it, supercluster CD is less defined by photosynthesis. The proximity of this supercluster to CB in the cluster cloud may point to a commonality of their cellular identity (parenchyma). Conversely, the reduced photosynthesis-related expression profile in CD points to a distinction similar to that predicted for the epidermis, such that supercluster CD captures the below-water parenchymal cells. Indeed, the highly vacuolated cells that form the bulk size of the frond would share features with their photosynthetically active counterpart but contain fewer chloroplasts as their submerged position makes them less amenable to photosynthesis (Fig. 1A). Further among DEGs distinguishing cells in this supercluster are orthologs of a cytochrome P450 and several WRKY and ethylene response factor (ERF) TF genes, whereas GO terms enriched among DEGs are principally related to biotic- and abiotic-stress responses (Supplemental Data Set S1). For example, homologs of pathogenesis-related (PR) proteins and WRKY60 are typically pathogen-induced, whereas MYB36 responds to both biotic- and abiotic-stress stimuli (Fig. 1D; Supplemental Fig. S2C; Dong et al. 2003; Liu et al. 2022). In addition, genes in the ABA response pathway, for example, ABI1 and AHG3, implicated in both stress-related responses and development are preferentially expressed in cells of CD (Supplemental Data Set S1; Yoshida et al. 2006; Pasaribu et al. 2023). Finally, a number of DEGs for this supercluster have predicted functions in development and morphogenesis, including in cell homeostasis, cell wall architecture/composition, and cytoskeleton organization, or encode developmental TFs. This latter finding is in line with the presence of meristematic activity in a defined region within the underwater parenchymal cell population (Li et al. 2023).
These results indicate that Wolffia is largely composed of cells with either an epidermal or parenchymal origin. However, there is specialization within this broad classification as evidenced by distinct separations of superclusters and clusters in the atlas. A previous annotation of the Wolffia genome identified several gene orthogroups specific to Wolffia and related duckweed species (Michael et al. 2021). Their predicted functions are associated with just four GO terms including sphingolipid biosynthesis, photomorphogenesis, and wax biosynthesis. It is of note that these terms are prominently reflected in the characteristics of three of the superclusters, CC, CB, and CA, respectively. A fourth major GO term, “cysteine-type endopeptidase,” which ordinarily connects to the processing of signaling peptides or antimicrobial peptides (AMPs) in the case of defense, does not define one specific supercluster, likely because of the broad spectrum of biological processes it affects. The fact that the Wolffia-specific orthogroups describe main superclusters identified highlights these as the key adaptations to the partially submerged environment in which Wolffia thrives.
Submergence is a key distinguishing characteristic within epidermal and parenchyma cells
A distinctive aspect of duckweeds such as Wolffia is that they live partly submerged, floating on the surface of water bodies. As noted above, superclusters CA and CC may distinguish the above- and within-water (i.e., top and bottom) portions of the epidermis, whereas superclusters CB and CD may comprise the top and bottom parenchyma, respectively (Fig. 1A). To further assess this possibility, we performed a bulk RNA-seq analysis on Wolffia plants manually bisected into above- and below-water parts, discernable by the stark difference in chlorophyll content and morphology (Fig. 1A; Supplemental Fig. S2D). In total, 262 DEGs (log2 fold change > 1, adjusted P-value < 0.05) between these regions were identified. Projecting the average relative expression of these DEGs onto the data set UMAP showed they are overwhelmingly expressed in the epidermal clusters and map specifically to superclusters CA and CC, as expected (Fig. 2A; Supplemental Data Set S2). The epidermis of course interacts directly with the water or air environment, but this finding predicts that this interaction triggers a strong local response that drives distinctive transcriptome landscapes primarily in the epidermis.
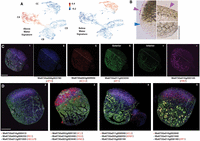
Tissue type–specific responses to life in air versus water are validated by PHYTOMap multiplex in situ RNA detection. (A) UMAP projections of “gene set activity” (Seurat) profiles of above- and below-water enriched DEGs as determined by bulk RNA-seq show the response to submerge is primarily detected in the epidermis. (B) Histological sections of Wolffia stained with Sudan IV show stained cuticle (purple arrowhead) on the exterior of the above-water epidermis (top) that is lacking from the below-water epidermis (bottom). Blue arrowhead indicates the approximate position of the waterline on the flank of the plant. (C) Select supercluster markers imaged using PHYTOMap show strong cell type specificity in line with scRNA-seq predictions. Four genes are imaged in the same plant in four fluorescence channels. From left to right: combined expression profiles; images in the blue, red, green, and magenta channels. White dashed lines indicate the approximate water line; (§/+) identical exterior or interior sections across different imaging channels. (D) PHYTOMap images showing the combined expression profiles of the genes indicated below. Composite images are a projection of 13–15 z-stack sections of 2–3 µm each. Information on genes examined can be found in Supplemental Table S2 and Supplemental Figure S2. Scale bar, 250 µm. When available, names of Arabidopsis orthologs are given (see Supplemental Data Set S1).
Even though the above- versus within-water response is less evident in the parenchymal cells at the bulk RNA-seq level, the single-cell data provide a unique opportunity to discern tissue type–specific adaptations to being in an air versus water environment. We therefore determined genes differentially expressed between cells of supercluster CB versus CD, as well as CA versus CC (Supplemental Data Set S3). Confirming the above hypothesis, photosynthesis-related functions associated with the chloroplast-rich, above-water tissues are enriched in CB, whereas supercluster CD matches below-water features. Notably, DEGs between these superclusters are overwhelmingly upregulated in the submerged cells (241 vs. 38). This may be explained in part by the developmentally active cells of the meristem and newly emerging daughter fronds in the submerged portion of the plant, but it also indicates that parenchymal cells primarily activate defined pathways upon submergence. Prominent among these are genes linked to responses to abiotic-stress stimuli, in line with the principal characteristic of supercluster CD (Supplemental Data Set S3). Numerous genes involved in ABA signaling and osmotic-stress responses are upregulated in the below-water cells. Additional signatures of the submergence response include a reduced response to GA, as well as the induction of ERFs. The latter points to a highly conserved role for ethylene in the response to submergence in plants (Raskin and Kende 1984; Fukao et al. 2006). The submerged cells are further characterized by a heightened defense response, which seems fitting given that the often-stagnant ponds in which Wolffia grow are likely to carry higher microbial loads and thus may require heightened defense functions. Of note, there is no evidence for a switch from respiration to production of energy through anaerobic pathways, such as ethanolic or lactic acid fermentation, in the below-water cells (Supplemental Data Set S3). This is perhaps explained by the relatively shallow submergence of Wolffia and the morphology of its below-water parenchyma, which has larger and less tightly packed cells with additional air spaces to provide increased buoyancy to the plant in the absence of aerenchyma (Bernard et al. 1990).
In contrast to the parenchyma, cells in both the top and bottom epidermis show environment-associated specializations, with 168 and 109 upregulated DEGs in superclusters CA and CC, respectively (Supplemental Data Set S3). Enriched GO terms for wax biosynthetic processes in the above-water epidermis (supercluster CA) (Fig. 1D) point to the top of the plant requiring additional hydrophobicity provided by the waxy cuticle to repel water and keep the organism floating in the correct orientation and to present a more hardened barrier against microbial and insect pests (Borisjuk et al. 2018). The presence of cuticle exclusively on the above-water epidermis was confirmed by Sudan IV staining, which indicates a fairly abrupt transition between the top and bottom epidermis (Fig. 2B). In addition, the set of DEGs upregulated in the top epidermis indicates cells responding to desiccation (see above), oxidative stress, and light stress (evidenced by induced expression of genes in the anthocyanin pathway) (Kovinich et al. 2015), which mirror responses in epidermal cells of Arabidopsis leaves (Galvez-Valdivieso et al. 2009). In contrast, the below-water epidermis is more involved in coordinating the transport of (micro-)nutrients from the water environment into the plant and shows increased expression of a number of genes with distinct transport-related and plasmodesmatal functions (Supplemental Data Set S3). These cells also show aspects of the submergence response seen in below-water parenchyma, including upregulation of genes predicted to mediate abiotic- and biotic-stress responses, although this expression signature is mostly distinct from that observed in the below-water parenchyma (Supplemental Data Set S3). The most prominent and distinctive feature of the submerged epidermal cells is in the biosynthesis of sphingolipids. The prominence of sphingolipid-related pathways in Wolffia has been suggested to indicate a trading of terpenoids with sphingolipids for defense, perhaps because the aquatic environment favors the latter (Michael et al. 2021; Glenz et al. 2022).
Taking these analyses together, the principal divisions in the Wolffia scRNA-seq atlas appear to reflect the two major tissue types, parenchyma and epidermis, divided by their relative location as the plant lives at the air/water interphase (Fig. 1A). Although these divisions are defined by relatively few DEGs, notable is the strong submergence response evident in parenchymal cells at the scRNA-seq level, which was mostly undetected in bulk transcriptomic analysis of above- and within-water regions. However, consistent with its direct interaction with the plant's environment(s), the epidermis is particularly responsive to life in air versus water.
Multiplex in situ RNA localization as validation of cell annotations
To further validate the cell annotations and their interpretation, the expression of select marker genes for the superclusters were imaged using PHYTOMap, a recently developed methodology for multiplexed spatial analysis of transcripts within whole-mount plant tissues (Nobori et al. 2023). PHYTOMap provides an affordable alternative to many spatial transcriptomic techniques and avoids the difficulties inherent with transforming Wolffia to produce tissue-specific reporter lines, approaches commonly used for validation of scRNA-seq annotation (e.g., Denyer et al. 2019; Zhu et al. 2023). Marker genes for supercluster CA were found to strongly express at the above-water region of the plant, in the epidermis as expected (e.g., Wa8730a016g000410, Wa8730a002g009150, and Wa8730a010g002840) (Fig. 2C; Supplemental Table S2). In contrast, marker genes for supercluster CB are predominantly expressed in the below-water epidermis (e.g., Wa8730a011g003030), whereas transcripts for Wa8730a005g008250, Wa8730a010g000100, and Wa8730a005g006050 are principally detected in the below-water mesophyll, in line with expression in cells of supercluster CD (Fig. 2C). Altogether, we examined 20 genes with varying degrees of tissue specificity (Fig. 2C,D; Supplemental Fig. S3; Supplemental Table S2). Of these, just four produced no defined data, largely owing to the low resolution of, particularly, the magenta fluorescence channel. In addition, we observed a degree of autofluorescence from the plant tissues largely in the green channel (Supplemental Fig. S3). However, despite this, almost all genes selected showed expression in line with expectations. These results validate our annotation of the scRNA-seq atlas and additionally highlight the utility of the PHYTOMap methodology for high-throughput examination of gene expression patterns in organisms such as Wolffia that are less amenable to transformation.
Cell clusters identify functional specialization within epidermal and parenchymal cells
Obvious subdivisions are present within each supercluster (Supplemental Table S1). This points to distinctions beyond transcriptomic differences stemming from life predominantly in water or air and suggests that epidermis- and parenchyma-derived tissues each encompass several discernable cell types or states. To clarify these distinctions, we identified DEGs describing each cluster by comparing its transcriptome to that of all other cells in the data set (Supplemental Data Set S4). As noted above for the superclusters, cluster distinctions reflect expression variation in comparatively small sets of genes, and these were often assigned widely diverse cellular functions. Within the below-water epidermis, for example, clusters C0 and C8 share 39 out of a total of 147 DEGs, which indicates only a subtle differentiation between these cell populations. Likewise, no cellular processes stand out as a defining characteristic for cells in clusters C1 and C7 within the above-water epidermal supercluster. However, a distinction is seen in cluster C3. Cells in this cluster show strong differential expression (DE) of genes with cuticle-related functions (Supplemental Data Set S4). Indeed, orthologs of CER genes involved in cuticle biosynthesis are particularly strongly expressed in C3 compared with other clusters (Fig. 1D). Likewise, several 3-KETOACYL-COA SYNTHASE (KCS) genes, as well as MYB31, known to regulate cuticle biosynthesis in tomato (Xiong et al. 2020), are DEGs for C3 (Figs. 1D, 3A).
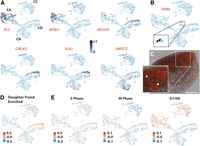
Specialized cell types and developmental stages are distinguished within major tissue type divisions. (A) UMAP projections showing normalized expression profiles of select DEGs within the atlas showing cluster specificity. Names of Arabidopsis orthologs are given (Supplemental Data Set S10). (B) UMAP showing that expression of Wa8730a007g001710, ortholog of Arabidopsis FAMA, is concentrated in a small subset of cells within supercluster CD. (C) Stomata, marked by autofluorescence (green), are dispersed across the upper, above-water, epidermis. White arrows highlight select guard cells. Scale bar, 250 µm. (D) UMAP visualization of “gene set activity” (Seurat) of daughter-enriched DEGs determined by bulk RNA-seq shows that developmentally active daughter cells are localized primarily within specific clusters, particularly within a region of C6. (E) UMAP visualizations of “gene set activity” of genes associated with distinct phases of the cell cycle (Supplemental Data Set S11) show that although cells undergoing DNA replication are largely dispersed across the atlas, mitotic cells primarily localize to C6 and, to a lesser extent, C4, mirroring the distribution of daughter cells.
Between C4 and C6, distinct clusters for the submerged parenchymal cells (Supplemental Table S1), the cells in C6 are distinguished by DE of a relatively large number (264) of genes. Although the only significantly enriched GO terms for this cluster relate to functions in stress responses, among the DEGs are a substantial number of orthologs for TFs and other genes with roles in plant development (Supplemental Data Set S4). Examples of this include those encoding NAC and LBD TFs, an auxin efflux carrier, a homolog to the receptor kinase CRINKLY4, several cell wall biosynthesis and modifying enzymes, and VLN2 and VLN3, linked to directional growth in Arabidopsis (Fig. 3A; van der Honing et al. 2012). We note that generations of Wolffia were profiled together, capturing the spectrum of developmental progressions; it is possible that this is resolved in the data, and the prominence of development-related DEGs here would indicate a notable concentration of less mature cells within C6. This point is explored in more detail below.
As expected, the DEGs for clusters C2 and C5 in supercluster CB primarily have functions relating to photosynthesis and light responses (Supplemental Data Set S4). Other marker genes for C5 point to functions in water transport, cell-to-cell communication, and cytoskeletal reorganization. Of particular interest, a gene orthologous to the SUGARS WILL EVENTUALLY BE EXPORTED TRANSPORTER genes (SWEET2 and SWEET6) is a prominent marker for a localized set of cells in C5 (Fig. 3A). Within vascular plants, these SWEET transporters are uniquely expressed in phloem-associated cells to facilitate the source-sink distribution of photosynthates (Kim et al. 2021). Within the minimal body plan of Wolffia, which lacks recognizable phloem cells (Fig. 1A), photosynthates must be distributed across cell types (Ware et al. 2023). Perhaps, the subset of SWEET-expressing cells could be acting as surrogates for phloem cells to transport photosynthetically derived sugars to the rest of the Wolffia plant.
The cell specificity seen for the SWEET2/6 gene orthologs prompted us to probe for additional examples of genes with expression confined to a localized subpopulation of cells in a cluster. To this end, we filtered the cluster DEGs to those detected in <25% of cells in a given cluster (PCT1 < 0.25) and in <5% of cells outside of that cluster (PCT2 < 0.05). Scrutiny of UMAPs of these genes resulted in a list of 32 diverse DEGs showing highly localized expression in one of five clusters (no such examples were found for C0, C1, C5, or C8) (Supplemental Data Set S4). C2 presented just one example. Expression of Wa8730a002g009940, the ortholog of Arabidopsis KRATOS, which restricts cell death during stress and vasculature development (Escamez et al. 2019), is localized to the tip of cluster C2 (Supplemental Fig. S4). Similarly, the expression of many of the “cuticle” genes is largely confined to cells within a subregion of C3 (Supplemental Fig. S4). Almost all mark the same strip of cells on the flank of the cluster. The localized expression would indicate that the upper epidermis contains specialized cells to produce cuticular waxes. However, this region is not exclusively defined by cuticle-related genes, and other examples with this localized expression include orthologs for a cytochrome P450 and bHLH134 (Supplemental Fig. S4). The linear arrangement of this group of cells is curious as it mirrors a dynamic commonly seen in scRNA-seq cluster projections that captures a developmental trajectory (Denyer et al. 2019; Shahan et al. 2022). However, given the small number of genes involved in this example, such a trajectory would likely reflect the differentiation of a specific specialized function, which we were unable to resolve with confidence.
Within the same supercluster, five genes mark two distinct regions within C7, four of which express in the same subset of cells, whereas the expression pattern of the other is quite distinct (Supplemental Fig. S4). Their predicted functions hint at localized signaling processes. Wa8730a010g003880, for example, is linked to oxidation of brassinosteroids, suggesting differential compositions of growth hormones across cells (Shimada et al. 2001). Most of the highly localized gene expression was seen in C4 and C6. In the former, expression is preferentially localized to the tip of the cluster, whereas in C6, a host of stress-related genes including those linked to ABA and drought responses, defense against fungi, and DNA repair upon UV damage, as well as a core circadian clock gene PSEUDO RESPONSE REGULATOR 5 (PRR5), are expressed in distinct subregions along the cluster (Supplemental Fig. S4; Supplemental Data Set S4; Nakamichi et al. 2005). In general, although the distinctive patterns of expression can be subtle, the strong localizations across the cluster cloud point to true complexities within the clusters. The “localized” genes also relate to a wide range of processes, implying distinct specializations within cells with otherwise similar transcriptome profiles in what would appear to be a highly streamlined organism.
High conservation of guard cell transcriptomes, even at broad evolutionary distance
Besides these observations, notable is the highly localized expression of Wa8730a007g001710 within a small group of cells close to the periphery of C4 (Fig. 3B). Its protein shares homology with FAMA, a TF that drives guard cell formation in Arabidopsis (Smit and Bergmann 2023). Consistent with this classification, several additional Arabidopsis stomatal genes have a Wolffia ortholog showing strong, if not exclusive, expression in this small subgroup of cells. This includes EPIDERMAL PATTERNING FACTOR 2 (EPF2), expressed in proliferating stomatal meristemoids (Hunt and Gray 2009), the guard cell maturation gene DOF4.7 (SCAP1), and SLAC1 and MYB10, which regulate stomata opening and closing (Cominelli et al. 2005; Negi et al. 2013; Deng et al. 2021). However, transcripts for orthologs to the early stomatal development genes MUTE and SPEECHLESS, which are known to be more transiently expressed, are not found in these cells or elsewhere in the atlas, suggesting that the stomatal cells captured are mostly mature in nature. The expression profiles of FAMA, SCAP1, SLAC1, and MYB10 support this idea, although the point is somewhat countered by the expression of an earlier-stomatal gene, EPF2 (Hunt and Gray 2009; Adrian et al. 2015). However, it is possible that the latter signaling peptide may serve additional, divergent roles in Wolffia, such as interaction with receptor-like kinases in the ERECTA gene family and receptor-like proteins to regulate epidermal cell size (Meng et al. 2015).
Despite relatively few being isolated, the presence of guard cells in the atlas further underscores the depth of our scRNA-seq data set, because Wolffia plants are estimated to develop just around 30 stomata in their upper epidermis (Fig. 3C; Lam and Michael 2022; Li et al. 2023). Their capture also provides an opportunity to assess conservation of stomatal expression profiles. We therefore generated a transcriptome for the Wolffia guard cells and compared this to an equivalent transcriptomic data set derived from Arabidopsis stomata (Lopez-Anido et al. 2021). Genes preferentially expressed in stomatal cells over other cells in the Wolffia atlas (Supplemental Data Set S5) reveal a general commonality with Arabidopsis stomata. Indeed, ∼36% of Wolffia guard cell–associated DEGs have an Arabidopsis ortholog expressed in the stomatal lineage (Supplemental Data Set S5). These data point to high conservation between Wolffia and Arabidopsis guard cells, even at this evolutionary distance, which is in line with the early origin and conserved function of this specialized cell type (Chen et al. 2017). Lastly, despite being epidermal in origin and location, the Wolffia guard cells cluster more closely with the similarly photosynthetic parenchyma. Given the streamlined developmental and environmental expression signatures observed thus far, gene expression associated with photosynthesis in guard cells may override that of their epidermal origin.
The Wolffia atlas captures developmental time between mother and daughter fronds
Wolffia reproduces through budding, and in some species within this genus, this can happen almost daily. Central to this, Wolffia plants contain a distinctive conical cavity, also called a “pocket,” just below the upper parenchymal layer toward one side of the frond (Fig. 1A; Lam and Michael 2022). The below-water parenchymal cells forming the floor of this cavity are distinguished by an enlarged nucleus, electron dense cytoplasm, and fewer plastids, features characteristic of meristematic cells (Li et al. 2023). Their apparent asymmetric growth pattern suggests that they continuously give rise to new primordia that develop into daughter fronds supported by a stipe (or “branch”) (Li et al. 2023), which, by extension, pushes out the new plantlet at a distal “exit.” This daughter will often have initiated a granddaughter frond before it separates from the mother, taking this subsequent generation with it (Fig. 1A). As such, the below-water parenchymal cells may reveal biological distinctions reflecting the continual production of daughter fronds, in addition to capturing the large vacuolated parenchyma cells that give the plant its buoyancy.
We next sought to discern whether the developmental progression between daughter and mother fronds was captured within our data set. We first identified DEGs via bulk RNA-seq analysis on surgically separated mother and daughter fronds (Supplemental Data Set S6; Supplemental Fig. S5). Given the range of sizes that daughter fronds can take (Fig. 1A; Supplemental Fig. S5A), the two samples would likely show strong commonalities. However, the daughter frond sample would be expected to additionally contain more developmentally active cells undergoing rapid growth, cell division, and differentiation. Indeed, we identified 114 genes in our data set that show significant DE between daughter and mother fronds. Among them, genes predicted to encode signaling components, including via auxin and cytokinin, and cell wall–modifying enzymes stand out. A “gene set activity plot” (Seurat) (Satija et al. 2015) shows expression of daughter-enriched DEGs primarily in regions of clusters 4, 6, and 8 (Fig. 3D). As expected, these clusters encompass the two major tissue types (Supplemental Table S1), although proportionately more daughter cells are of a seeming parenchymal origin (C6). This may in part have a technical basis, as smaller-sized cells are captured more efficiently in the scRNA-seq process over the ordinarily large below-water parenchymal cells. Nonetheless, the pronounced daughter cell expression signature within C6 is in line with the aforementioned DE of developmentally relevant genes in this cluster (Supplemental Data Set S5). Its prominence particularly at the tip of C6 may signify a developmental trajectory from the tip to the center of the cluster cloud that captures maturation of daughter-to-mother cell states. However, at this time, putative trajectories and rare cells of the meristem inside the pocket region could not be confidently distinguished.
Wolffia orthologs of Arabidopsis genes marking distinct phases of the cell cycle show generally broad expression in cells across the atlas, suggesting that a daughter-to-mother state transition is not explained easily by cell cycle activity (Supplemental Data Set S11). Indeed, cell cycle–related genes are not prominent among DEGs for any particular cluster. However, there are some distinctions of note. Genes connected to the G1/G0 transition, for example, may form an exception. Gene set activity plots of these genes show that epidermal cells are primarily captured in this more quiescent phase of the cell cycle (Fig. 3E). In addition, genes linked to mitosis show somewhat stronger expression in cells at the tips of particularly C6, possibly in line with the daughter state (Fig. 3E). However, cells displaying S-phase activity do not localize in one particular point of the UMAP, which may reflect a need for genome endoreduplication in expanding cells.
TOD sampling demonstrates a streamlined system incorporating cell type–specific responses
Aside from submergence, TOD is a critical environmental input to which cells in plants must respond (Steed et al. 2021; Swift et al. 2022). In previous work, 13% of Wolffia genes show a robust TOD response, a percentage far below that seen in other plant species (Filichkin et al. 2011; Ferrari et al. 2019; Michael et al. 2021; Michael 2022b). We reasoned that performing bulk RNA-seq on whole plants may mask TOD signals that are variable across cell types, especially for a plant with such a simple body plan, and thus could significantly underestimate the true number of genes under TOD control. As such, we generated an additional scRNA-seq atlas for plants collected at dawn, 12 h separated from the time analyzed above (dusk). The dawn data set, after applying the same filter parameters as for the dusk data, comprises 2435 cells with a mean of 1215 genes per cell and 12,565 genes in total detected (mean, 4417 UMIs/cell). The two replicates within this atlas are also highly congruent (R > 0.99) (Supplemental Fig. S5B).
The dawn data set shows a largely similar cluster landscape to that observed at dusk, with eight clusters arranged into four distinct superclusters (Fig. 4A; Supplemental Data Set S8; Supplemental Interactive 3D UMAP S2). Integration of the dawn and dusk atlases into a joint UMAP shows that cells from the two TOD data sets form separate clusters that are near-mirror images of each other (Fig. 4B; Supplemental Interactive 3D UMAP S3). Although the dawn and dusk superclusters clearly separate, the mirroring suggests they represent similar cell types. Indeed, overall gene expression in cells of each supercluster is highly correlated between dawn and dusk (Fig. 4C), and the DEGs characterizing the four superclusters show substantial overlap across the dawn and dusk TOD conditions (Fig. 4D; Supplemental Data Sets S1, S7). As such, the defining characteristics of the four superclusters, for example, cuticle and sphingolipid biosynthesis, do not change by TOD, except that in the chlorophyll-rich above-water parenchyma (CB), light responses are much enhanced within the dusk data set, as might be expected following an extensive period of light (Sanchez and Kay 2016; Oravec and Greenham 2022). Accordingly, the following broad cell identities could be assigned to the dawn superclusters: CA, above-water epidermis; CC, below-water epidermis; CB, above-water parenchyma; and CD, below-water parenchyma (Supplemental Table S1). The separation of the dawn and dusk clusters highlights that TOD plays a prominent role in defining the transcriptome, and although this information does not override cell identity, sampling across TOD provides a more comprehensive view of tissue type–dependent expression and identifies marker genes, otherwise missed (Supplemental Data Sets S1, S7).
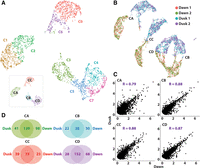
Dusk and dawn transcriptomes are well conserved, although many genes show TOD responses. (A) UMAP visualization of the dawn scRNA-seq atlas reveals eight distinct clusters, organized around four superclusters (inset). (B) Integration of dusk and dawn data sets reveals a mirror image arrangement of superclusters, indicating a strong conditional transcriptome distinction that does not override cell identity. (C) Gene expression correlation between dusk and dawn replicates of distinct superclusters reveals high correlation. Genes expressed in >10% of the cells in each supercluster were used in each instance. (D) Venn diagrams showing just a partial conservation of DEGs between superclusters of dusk and dawn conditions, indicating that sampling across TOD identifies unique tissue-specific DEGs. Identities of superclusters CA, CB, CC, and CD are matched across data sets.
To discern features of TOD regulation across Wolffia cells, supercluster transcriptomes were compared across dawn and dusk data sets (Fig. 5A; Supplemental Table S3). Even with a strict cutoff (Log2 FC > 1 and adjusted P value < 0.05), a considerable number of TOD-responsive DEGs were identified between equivalent cell populations: 364, 164, 157, and 157 for CA, CB, CC, and CD, respectively (Supplemental Data Set S9). The above-water epidermis is the most TOD-responsive, with considerably more DEGs between dawn and dusk than even the key photosynthetic cells of the parenchyma. This would perhaps suggest a dynamic protective role of the above-water epidermis to the change of light during the light/dark cycle. Of the TOD-responsive genes detected, 76 are TOD regulated in all four superclusters. These 76 genes display the same TOD expression pattern across each of the superclusters, indicating that at least in Wolffia, some genes show a coordinated TOD response across the organism regardless of the cell type and growth environment (Supplemental Data Set S9; Fig. 5A; Supplemental Table S3). Similarly, genes that are TOD regulated in two or three superclusters share either a day- or night-dependent pattern of expression. Consistent with the findings from bulk RNA-seq (Michael et al. 2021), as well as from other plant species (Ferrari et al. 2019), across the four main cell clusters, dawn DEGs are enriched for GO terms relating to photosynthesis and light responses, whereas dusk DEGs are enriched for GO terms relating to ribosomal and polysome activity, which would point to increased translation late in the day (Supplemental Text; Supplemental Data Sets S8, S9; Dodd et al. 2005; Ferrari et al. 2019). Further, whereas the effects of light on photosynthesis and plastid development are seen in all superclusters, this signature is most prominent in cells of the above-water tissues.
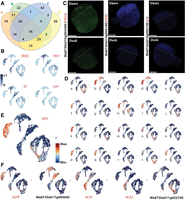
TOD responses vary depending on cell type and on life in water versus air. (A) Venn diagram illustrating the overlap of TOD DEGs across superclusters; 76 genes are TOD regulated in all four supercluster comparisons, but many genes show a tissue-dependent TOD response. (B) UMAP projections of normalized expression for the core circadian clock genes REVEILLE (RVE), GIGANTEA (GI), FLAVIN-BINDING, KELCH REPEAT, FBOX (FKF1), and LATE ELONGATED HYPOCOTYL (LHY) reveal the expected strong TOD responses. (C) Expression of select dawn or dusk markers imaged at dawn (top) and dusk (bottom) using PHYTOMap shows strong preferential expression at dawn (left two), or dusk (right). Composite images are projections of 13–15 z-stack sections of 2–3 µm each. For information on genes examined, see Supplemental Table S2. Scale bars, 250 µm. (D) UMAP projections of example genes showing differential expression (DE) between dusk and dawn conditions as well as a level of cell type specificity. Expression for each gene is plotted on an integrated plot of data sets: (a) Wa8730a017g001020, (b) Wa8730a016g003000, (c) Wa8730a017g003930, (d) Wa8730a005g008830, (e) Wa8730a006g006550, (f) Wa8730a003g006530, (g) Wa8730a006g005980, (h) Wa8730a019g001460, (i) Wa8730a020g001730, (j) Wa8730a010g005400, (k) Wa8730a012g004550, (l) Wa8730a002g000180, (m) Wa8730a006g005670, (n) Wa8730a003g004150, (o) Wa8730a002g007540, (p) Wa8730a002g007630, (q) Wa8730a015g001450, (r) Wa8730a018g000010, (s) Wa8730a002g010730, and (t) Wa8730a010g003830. (E) UMAP projection of IRT2 (Wa8730a011g002740) expression in the integrated dusk/dawn atlas showing a strong tissue-specific TOD response. (F) UMAP projections of further genes showing tissue-specific TOD responses. (SGPP) 8730a011g001940, (ACA1) Wa8730a020g000810, and (ACA3) Wa8730a017g000040.
Among the TOD coregulated genes are core components of the plant circadian clock, which forms a negative feedback loop that interacts with an array of light signaling and developmental genes to control global TOD expression in plants (Supplemental Data Set S11; Sanchez and Kay 2016; Michael 2022a; Oravec and Greenham 2022). At its core, the circadian clock is regulated by the SHAQKYF-type-MYB (sMYB) TFs, LATE ELONGATED HYPOCOTYL (LHY), and REVEILLE (RVE), whose transcripts are highly expressed in cells of the dawn but not the dusk clusters, as expected (Fig. 5B; Oravec and Greenham 2022). In contrast, the clock genes GIGANTEA (GI) and FLAVIN-BINDING, KELCH REPEAT FBOX (FKF1) are primarily expressed in cells collected at dusk, as seen in bulk RNA-seq (Fig. 5B). Further, validating detection of TOD-specific expression behaviors in the scRNA-seq data sets, in situ imaging of RVE expression using PHYTOMap resulted in a strong expression signal only at dawn, as seen in other species as well as in bulk RNA-seq (Fig. 5C). Likewise, transcripts for Wa8730a010g005400, an ortholog of Arabidopsis FATTY ACID DESATURASE 2 (FAD2) identified as strongly upregulated in the dawn data set, were preferentially detected in morning samples, whereas the Wa8730a010g000190 expression signal was detected primarily at dusk, as predicted (Fig. 5C; Supplemental Table S2). However, several “core” circadian genes (Supplemental Data Set S11) were found to be differentially expressed in one or more clusters, suggesting that the circadian machinery may be distinct per tissue/cell type. Orthologs for clock-associated pseudoresponse regulators (PRRs) and for members of the ZEITLUPE (ZTL) family (Wa8730a009g001000, Wa8730a003g007690), for example, cycle exclusively in CA and CA and CB, respectively (Supplemental Data Set S9; Somers et al. 2000; Para et al. 2007). This finding that both core circadian genes as well as diurnal-regulated genes can show cell type specificity is consistent with results from both a different duckweed system and a range of other plant species (Endo 2016; Watanabe et al. 2021; Swift et al. 2022).
Of the 456 TOD-responsive genes detected by scRNA-seq, 289 (>60%) do not overlap with high-confidence “cycling” genes detected by bulk RNA-seq (cutoff R > 0.8; Michael et al. 2021). The considerable excess of TOD-responsive genes identified by scRNA-seq highlights the value of this approach for detecting tissue-dependent environmental responses (Jean-Baptiste et al. 2019; Shulse et al. 2019). On an individual basis, 233, 60, 68, and 58 DEGs from CA, CB, CC, and CD, respectively, were not previously detected at the bulk RNA-seq level. Beyond these novel TOD-regulated genes, each supercluster harbors 195, 22, 32, and 26 DEGs, respectively, that are specific to that supercluster (Fig. 5A; Supplemental Data Set S9), demonstrating responses to TOD input that are not only tissue type specific but interact uniquely with environmental cues stemming from life within or above water (Greenham et al. 2017). These supercluster-specific TOD responses can be considerable (e.g., Fig. 5D). A prominent example is Wa8730a008g003780 (IRT2), which unlike the aforementioned IRT1 ortholog (Supplemental Fig. S2C), is expressed predominantly in the above-water epidermis at dawn (Fig. 5E). BROAD-RANGE SUGAR PHOSPHATE PHOSPHATASE (SGPP, Wa8730a011g001940) shows a similar expression profile, whereas Wa8730a017g000040 displays the opposite behavior in the epidermis (Fig. 5F). Other genes showing strong distinctions in this regard include the dawn-upregulated orthologs of Arabidopsis ALPHA CARBONIC ANHYDRASE1 (ACA1) and ACA3 (Wa8730a020g000810 and Wa8730a012g002610), specifically cycling in the above- and within-water epidermis, respectively (Fig. 5F). The epidermis-specific expression of these ACA genes at dawn highlights the important role this enzyme plays in facilitating carbon capture from the air and water environments through conversion of CO2 into carbonic acid. Other DEGs in the above-water epidermis at dawn are associated with stress and temperature responses, whereas the below-water epidermis shows regulation of the response to ultraviolet light (Supplemental Text; Supplemental Data Sets S8, S9).
Fewer genes cycle specifically in the parenchyma, although Wa8730a011g002740 stands out in that it is upregulated in the below-water parenchyma at dawn (Fig. 5F). Also, the karrikin response appears to be TOD regulated in this way, whereas the above-water parenchyma has DEGs with functions in various metabolic and biosynthetic pathways at the same TOD (Supplemental Data Set S9). Importantly, aside from these select examples, the genes that are TOD regulated in individual superclusters have diverse functions, indicating TOD effects on a range of processes, rather than explicit up- or downregulation of overt regulatory networks.
Taken together, this single-cell-level analysis of TOD-responsive gene expression is consistent with the relatively streamlined nature of Wolffia gene regulation. It also highlights the benefits of the methodology for discerning expression signatures distinguishing cell types and their responses to environmental cues, as well as demonstrating the considerable cell type–specific changes in this regard. Indeed, although select biological processes are generally controlled in a TOD fashion as expected (Supplemental Text; Supplemental Data Sets S8, S9), many genes cycle in a manner dependent on cell type and/or life in water versus air. These are associated with widely diverse cellular functions, suggesting a pleiotropic response (Supplemental Text; Supplemental Data Sets S8, S9). That the above-water epidermis is the most responsive of all cell types was unexpected and might form a feature linked to the very specific habitat and lifestyle of Wolffia.
Discussion
Wolffia holds great promise as a key model organism for the exploration of manifold biological questions. The experimental and practical opportunities offered by Wolffia as a biofuel crop and novel food, for example, are in no small way connected to its basic body plan, small size, and rapid regeneration (Lam and Michael 2022). Further, its compact genome offers opportunities to discover basic cellular processes defining cell types and how these respond to environmental change. In particular, the half-submerged lifestyle of Wolffia offers a novel opportunity to examine how a plant responds to critical environmental inputs stemming from life in air versus water. We examined this, and the impact of TOD, on individual cell types of the entire plant using single-cell-resolution transcriptomics. The division of Wolffia cells into two major tissue types, specialized by the submerged water environment, demonstrates the most fundamental divisions of this minimalist plant. Transcriptomic comparisons between these cell populations reveals a very streamlined organism with relatively few DEGs between cell types, between submerged and above-water tissues, between mother and daughter fronds, or across different TODs. As we detected transcripts for most annotated genes in the genome, we can surmise that the majority are broadly and evenly expressed “housekeeping” genes. Nonetheless, DEGs between these various divisions identify basic cellular functions underlying the distinct biology of these tissue types. Most notably, we see that gene orthogroups specific to the duckweeds are linked to specific tissue-dependent adaptations for a species that floats on water.
Beyond the fundamental tissue type divisions, the atlas captures additional cellular complexity and specialization, including cells with a strong developmental signature within the below-water parenchyma. Developmental activity is clear from the abundant expression of TFs and other key genes underlying patterning and growth, as well as a strong daughter frond and mitotic “signal,” particularly in cluster C6. Its cells likely include those of the youngest daughter fronds and perhaps the rare cells of the meristem and stipe. Further, expression dynamics across C6 seemingly point to the capture of a developmental progression within the cluster structure. Although further scrutiny of putative trajectories and rare cell types would benefit from increased sequencing depth and cell numbers, the current data offer a first opportunity to explore development in this minimalist plant, unique as it is in the rapidity of its replication and growth.
Aside from developmental progression, the atlas identifies transcriptomes for guard cells of the stomata and particular epidermal cells with a penchant for cuticle production. In addition, we see evidence for potential subspecializations that could compensate for the simple body plan of Wolffia, relative to other plants. The highly localized expression of SWEET transporters within the parenchyma, for example, potentially compensates for the lack of a vascular system. Similarly, defense genes in cells of the below-water parenchyma and epidermis point to an acute response to potential waterborne antagonists. The stomatal transcriptome compiled from our data highlights the conservation of certain cell types across species as distantly related as the model eudicot Arabidopsis and this nongrass monocot. However, it also exposes the power of looking at new model systems representing unique biology and positions in the green lineage, with the Wolffia superclusters displaying unique gene networks adapted to the aquatic environment.
Our analysis further highlights the importance of taking TOD responses into consideration when developing an understanding of the system-level organization of cells, as well as distinct aspects of networks resulting from the primary driver of plant biology, the sun. The observed TOD expression changes predict that the clock changes from tissue type to tissue type even in this minimal plant system as in other systems (Endo 2016; Swift et al. 2022) and that responses do so as well. Photosynthesis, plastid development, and translation are major processes TOD regulated across the plant, but many genes respond to TOD in a manner dependent on cell type and life in water versus air. These genes are predicted to have widely diverse functions, suggesting effects of TOD on a broad range of specialized cellular processes. In particular, the above-water epidermis is considerably more responsive to TOD than other cell types. This could be a consequence of this cell type being at the vanguard of the changing light and highlights the relative prominence of local protective measures to this environmental cue.
Despite its virtues, a limitation of Wolffia as a model arises from current challenges in its routine and rapid transformation for molecular analyses. Therefore, to validate our cluster annotation, a critical element of scRNA-seq studies, we utilized PHYTOMap, a recently developed, multiplexed, in situ hybridization methodology (Nobori et al. 2023). Besides the limitation of select fluorescence channels to vividly capture rarer- or lower-expressed transcripts, this proved highly successful, offering a practical option going forward to examine spatial patterns of expression in model organisms like Wolffia that are less amenable to analysis of transgenic reporters.
Global studies of gene regulation at the system level in multicellular organisms are extremely complex owing to, in part, the intricate networks of TF activity and the myriad tissue types involved. Notwithstanding the aforementioned limitations of sequencing depth in resolving the rarest dynamics within this species, the streamlined gene expression variation identified in this work points to Wolffia being a tractable model for studies of regulatory dynamics at the organismal level with examination of chromatin dynamics at single-cell resolution, for example, as an attractive next step. This model equally offers amenable opportunities for integrating metabolic or proteomic examination, utilizing the amalgamation of nascent technologies and methodologies (Clark et al. 2022; Zhao and Rhee 2022). Such complex, layered data transposed onto an organism of such elementary structure, but harboring fascinating generational dynamics and a unique subaquatic lifestyle convenient for environmental studies, offer an opportunity to examine complex dynamics. The atlas we present here, of 5604 high-quality cells, broadly representative of the entire organism, offers a strong reference to explore a wealth of fundamental questions going forward. Key curiosities in this regard are the differences between above- and below-water-situated tissues and the varying roles the different tissues undertake in these contexts. Particularly interesting, perhaps, are the variable defense mechanisms of the below-water parenchyma and epidermis, as well as the differential responses to TOD. Similarly, there is clearly more to be learned regarding the unique development of Wolffia. To this end, the atlas can be easily explored on our interactive browser at https://www.zmbp-resources.uni-tuebingen.de/timmermans/plant-single-cell-browser/ (Ma et al. 2020).
Methods
Plant growth
W. australiana line 8730 (New South Wales, Australia) was used for all experiments. This line was chosen because it has the best genome assembly available at the time and because of the availability of bulk RNA-seq TOD time course data (Michael et al. 2021). Plants were grown at intermediate days of 12 h light (100 µE)/12 h dark cycles at a constant 24°C in 100 mL nutrient medium (0.5 × Schenk & Hildebrandt [Duchefa], 0.1% sucrose at pH 6.7). Plants were subcultured weekly by transferring ±10 fronds to fresh medium.
Protoplast isolation for scRNA-seq library construction
Approximately 200–300 plants, <1 month old, were collected for each sample. Dusk and dawn samples were collected in parallel from separate growth chambers with contrasting light/dark cycles. Dusk cycles were collected at the end of the light period (lights off, Zeitgeber [ZT] 12) and dawn cycles at the end of the dark period (lights on; ZT0). To check TOD dynamics were retained despite this, gene sets previously shown to cycle at particular times in Wolffia were assessed in the resultant data (Supplemental Fig. S5C; Michael et al. 2021). To avoid problems associated with the thick, waterproof cuticle of the plants, individual samples were diced with a razor blade to improve access the innermost cells. Diced tissue samples were placed into a freshly made “protoplast enzyme mix” with a higher concentration of commonly used cell wall digestion enzymes (0.1 M KCl, 0.02 M MgCl2, 0.1% BSA [Sigma-Aldrich], 0.08 M MES [Duchefa], 0.6 M mannitol [Duchefa] at pH 5.5, adjusted with Tris, 1.5% cellulase R-10, 1% maceroenzyme, 0.5% pectolyase [all enzymes Duchefa]). Tissue samples were digested on an orbital shaker set at <200 rpm for maximum 90 min to minimize transcriptional noise. Dawn samples were kept in darkness for this step. Digested cells were passed through a 100 µm sieve followed by a 40 µm cell strainer (pluriSelect) and resuspended in 10 mL “wash buffer” (“protoplast enzyme mix” without enzymes) and centrifuged (1000g, 20°C). The supernatant was then removed, and the pellet was resuspended in 10 mL wash buffer. This was centrifuged again (500g, 20°C), the supernatant removed, and the pellet resuspended in 500 µL wash buffer. By necessity, wash steps for both samples were performed in light with the whole process minimized to 30 min. Protoplasts were validated under a light microscope and quantified using a hemocytometer. Concentrations were adjusted with wash buffer to a density of ∼800–900 cells per milliliter.
scRNA-seq library preparation and sequencing
scRNA-seq libraries were prepared from fresh protoplasts according to the 10x Genomics single-cell 3′ reagent kit v 3.1 protocol. Cells were loaded onto the 10x Chromium controller within 1 hour of the end of digestion. About 60% of excess cells over target were added, as per the 10x protocol. Eleven cycles were used for cDNA amplification and 12 for final PCR amplification of the adapter-ligated libraries. Final library size and quality were checked on a DNA high-sensitivity Bioanalyzer chip (Agilent), and libraries were quantified using the NEBNext library quantification kit for Illumina and sequenced to a depth of more than 20,000 reads per cell.
RNA-seq library preparation and sequencing
For bulk RNA-seq comparisons of above- and below-water tissues and of mother–daughter fronds, plants were bisected with needles under a light microscope, and appropriate tissues were collected in liquid nitrogen and ground to a fine powder. RNA was extracted using TRIzol (Thermo Fisher Scientific), following the manufacturer's protocol. RNA samples were quantified by NanoDrop and were quality-assured based on RNA bioanalyzer chip traces (Agilent). mRNA was enriched by Oligo(dT) pull-down using the NEBNext Poly(A) mRNA magnetic isolation module, and RNA libraries were constructed using the NEBNext Ultra II directional RNA library prep kit for Illumina with NEB Multiplex oligos. Final library size and quality were checked on a DNA high sensitivity bioanalyzer chip, and libraries were quantified using the NEBNext library quantification kit for Illumina and sequenced to a depth of 10 million reads per sample.
PHYTOMap sample preparation and image capture
Wolffia was grown under 12 h/12 h light cycles, with lights on at 7:00 AM and off at 7:00 PM. Live fronds were used for the experiment. The dawn set was harvested at 8:20 AM and the dusk set at 5:00 PM, immediately starting the initial FAA fixation step upon harvesting. The protocol by Nobori et al. (2023) was followed with some modifications to accommodate the increased volume of sample required for each set of probes used, as well as increased incubation times to allow solutions to fully penetrate the fronds. Yakult enzymes in the cell wall digestions steps were substituted with cellulase (Sigma-Aldrich SAE0020), macerozyme (PlantMedia 21560017-1), and pectinase (Sigma-Aldrich P2401-1KU). Additionally, the fronds did not stick to poly-D-lysine-coated dishes, so the tissue was processed in 1.5 mL tubes for all incubation steps. The SNAIL probe concentration was also increased from 10 nM in the original protocol to 500 nM per oligo in the final pool to account for the larger volume of Wolffia fronds. Samples were prepared for imaging on microscope slides with a 22 mm × 50 mm coverglass. The additional size of the coverglass allowed better adhesion to the slide. Smaller coverslips were more easily disrupted owing to the thickness of the duckweed fronds. Imaging was performed on an Olympus FV3000 confocal microscope. Images were taken using a 20× zoom with image resolution of 254 × 254 pixels per tile. Four tiled images per frond were taken and stitched together for a total of 1019 × 1019-pixel image sizes to encompass a whole frond in one image. Z-stacks were generated for each set of tiles to span the entire depth of each frond imaged and number of layers dependent on frond thickness. The following channel settings were used for each fluorophore: AF488, 499 nm excitation and 504–554 emission; Cy3, 554 nm excitation and 559–650 emission; Cy5, 649 nm excitation and 657–735 nm emission; and AF740, 752 nm excitation and 760–839 nm emission.
Histological staining
Wolffia plants were embedded in 7% agarose and sectioned to a depth of 75 µM by Vibratome (Leica). Staining was performed with 0.1% Sudan IV (Sigma-Aldrich) for ∼10 min, as per the method of Nadiminti et al. (2015).
Bulk RNA-seq analysis
Sequence reads (paired end, 150 bp) were aligned to the W. australiana line 8730 (Wa8730.asm201904v2.fasta) reference genome with STAR (version 2.7.10a) (Dobin et al. 2013; Ernst et al. 2023). All common names and accession of genes referred to in this paper can be found in Supplemental Data Set S12. Read counts of genes were calculated on uniquely mapped reads using “featureCounts” (version 2.0.1) (Liao et al. 2014) with W. australiana annotation (Wa8730.gtf). DE analysis was conducted using DESeq2 (version 1.34.0) (Love et al. 2014). DEGs were identified by having a Log2 fold-change > 1 and an adjusted P-value < 0.05. For correlation analysis of gene expression between bulk RNA-seq and scRNA-seq, Log2 (mean RPM + 1) expression values were calculated for each gene and the Pearson correlation coefficient determined in R (v4.1.2) (R Core Team 2021).
Generation of single-cell expression matrices
Cell Ranger version (5.0.1; 10x Genomics) was used for initial scRNA-seq data processing (default settings). CellRanger Count was used to align sequencing reads to the W. australiana reference genome. Cells were filtered with a quality cutoff of >650 UMIs/cell. Gene expression matrices were created for each readset (dusk-1, dusk-2, dawn-1, dawn-2), and readsets were merged with cellranger aggr. The final H5 file was converted to a CSV expression matrix via CellRanger mat2csv and used for further analysis. For further metrics pertaining to scRNA-seq data analysis and quality, see Supplemental Table S4.
Dimensionality reduction, UMAP visualization, and cell clustering analysis
All standard bioinformatic analyses were performed using R Statistical Software (v4.1.2) (R Core Team 2021). The Seurat R package (version 4.3.0) (Satija et al. 2015) was used for downstream analysis with default parameters unless otherwise specified. Gene expression values were normalized with the “LogNormalize” function. The top 2000 highly variable genes were identified with “FindVariableFeatures” and used for PCA calculation. The first 10 PCs were used for Louvain clustering of the cells and UMAP dimensionality reduction. Interactive 3D UMAPs for the dusk, dawn, and combined data sets can be found in the Supplemental Materials. Superclusters were manually assigned by analyzing UMAP visualizations. Select clusters were subclustered using 50 PCs. The Pearson's correlation of gene expression between the dusk and dawn was calculated on genes expressed in >10% of cells in both superclusters. DEGs were identified with “FindAllMarkers” with a Log2 Fold change threshold of >1, adjusted P-value < 0.05, and PCT1 (percentage of cells) >10%. Gene set activity plots were calculated using “AddModuleScore.” For integration and comparison of dusk and dawn data sets, Seurat objects were generated independently and then integrated using a Seurat integration pipeline using 2000 anchor features and the canonical correlation analysis (CCA) integration method. After integration, 20 PCs were used as input features for the “RunUMAP” and “FindNeighbors” functions. TOD DEGs between dusk and dawn of each supercluster was performed with “FindMarker” using integrated data with Log2 fold-change >1, adjusted P-value < 0.05, and PCT >10% in either PCT1 or PCT2.
Stomatal transcriptome analysis
The cluster of cells expressing the stomatal marker Wa8730a007g001710 (AT3G24140, FAMA) was manually selected. DE analysis between stomatal cells and remaining cells in the dusk/dawn integrated data object was conducted using “FindMarkers” with the same parameters as above.
GO enrichment analysis
GO term overrepresentation analysis was carried using the Wolffia gene annotation and GOATOOLS (Klopfenstein et al. 2018). Significant GO terms (P > 0.05) were identified from specific gene lists with the following flags: ‐‐pval = 0.05 ‐‐method = fdr_bh ‐‐pval_field = fdr_bh.
Identification of Wolffia orthologs
Orthology was determined with OrthoFinder (Emms and Kelly 2019). Marker genes from model species based on existing literature were used to search the orthogroups for lm5633 orthologs. These marker genes in model organisms were compared with the OrthoFinder gene table, and a corresponding lm5633 ortholog was observed. Generally, annotations with a geneID from eggnog mapper were more reliable than observing marker genes in larger gene families (more than three copies per genomes).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) database under accession number PRJNA1124135. For individual SRA accession numbers, please see Supplemental Table S5. The scRNA-seq atlas can be easily explored via the interactive browser at https://www.zmbp-resources.uni-tuebingen.de/timmermans/plant-single-cell-browser/. The scRNA-seq Seurat objects and associated matrices are accessible via the same Browser.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the Salk Sequencing Core for high-throughput sequencing and Detlef Weigel for cosupervision of P.-J.W. We also thank members of all laboratories who provided critical input. This work was supported by a grant from the Alexander von Humboldt Foundation to M.C.P.T. from the Tang Fund to T.P.M. and by the Max Planck Society (P.-J.W., through funding to Detlef Weigel). Research on duckweed in the Lam laboratory was supported by the U.S. Department of Energy, Office of Biological and Environmental Research program under award number DE-SC0018244. In addition, this work was supported by a Hatch project (12116) and a multistate capacity project (NJ12710) from the New Jersey Agricultural Experiment Station at Rutgers University (E.L., Z.P.). This work was also supported by the Waitt Advanced Biophotonics Core Facility of the Salk Institute with funding from the National Institutes of Health (National Cancer Institute, Cancer Center Support Grants: P30 CA01495, National Institute on Aging, San Diego Nathan Shock Center P30 AG068635) and the Waitt Foundation.
Author contributions: E.L., T.P.M., M.C.P.T., and T.D. conceived and designed the study. T.D. produced all libraries and performed all wet laboratory experimentation, except for the PHYTOMap analysis, which was carried out by K.C., A.M. in the lab of J.R.E. In addition, T.N., K.C., Z.P., and B.W.A. carried out sequencing and performed an initial analysis of the dusk single-cell data. P.-J.W. performed the single-cell and bulk RNA-seq data analyses. T.P.M. performed the OrthoFinder and TOD analyses. P.S. generated the single-cell browser interface for accommodating the Wolffia single-cell atlases. T.D. and M.C.P.T. wrote the manuscript with contributions from E.L. and T.P.M., who also proofread the final version.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279091.124.
-
Freely available online through the Genome Research Open Access option.
- Received February 9, 2024.
- Accepted June 20, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








