
Accurate allocation of multimapped reads enables regulatory element analysis at repeats
Abstract
Transposable elements (TEs) and other repetitive regions have been shown to contain gene regulatory elements, including transcription factor binding sites. However, regulatory elements harbored by repeats have proven difficult to characterize using short-read sequencing assays such as ChIP-seq or ATAC-seq. Most regulatory genomics analysis pipelines discard “multimapped” reads that align equally well to multiple genomic locations. Because multimapped reads arise predominantly from repeats, current analysis pipelines fail to detect a substantial portion of regulatory events that occur in repetitive regions. To address this shortcoming, we developed Allo, a new approach to allocate multimapped reads in an efficient, accurate, and user-friendly manner. Allo combines probabilistic mapping of multimapped reads with a convolutional neural network that recognizes the read distribution features of potential peaks, offering enhanced accuracy in multimapping read assignment. Allo also provides read-level output in the form of a corrected alignment file, making it compatible with existing regulatory genomics analysis pipelines and downstream peak-finders. In a demonstration application on CTCF ChIP-seq data, we show that Allo results in the discovery of thousands of new CTCF peaks. Many of these peaks contain the expected cognate motif and/or serve as TAD boundaries. We additionally apply Allo to a diverse collection of ENCODE ChIP-seq data sets, resulting in multiple previously unidentified interactions between transcription factors and repetitive element families. Finally, we show that Allo may be particularly beneficial in identifying ChIP-seq peaks at centromeres, near segmentally duplicated genes, and in younger TEs, enabling new regulatory analyses in these regions.
High-throughput sequencing technologies underlie the study of various regulatory genomic phenomena, including gene expression (RNA-seq), protein–DNA interactions (ChIP-seq), and chromatin accessibility (ATAC-seq). Although longer read sequencing techniques like Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) have been established, many regulatory genomics assays continue to use short-read sequencing technologies owing to the higher sampling rate (i.e., higher numbers of reads) and constraints with various preparation steps prior to sequencing. For example, the immunoprecipitation step in ChIP-seq is unlikely to allow the pull down of long stretches of chromatin and thus produces DNA fragments that are most compatible with short-read sequencing. However, repetitive regions pose problems for short-read alignment. Any sequence that is repeated in the genome and is longer than the sequencing read length will create multimapped reads (MMRs). Using the common read lengths (35–100 bp) seen in many studies and public repositories, up to 30% of sequenced reads are not uniquely mappable (Derrien et al. 2012). MMRs, because of their ambiguous nature, are generally removed during preprocessing in most regulatory genomics pipelines, including those used by the ENCODE Consortium (The ENCODE Project Consortium et al. 2020). For these reasons, repetitive regions have been largely overlooked in most gene regulatory analyses.
Several studies have demonstrated that repetitive regions contain transcription factor binding sites, suggesting that they play active roles in gene regulation (Sundaram et al. 2014; Imbeault et al. 2017). One study of 26 orthologous transcription factors in mouse and human showed that ∼20% of all binding sites were derived from transposable elements (TEs) (Sundaram et al. 2014). TEs have been shown to play a role in evolutionary adaptation, a prominent example of which is the insertion of a TE within the cortex gene of peppered moths (van't Hof et al. 2016). This mutation increased transcription of the cortex gene, which allowed for darker wing colors and better camouflage during the Industrial Revolution. TEs have also been implicated in the evolution of mammalian pregnancy development (Lynch et al. 2011), interferon response (Chuong et al. 2016), and pluripotency maintenance (Wang et al. 2014). TE mobilization has also been shown to perturb gene regulation and create disease states such as breast cancer (Jiang and Upton 2019). Most of these studies did not consider MMRs in their analyses and thus only investigated repetitive elements that were uniquely mappable. Therefore, the representation of TEs and other repetitive elements in regulatory processes is likely undercharacterized.
Various methods have been proposed to deal with MMRs in gene regulatory data (Hashimoto et al. 2009; Chung et al. 2011; Jin et al. 2015; Schmid and Grossniklaus 2015; Zeng et al. 2015; Consiglio et al. 2016; Kahles et al. 2016; Zytnicki 2017; Sun et al. 2018; Zheng et al. 2019; Shah and Ruthenburg 2021; Almeida da Paz and Taher 2022). Some of the first methods for ChIP-seq MMR analysis directly aligned reads to the consensus sequences of TEs and other repetitive regions (Sun et al. 2018; Almeida da Paz and Taher 2022). Although consensus sequence mapping provides family-level associations, these approaches cannot identify regulatory events at individual repetitive elements. Another set of approaches were developed to specifically allocate MMRs in the genome by using uniquely mapped read (UMR) counts in the vicinity of possible MMR mapping locations (Hashimoto et al. 2009; Chung et al. 2011; Shah and Ruthenburg 2021). The intuition underlying these approaches is that a region containing UMRs is more likely to also have generated MMRs compared with alternate mapping locations. For example, MuMRescueLite (Hashimoto et al. 2009) used the UMR counts to create a simple probabilistic mapping of MMRs; MMR reads are allocated to specific locations based on the ratio of UMR counts at each of their possible mapping locations. Another method, ChIP-seq multiread allocation using expectation maximization (CSEM), combined probabilistic mapping with iterative reweighting using expectation maximization (Chung et al. 2011). Partial read counts were assigned based on this mapping in each iteration. Unfortunately, both CSEM and MuMRescueLite are, at time of writing, unusable owing to uncompilable code and execution errors, respectively. The recent SmartMap method revived the probabilistic MMR mapping approach using a Bayesian model (Shah and Ruthenburg 2021). Because of the use of a Bayesian model over the entire genome, even fully mappable regions are modeled, and thus, UMR counts can be affected, introducing unnecessary error in the read coverage landscapes output by SmartMap. Additionally, SmartMap cannot be applied to single-end sequencing data without modifying the source code as it relies on the insertion sizes of paired-end reads to make allocation predictions (Shah and Ruthenburg 2021).
Finally, all probabilistic MMR mapping methods to date have outputs that are not easily integrated into most regulatory genomic pipelines as they output custom file types (Hashimoto et al. 2009) or file types not accepted by commonly used peak callers (Chung et al. 2011; Shah and Ruthenburg 2021). In this work, we address the drawbacks of current approaches while also increasing the accuracy of MMR allocation. Our method, Allo, combines probabilistic mapping based on UMR counts with a convolutional neural network (CNN) that has been trained to identify the appearance of peak-containing regions. Allo is applicable to both single-end and paired-end sequencing data, and its SAM/BAM format output is easily integrated into any analysis pipeline.
Results
Allo: MMR allocation combining probabilistic read allocation and image-based peak detection
Regulatory genomics data sets contain substantial proportions of MMRs; for example, up to 30% of mappable reads in ENCODE ChIP-seq experiments are MMRs (Fig. 1A; Supplemental Fig. S1A). Previous methods to allocate MMRs focused solely on leveraging UMR counts in regions around MMRs, as illustrated in Figure 1B. Our method, Allo, implements a probabilistic MMR mapping approach similar to that deployed by MuMRescueLite (Hashimoto et al. 2009), wherein regions with more UMRs will have a higher probability of being allocated an MMR. However, Allo combines probabilistic MMR mapping with a separate neural network module that predicts whether each possible MMR mapping location has read distributions consistent with a ChIP-seq peak (Fig. 1C). The intuition behind this second module is that ChIP-seq reads are more likely to be generated from peak regions than from alternative nonpeak locations on the genome.
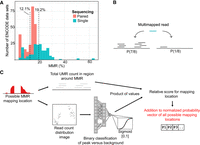
Overview of the prevalence of multimapped reads (MMRs) in ChIP-seq data sets and Allo's algorithm for rescuing MMRs. (A) Proportions of reads that are multimapped across 481 K562 ENCODE TF ChIP-seq data sets. (B) Overview of probabilistic MMR allocation by proximal uniquely mapped read (UMR) count. (C) Overview of the Allo algorithm combining UMR counts and image classification of read distribution.
Allo's neural network takes the form of a CNN that is trained on images of UMR distributions at peaks (Supplemental Fig. S1B). The CNN examines each possible mapping location of a given MMR and outputs scores for each ranging from zero to one, with higher values corresponding to CNN predictions that a region contains a UMR distribution consistent with ChIP-seq peaks. The total UMR count in each region is then multiplied by the corresponding CNN output score. Thus, areas with more UMRs and a more peak-like distribution of reads will have higher combined scores. Allo normalizes across the scores for each possible MMR mapping location to create a relative probability vector. The MMR is then allocated to a single possible mapping location by rolling a dice weighted according to the probability vector; namely, locations with higher normalized scores are more likely to be allocated the MMR. A summary of Allo's algorithm is shown in Figure 1C.
Training a neural network to predict partially mappable peaks based on images of UMR distributions
ChIP-seq reads are distributed around protein–DNA binding events according to a characteristic shape, which is apparent at individual binding sites in successful ChIP-seq experiments given high enough sequencing coverage. Image-based classification approaches that leverage the shape properties have been previously used in multiple ChIP-seq peak calling applications (Strino and Lappe 2016; Hentges et al. 2022). We hypothesized that adding a measure of peak potential based on UMR distribution would increase the accuracy of MMR allocation within ChIP-seq data sets. To train a neural network to predict peak potential, we obtained 10 human transcription factor ChIP-seq data sets and nine human histone ChIP-seq data sets from ENCODE (Supplemental Table S1). The former were used to train a neural network on narrow peaks, and the latter were used to train a neural network on mixed peaks. Switching between narrow peak and mixed peak mode is a simple one argument option in Allo.
To create a training set with known labels, we needed MMR-containing regions in which the peak status (i.e., peak vs. nonpeak) is known. We achieved this by artificially shortening ENCODE ChIP-seq reads to create MMRs. The starting and shortened read lengths of the data sets varied and can be found in Supplemental Table S1. The ground-truth peak regions were defined as 500 bp windows centered on MACS2 peak midpoints that were called from the full-length data set. The 5′ read count distributions of artificially shortened UMRs in these peak windows were used to create 100 pixel–by–100 pixel images for CNN training. Treating the ChIP-seq data as images allows the CNN's convolutional layer to find shape-related features at peaks without dependence on ChIP-seq signal levels. To make images for the negative set, we used an equivalent number of randomly selected MMR-containing regions from the background. A summary of the CNN training set generation process is shown in Figure 2A.
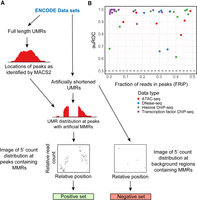
Allo's neural network training process and accuracy testing. (A) Overview of Allo's neural network training process on ENCODE data sets. (B) auROC of Allo's neural networks compared with the fraction of reads in peaks (FRiP) score of the full-length data sets. The color of the points indicates the assay type analyzed.
To evaluate our neural networks’ abilities to classify peaks in MMR-containing regions, we gathered 40 data sets with diverse properties (Cusanovich et al. 2018; Li et al. 2018; Shi et al. 2019; Zhang et al. 2020; Kaushal et al. 2021; You et al. 2021; Delaney et al. 2022; Shang et al. 2022; Abay-Nørgaard et al. 2023; Chen et al. 2023; Ellison et al. 2023; Sanchez et al. 2023; Zhao et al. 2023; Zheng et al. 2023). Although the training sets only included human samples, the test sets included data from human, mouse, Arabidopsis thaliana, Drosophila melanogaster, Caenorhabditis elegans, and Saccharomyces cerevisiae (Supplemental Table S2). We also included ATAC-seq and DNase-seq data sets in the test set, reasoning that our peak shape detection approach would also be applicable to these data types. Note that we initially trained additional CNNs to specifically recognize ATAC-seq and DNase-seq peaks, but the performance of these specialized models had mixed results compared with the performance of the mixed peak CNN (Supplemental Table S3). Consequently, as the mixed peak CNN showed high performance on most DNase-seq and ATAC-seq data sets, we applied the narrow peak CNN to the TF ChIP-seq test data sets and the mixed peak CNN to the histone ChIP-seq, ATAC-seq, and DNase-seq data sets. The specialized DNase-seq and ATAC-seq CNNs remain available for use within Allo, and users can test their data sets with the various CNNs if desired.
Figure 2B summarizes the performance of the CNNs in classifying peaks from nonpeaks in MMR-containing regions; the auROC values are well above random classification for most tested data sets. The handful of data sets with CNN auROC scores lower than 0.70 contain more dispersed read distributions at peaks, potentially making it more difficult for the CNNs to make accurate classifications. Additionally, CNN classification performance does not appear to strongly depend on the fraction of reads in peak (FRiP) score in the relevant test data set (Fig. 2B) or on the source species of each test data set (Supplemental Fig. S1C). Finally, we investigated whether the test set read length has an impact on CNN classification performance. We found only a minimal effect on peak classification performance in data sets with progressively shortened read lengths (Supplemental Fig. S1D; Supplemental Table S4); some decay in performance is to be expected as the read length gets shorter, because the CNN has fewer UMRs available to enable predictions for proximal MMRs.
A CNN trained on read count distribution features increases accuracy of MMR allocation
To test the overall accuracy of Allo's MMR allocation against other methods, we developed an approach that provided knowledge of ground-truth locations for a subset of MMRs in real ChIP-seq, ATAC-seq, and DNase-seq data sets. Briefly, we used a standard UMR-based pipeline to align reads and perform peak calling on a series of publicly available data sets. We then artificially shortened the reads to 30 bp (Fig. 3B) and realigned them to the genome. Some of the reads that were uniquely mappable at full length then became multimapped at this new shortened length. For reads in this category, we had a ground-truth location. Following alignment of the shortened reads, we used three separate methods to allocate MMRs: random allocation, read count–only (similar to MuMRescueLite and also an option in Allo), and Allo using read count information and the CNN. We were unable to test CSEM (Chung et al. 2011), as the software was no longer executable. We were also unable to test SmartMap (Shah and Ruthenburg 2021) using this methodology, as it only outputs a bedGraph file, which would not enable us to calculate accuracy on a per-read basis.
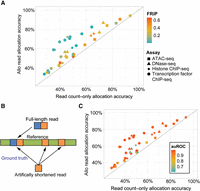
Allo outcompetes other methods for MMR allocation. (A) Accuracy of MMR assignment within testing data set peaks using Allo or read count–only methods. The color denotes the FRiP from the UMR peak calls, and the shape denotes the assay being tested. (B) An overview of how the test data sets were generated by artificially shortening full-length data sets. (C) The accuracy of Allo's read allocation algorithm compared with the read count–only algorithm. Color denotes the CNN's auROC score when detecting peaks in each data set.
To ensure Allo is applicable across data sets, the set of transcription factors used in training was excluded from the training set, and we also chose data sets from different species as explained above (Supplemental Table S2). We included data sets of various transcription factors known to bind to repetitive elements. Examples included EDM2 in Arabidopsis (Tsuchiya and Eulgem 2013), YY1 in humans (Becker et al. 1993), ZFP57 in mice (Shi et al. 2019), and DOT1L in mice (Zhao et al. 2023). Additionally, we included CTCF data sets from mouse, human, and Drosophila, as CTCF has been previously shown to bind TEs (Sundaram et al. 2014). Allo outperforms random allocation and the read count–only method on every data set tested (Fig. 3A; Supplemental Fig. S2A). Allo allocation accuracy does not appear to strongly depend on the data set species or the proportion of MMRs (Supplemental Fig. S2B). Allo appears to offer higher benefits when analyzing transcription factor ChIP-seq data sets compared with the other data types tested (Fig. 3A). TF ChIP-seq data sets have more punctate peaks with more concentrated read distributions and a stronger characteristic shape. Thus, Allo's incorporation of peak detection may offer higher benefits than read count–based allocation alone in cases in which the peak shape is more clearly detectable. Indeed, although the primary determinant of Allo's overall allocation accuracy is the accuracy of the read count–only module, the accuracy of the CNN's ability to predict peaks is a driver of increased performance (Fig. 3C).
Allo's allocation accuracy and performance is competitive with that of SmartMap
As previously noted, we were unable to directly compare the read allocation accuracies of Allo and SmartMap, as it was not feasible to extract individual read mapping locations from the bedGraph files output by SmartMap. We were also unable to compare the two methods on a peak level basis, as MACS2 was unable to properly call peaks on SmartMap bedGraph files. Applying the MACS2 bdgbroadcall function to the SmartMap bedGraph files, as suggested in the SmartMap paper, resulted in over a million peaks called for each data set tested. Even if MACS2 had functioned as anticipated, the bdgbroadcall function does not allow input of a control file and thus is not ideal for statistically valid analysis of ChIP-seq data sets.
To compare the performance of Allo and SmartMap, we instead relied on our method of shortening full-length data sets and compared the average read depth resulting from each method across MMR-containing peaks. We first removed all MMRs in the shortened data sets that were not UMRs in the full-length data sets, meaning all MMRs should have a ground-truth location. We then ran SmartMap and Allo on these filtered alignment files. The BEDTools map function was then used to calculate the average read depth at peaks, analogous to the comparison procedure performed in the SmartMap paper (see Methods). We note that SmartMap only functions on paired-end data sets at time of writing, so we can only compare performances on the subset of test data sets that are paired-end sequenced (38 data sets). We were unsuccessful in our attempts to rewrite SmartMap's processing script to allow single-end data processing.
As shown in Figure 4A, Allo had similar or lower percentage error in allocated read depth at peaks on all but three of the 38 tested data sets. As in the comparisons with the read count–only approach, Allo appears to offer particular improvements over SmartMap in TF ChIP-seq data sets with lower FRiP scores (Fig. 4A). Because both Allo and SmartMap rely on probabilistic assignment using UMR counts, we reasoned that peaks containing fewer UMRs would be underallocated MMRs. In Figure 4, B and C, we show the percentage error at each peak (combining all target samples used above) as a function of the total UMR count at each peak. As expected, regions with fewer UMRs are more likely to be underallocated MMRs by both methods. However, this effect dissipates more quickly in Allo compared with SmartMap. It also appears that Allo is more likely to overallocate reads overall, whereas SmartMap is more likely to underallocate reads. The overallocation by Allo within peaks may be a result of Allo's focus on adding reads to peak regions specifically through the use of its neural networks. Nevertheless, Allo displays reduced error across the tested peaks as seen by the overall lower discrepancy from the ground-truth values.
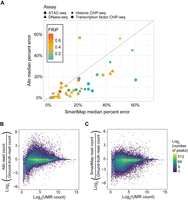
Allo outcompetes SmartMap in overall MMR allocation. (A) Allo's median percentage allocation error versus SmartMap's median percentage allocation error across peaks in 38 testing data sets. The shape indicates data set assay, and the color indicates the FRiP score. (B) Log2 of Allo read counts divided by ground-truth read counts within peaks compared with the log2 of UMR counts in those regions. (C) Log2 of SmartMap read counts divided by ground-truth read counts within peaks compared with the log2 of UMR counts in those regions.
We next employed a ChIP-seq simulator to compare the performance of Allo and SmartMap more comprehensively. In a simulated data set, we have a true location for every read, and we can more accurately study the effects of noise level, read length, and MMR rates on overall allocation accuracy. Allo retains lower read allocation error at all tested levels of simulated noise and at all simulated read lengths (Supplemental Fig. S2C,D). We found that the level of noise in the simulated data sets had more of an impact on median percentage error than did read length or MMR proportion (Supplemental Fig. S2C,D; Supplemental Table S5), with both Allo's and SmartMap's error rate increasing at ∼96% noise. Additionally, we found an inverse correlation between the percentage of MMRs and allocation error rates of both Allo and SmartMap (Supplemental Table S5). As the read length increased, the percentage of MMRs decreased as expected. The areas that contained MMRs in these longer read data sets are more likely to be completely non-uniquely-mappable over a larger span and thus pose a more difficult problem for both methods, because there are few to no surrounding UMRs to enable allocation. To investigate further, we calculated the median length of contiguous non-uniquely-mappable regions in hg38 at different k-mer lengths using GenMap (Pockrandt et al. 2020). We found that the length of non-uniquely-mappable windows in hg38 was highly positively correlated with the length of the k-mers used (Supplemental Fig. S2E). Thus, the longer non-uniquely-mappable windows observed at longer read lengths lead to difficulty in assigning MMRs.
Finally, we compared the computational requirements of the two methods using the simulated data sets described above. As shown in Supplemental Figure S2F, Allo uses more CPU time than SmartMap per 1 million reads allocated. This is likely because Allo is implemented in Python, whereas SmartMap is implemented in C/C++. To mitigate the performance disparity, we implemented a multithreading approach in Allo. Across the data sets tested above, Allo has a lower execution wall-time compared to SmartMap when using six processes. We also found that Allo uses significantly less memory than SmartMap. Allo uses a seventh of the memory used by SmartMap on average at six processes. It is important to note that we ran SmartMap with the default settings, which includes only one algorithm iteration. We would expect that including more reweighting iterations would increase the execution time of SmartMap and possibly also the RAM usage. Together, these results suggest that Allo is competitive with SmartMap in allocation accuracy as well as computational performance.
Allo peaks have similar characteristics to peaks derived from UMRs, reinforcing their validity
To demonstrate the utility of Allo and MMR-inclusive ChIP-seq pipelines, we focused on the properties of K562 CTCF peaks that are discoverable with the incorporation of Allo but are not detected using a traditional UMR-only ChIP-seq analysis pipeline. Allo's inclusion of MMRs uncovered 3114 CTCF peaks in addition to the 54,677 found using UMRs alone (Fig. 5A). Peaks only found using Allo are labeled as “Allo-only” peaks. The CTCF cognate motif was the highest ranked motif found by MEME-ChIP (Machanick and Bailey 2011) in both the UMR-derived peaks (Fig. 5B) and the Allo-only peaks (Fig. 5C), suggesting that these peaks are characteristic of true CTCF binding sites. The Allo-only peaks contained CTCF motif instances 58% of the time compared with 72% of UMR-derived peaks. The cognate motif was identified as similarly centrally enriched in both Allo-only and UMR-derived peaks using CentriMo (Fig. 5D; Bailey and Machanick 2012). Allo also increased the read depth significantly in Allo-only peak regions (Fig. 5E).
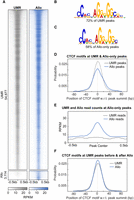
Allo results in the discovery of 3114 additional peaks in a CTCF ChIP-seq data set. (A) ChIP-seq heatmaps comparing K562 CTCF peaks called by MACS2 using UMRs only or UMRs plus Allo-mapped reads. (B,C) Top-ranked motifs from MEME-ChIP in the UMR-derived and Allo-only peaks, respectively. (D) Position of the CTCF motif with respect to the peak summit for UMR-derived peaks and Allo-only peaks. (E) Read depth at Allo-only peaks using UMR BAM files versus using Allo output. (F) Position of the CTCF motif with respect to the peak summit before and after the inclusion of Allo at UMR-derived peaks.
We next examined the overlap between UMR-derived peaks and Allo-only peaks at topologically associating domain (TAD) boundaries using TAD calls from the 3D Genome Browser (Rao et al. 2014). A TAD boundary was defined as being within 25 kb of the beginning or end of an annotated TAD. We found that there was a similar overlap between peak sets and TAD boundaries: 22.4% for the UMR-derived peaks and 19.1% for the Allo-only peaks. Figure 6A shows an example of two Allo-only CTCF peaks at TAD boundaries near the THOC3 gene. In this case, the MMRs present are a result of a duplicate pseudogene of THOC3.
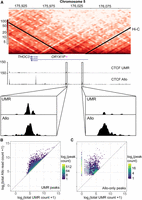
Allo increases the overall read counts at multimapped CTCF peaks and improves the resolution of uniquely mappable peaks. (A) Hi-C interaction heatmap showing two TAD boundaries on Chromosome 5 near the THOC3 gene. K562 CTCF read counts are also plotted for the UMR-only analysis and the Allo analysis. Bottom sections show the zoomed in version of two Allo-only peaks at these TAD boundaries. (B) Density scatterplot showing the total read count before and after the incorporation of Allo within CTCF UMR-derived peaks. (C) Density scatterplot showing the total read count before and after the incorporation of Allo within CTCF Allo-only peaks. Note that Allo was run using default settings, which allocates MMRs even in cases in which there are no nearby UMRs. This results in peaks called in regions with zero starting UMR counts as seen in panel C. The user can shut off this functionality using the argument “‐‐remove-zeros” to have more stringent criteria for MMR allocation.
Furthermore, we investigated the effects of Allo on peaks that were discoverable using the standard UMR-only pipeline, as some of these peaks additionally contain MMRs. Figure 6, B and C, shows the total read counts before and after using Allo, at UMR-derived peaks and Allo-only peaks, respectively. It is evident that many UMR-derived peaks gain substantial numbers of reads with the inclusion of MMRs. We also found that Allo slightly improved the precision of peak finding at UMR peaks, as the addition of the Allo reads resulted in tighter distribution of distances between peak summits and the CTCF motif (Fig. 5F). Therefore, Allo can increase peak resolution and ChIP-seq enrichment quantification accuracy at many peaks, even those that were already discoverable using UMR-only pipelines. Supplemental Figure S3A provides examples of the increased resolution at selected CTCF UMR-derived peaks after MMR inclusion.
Schmidt et al. (2012) previously demonstrated that many rodent-specific CTCF binding sites are associated with an expansion of SINE B2 elements in the rodent lineage. Because TEs are a common source of MMRs, we might expect Allo will lead to the discovery of additional SINE-associated CTCF binding sites. We thus deployed Allo on the Schmidt et al. CTCF ChIP-seq data sets from human and mouse liver samples. Analysis of Allo-only CTCF peaks shows an enrichment of SINE B2 class elements, consistent with the observations of Schmidt et al. (Supplemental Fig. S3B,C). Although the pattern of SINE binding remained similar between the UMR and Allo-only peaks, the use of Allo resulted in 1652 new mouse and 691 new human CTCF binding sites within SINE elements.
Allo supports the discovery of associations between TFs and repeat families in large-scale ChIP-seq analyses
To broadly survey how an MMR-inclusive pipeline might enable the discovery of additional protein–DNA binding events, we reanalyzed 481 ENCODE K562 TF ChIP-seq data sets using Allo. The use of Allo resulted in a median increase of 5.86% additional peaks compared with the standard UMR-only pipeline (Fig. 7A; Supplemental Table S6), yielding the discovery of 385,563 new TF binding sites over the entire collection. There were no data sets in which the inclusion of Allo did not result in additional peaks. To evaluate the quality of the new peak calls within these data sets, we plotted the distribution of reads at Allo-only peaks in a random subset of data sets (Supplemental Fig. S4), finding distributions that are characteristic of ChIP-seq peaks. Allo leads to an increase in peak read counts across many of the ENCODE data sets (Supplemental Fig. S5A). We also observed that the Allo-only peaks were widely distributed across the genome (Supplemental Fig. S5B), suggesting that they are not artifacts of specific chromosomal regions.
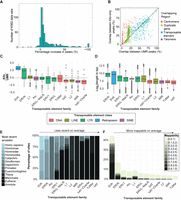
The use of Allo results in the discovery of additional peaks in 481 K562 data sets. (A) Percentage increase in peaks between the Allo-inclusive pipeline and the UMR-only pipeline across 481 ENCODE K562 ChIP-seq data sets. The dotted line represents the median increase in peaks (5.8%). (B) Percentage overlaps between Allo-only peaks and centromeres, telomeres, segmentally duplicated genes, and transposable elements (TEs). (C) The ratio between Allo-only peak overlap rates and UMR-derived peak overlap rates for each TE family. (D) Log2 read length of each TE insertion in hg38, grouped according to its respective repeat family. (E) Percentage of insertions within each TE subfamily that belong to each most recent ancestor. From left to right, the overall age increases. (F) Mappability score (UMAP K10069) of TE insertion sites, grouped according to their respective TE family. Mappability values equal to one (i.e., fully uniquely mappable) are not included.
Next, we evaluated the repetitive element content of the newly discovered Allo-only peaks by comparing with RepeatMasker (Tarailo-Graovac and Chen 2009) annotated repetitive regions (Supplemental Fig. S5C). Allo-only peaks overlap many repeat element classes at similar rates to UMR-derived peaks. Two exceptions are the satellite repeat and retroposon classes, which display higher enrichment rates in Allo-only peaks compared with UMR-derived peaks. Satellite repeats are commonly found within centromeric and telomeric regions (Thakur et al. 2021) and have been shown to regulate gene expression in eukaryotic organisms (Arunkumar and Melters 2020; Shatskikh et al. 2020; Horton et al. 2023). To further investigate the association with satellite regions, we compared the rates of overlap between Allo-only and UMR-derived peaks and centromeric or telomeric regions (Fig. 7B, each dot represents the overlap rate of one transcription factor data set with each annotation type). Allo is especially beneficial for finding peaks in centromeres. For many examined TFs, centromeric peaks are only discoverable using Allo (Fig. 7B; Supplemental Figs. S6, S7).
Another possible source of MMRs are segmentally duplicated genes. Roughly 70% of genes in the human genome have at least one paralog (Ibn-Salem et al. 2017), and more recent duplications are likely to retain high similarity and thus may produce MMRs. As with centromeres, Allo-only peaks are generally more likely than UMR-derived peaks to lie nearby or within segmentally duplicated genes (Fig. 7B; Supplemental Figs. S6, S7). Thus, Allo may enable new insights into the regulation of recently duplicated genes.
Although Allo-only peaks generally do not have a higher rate of overlap with TEs compared with UMR-derived peaks (Fig. 7B), two notable outlier families are SVA retroposons and ERVK LTR elements (Fig. 7C). We saw a similar increase in peaks at LTR elements using the Schmidt et al. CTCF data sets in mouse and human liver samples (Supplemental Fig. S5D,E). The top five transcription factors within the K562 data sets that show the highest gains in SVA-overlapping peaks are TAL1, ZNF740, TCF3, CBFA2T2, and TCF12 (Supplemental Table S7). Of these, only CBFA2T2 has been previously shown to bind to SVA elements (Smallegan et al. 2021). The top five transcription factors that show the highest gains in ERVK-overlapping peaks are TAL1, TFE3, ZBTB7A, RFX5, and KLF16, none of which have a previous association with ERVK elements. To understand why Allo-only peaks are particularly enriched in SVA and ERVK elements, we note that these element types are generally longer than members of other TE families (Fig. 7D). They are also among the youngest families of TEs (Fig. 7E). Longer TEs are generally less mappable as read lengths are less likely to extend past them into neighboring nonrepetitive DNA. Likewise, newer insertions should be less mappable as they have had less time to gain mutations with respect to their parent copy. Indeed, SVA and ERVK repeats are less mappable than other repeat families according to UMAP K100 scores (Fig. 7F). Thus, it appears that Allo is particularly useful for finding peaks in longer and/or younger TEs.
Discussion
Although repetitive elements have long been recognized to play a role in gene regulation (Britten 2010), and despite previous efforts to develop MMR allocation tools (Hashimoto et al. 2009; Chung et al. 2011; Shah and Ruthenburg 2021), MMRs have not been consistently included in standard regulatory genomics analysis pipelines. As a result, many regulatory elements within repeats remain ignored by regulatory genomics analyses. A major reason for this exclusion is that existing methods to allocate MMRs are not easily integrated into established analysis pipelines. For instance, the recent SmartMap method produces a bedGraph file, making it incompatible with standard pipelines such as those used by ENCODE (The ENCODE Project Consortium et al. 2020). Older methods, such as MuMRescueLite and CSEM, have not been maintained and are no longer functional, and they also lacked read-level output. Allo addresses these shortcomings by producing a SAM/BAM format alignment file, ensuring compatibility with various downstream analyses.
Besides increasing basic usability, Allo also outcompetes previous approaches. Random allocation is used frequently in pipelines as it is the easiest method to implement; both BWA and Bowtie perform random allocation by default. Allo significantly outcompetes random allocation, as shown in Supplemental Figure S2A. Allo also has greater read assignment accuracy compared with a strategy based entirely on UMR-weighted probabilistic mapping of MMRs (similar to the method implemented in MuMRescueLite) (Fig. 3A), and compared with the Bayesian mapping approach implemented in SmartMap (Fig. 4A). These comparative results demonstrate the advantages of Allo's integrated neural network peak classifier, which aims to take advantage of UMR distribution shapes in alternate mapping locations.
To emphasize the importance of including MMRs in ChIP-seq analyses, we analyzed 481 ENCODE data sets using Allo. The average total peak number increase was 15.7% (Fig. 7A), resulting in the identification of thousands of additional peaks in many of the samples. Five data sets in this sample actually doubled their peak numbers after the inclusion of Allo, including BRCA1, a well-known tumor-suppressor gene (Supplemental Table S6; Fu et al. 2022). In analyzing specific repeat element family associations (Fig. 7C), we also identified several other TFs involved in cancer progression, including TAL1 (Sanda and Leong 2017), TCF3 (Gui et al. 2021), CBFA2T2 (Chen et al. 2017), TCF12 (Yang et al. 2019), ZBTB7A (Singh et al. 2021), RFX5 (Guo and Liu 2022), and KLF16 (Supplemental Table S7; Bang et al. 2020). We additionally found that Allo was especially important for the discovery of binding sites in longer and younger TEs (Fig. 7C–F). Previous studies have identified primate-specific enhancers that may have contributed to the evolutionary development of humans (Klein et al. 2018; Uebbing et al. 2021). Enhancers that arose from repetitive elements are less likely to be categorized in these types of studies owing to issues with MMR inclusion. Thus, Allo could be used to further study the regulatory contributions of TEs in an evolutionary context.
Finally, we highlight the importance of including MMRs in the context of centromeric satellite repeats and recently duplicated genes (Fig. 7B). One caveat that applies to our analyses of satellite repeats is related to the limitations of genome assemblies. If satellite repeats were not sufficiently represented on the hg38 genome assembly, annotated copies could become a “sink” for allocated reads, thereby producing artifactual peak signals. Although we are confident that the Allo-only peaks discovered at centromeres display the shape properties of valid ChIP-seq peaks (e.g., Supplemental Figs. S6, S7), this issue remains a potential concern. The new Telomere-to-Telomere (T2T) genome assembly (Vollger et al. 2022) has more accurately sequenced and annotated satellite repeats, segmental duplications, and repetitive regions more broadly. In combination with Allo's allocation of MMRs, the T2T genome may thus provide even greater abilities to map regulatory elements at repetitive regions of the genome.
Together, the results of this work suggest that rescuing MMRs with Allo should be employed in the analysis of all regulatory genomics data sets, even those without prior hypotheses tied to repetitive regions. The inclusion of MMRs in future analyses will provide greater insight into regulatory activities at these overlooked areas of the genome.
Methods
Data sets
All ChIP-seq, ATAC-seq, and DNase-seq training and testing data sets were obtained from publicly available databases, including ENCODE and NCBI. Supplemental Table S1 lists the IDs for data sets used in training Allo's CNN. Supplemental Table S2 lists the IDs of all data sets used for testing. Supplemental Table S6 lists the IDs of all K562 data sets extracted from ENCODE in our large exploratory analysis.
Percentage of MMRs
To find the percentage of MMRs in ENCODE data sets, all FASTQ files were aligned using Bowtie 2 (Langmead and Salzberg 2012) v2.5.0, and multimapping rates were extracted from the output of Bowtie 2 upon alignment. Paired- and single-end data sets were separated and plotted with ggplot2 (Valero-Mora 2010).
CNN training and testing set generation
To train and test the neural networks, we first obtained 59 data sets from both ENCODE and NCBI from a mixture of assay types, including ChIP-seq, ATAC-seq, and DNase-seq. Nineteen of the human ChIP-seq data sets were used to train the narrow and mixed peak neural networks (Supplemental Table S1). The remaining 40 data sets were used for testing the neural networks as well as testing the allocation accuracy (Supplemental Table S2). The data sets were aligned to hg38 for human data sets, mm10 for mouse data sets, dm6 for D. melanogaster data sets, TAIR10 for A. thaliana data sets, ce10 for C. elegans data sets, and sacCer3 for S. cerevisiae data sets using Bowtie (Langmead et al. 2009) v1.0.0 with the arguments “‐‐best ‐‐strata -m 1 -k 1 ‐‐chunkmbs 1024.” These alignments represent our full-length read set, and MACS2 (Zhang et al. 2008) v2.2.7.1 was used to call peaks with all default parameters, generating our ground-truth peaks. We next artificially shortened these reads from the 3′ end using cutadapt (Martin 2011) v.4.2 with the parameters “-l 30” for testing data sets, and we used a mixture of cut sizes for the training set (Supplemental Table S1).
In the case of testing different trimming lengths, we used cutadapt with the same “-l” parameter but adjusted the value based on the total length of the read and our desired trimming percentage. We chose a random subset of data sets, two from each organism tested, to analyze in this way. For alignment, we used Bowtie instead of Bowtie 2 owing to Bowtie's ability to align very short reads more accurately after this artificial trimming. Next, we aligned the artificially shortened reads to each respective genome. For the UMR samples, we used the following arguments “‐‐best ‐‐strata -m 1 -k 1 ‐‐chunkmbs 1024.” For the MMR samples, the arguments were “‐‐best ‐‐strata -m 25 -k 25 ‐‐chunkmbs 1024.” We then used Allo's parser utility (argument ‐‐parser) to separate out the UMRs and MMRs in the shortened samples.
To find the 5′ read counts within these peaks and background regions, we created a custom script named “cnn_training_gen.py” (available in the Allo GitHub repository and the Supplemental Code). The user must supply the script with four arguments: the alignment (SAM) file, a BED file of peak regions, a BED file of background regions, and the output name. To make the positive and negative BED files, we first took the MMR-only artificially shortened SAM file (output of Allo's parser utility) and converted it into a BED file. We then overlapped this BED file using BEDTools (Quinlan and Hall 2010) v2.27.1 intersect -u with the ground-truth peak file. This gave us the positive regions. For the negative regions, we used BEDTools intersect -v to find areas with MMRs that did not overlap ground-truth peak regions. These regions have different lengths, but this is corrected to ±250 bp from the midpoint within the Python script. Additionally, the Python script also corrects the negative set size by only outputting an equal number to the positive set. The output is a text file with comma-separated arrays of 5′ read counts in 500 bp windows around the positive and negative regions. To obtain the auROC of each data set, “metrics.roc_auc_score” from scikit-learn (Pedregosa et al. 2011) was used. The bash script for this entire pipeline can be found in the Allo GitHub repository and the Supplemental Code named “cnn_training_examples.sh.” The same script was used to generate the testing sets.
CNN architecture and training
The neural networks used in Allo's allocation algorithm are image-based CNNs. To convert the 5′ read count text files explained in the above section into images, we used a custom Python function. This function can be found on the Allo GitHub repository and the Supplemental Code folder within the first code block of the Python markdown file “Narrow_train.ipynb.” Both neural networks have similar architectures (Supplemental Fig. S1B) and were trained using TensorFlow (Abadi et al. 2016) v2.11.0. The optimizer used was Adam, the batch size was 500, and the loss function used was binary cross entropy. The specific code used to train the neural networks can be found on the Allo GitHub repository and the Supplemental Code under the files named “Narrow_train.ipynb” and “Mixed_train.ipynb.”
Allo allocation algorithm
Allo's algorithm has two main phases. The first phase is the pileup of UMR within the genome. To do this, Allo first loops through the alignment file and parses uniquely mapped and multimapped reads. Depending on the aligner used, alignment files can contain locations that do not have the highest alignment score and thus require extra parsing. In addition, Allo identifies the correct pairs when using paired-end sequencing data. When Allo encounters a UMR, it adds it to a Python dictionary. The keys are the locations on the genome, and the values are the 5′ read counts in that specific location. A dictionary was used to increase the speed of data acquisition to create images of various regions on the genome in phase II. For paired-end reads, only the first read in each pair is used to construct this dictionary. The second in pair can be used instead through the argument “‐‐r2.” Also, during this phase, temporary files are constructed from MMRs.
In phase II, the MMRs in the temporary files are allocated. Allo analyzes one read at a time by grouping it with its possible locations. Two vectors are constructed. The first vector contains the total read count in 500 bp windows (±250 bp) around each location. In this vector, a pseudocount of one is added to all location UMR counts so that locations without UMRs are still considered. The second vector contains the output of the sigmoid function from Allo's neural network. The 5′ read counts in 500 bp windows around each read location are converted to a 100 × 100 bitmap matrix (representing the peak “image”) using the dictionary described above and a custom script to bin the counts. This script can be found on the Allo GitHub repository and the Supplemental Code within the file “Narrow_train.ipynb.” After constructing the image matrix, it is fed through the neural network, and the results are stored in the vector above. The result is two vectors with each index corresponding to a mapping location of the read being analyzed. These vectors are then multiplied together to get a final score vector. The final score vector is then normalized by dividing all entries by the vector sum, giving the final probabilities. The choice function in Python is then used to select the location based on the final probabilities.
To increase the speed of Allo's algorithm, a few extra steps were employed. First, Allo stores previously analyzed regions in a dictionary as reanalysis slowed down Allo considerably. In addition, Allo does not create an image matrix for regions with zero read counts as the result will be consistent across these regions. Rather, it stores the value for zero-count regions once. These regions are stored as NULL in the dictionary above. Finally, Allo also does not create an image matrix for regions containing fewer than three reads. The reasoning behind this was similar as described for zero-count regions. When we created 1 million randomized regions with three reads in a 500 bp window, the results were very similar from the neural network across the many images. We believe this is because low read depth regions do not have sufficient resolution to enable Allo's neural network to make useful predictions. The average score across 1 million random three-count regions was 0.0062 with a standard deviation of 0.00211 from the final sigmoid function. This average value is thus predefined as the neural network output for all regions with three or fewer total read counts.
Read allocation accuracy
To measure the allocation accuracy of various methods, we needed to create a peak set that contained MMRs but had a ground-truth read count. FASTQ files were downloaded from ENCODE for 40 experiments (Supplemental Table S2). To avoid problems with mixing single- and paired-end data, experiments that were paired-end were treated as single-end by only utilizing the first read file (this is only for the purposes of accuracy testing; Allo can properly handle paired-end data as described above). All files were aligned using Bowtie v1.0.0 with the parameters “‐‐best ‐‐strata -m 1 -k 1 ‐‐chunkmbs 1024.” Bowtie v1 was used because of its higher accuracy in aligning very short reads. The reads were then artificially shortened from the 3′ end using cutadapt v4.2 with the parameters “-u -LENGTH.” Read lengths for all testing data sets were trimmed to 30 bp. The shortened reads were then aligned with Bowtie v1.0.0 using the same parameters as above. The full-length alignments were used to call peaks using MACS2 v2.7.1 on all default parameters. This became the ground-truth peak set. Using the BEDTools v2.27.1 intersect, we extracted the reads in the full-length data set that fell within the ground-truth peaks. This gave us our final ground-truth alignment file needed for comparisons.
Allo (default), Allo (‐‐read-count), and Allo (‐‐random) were then used to allocate the reads in the artificially shortened samples. The locations of these allocations were compared with the alignment file from the full-length data set. Accuracy percentages were simply calculated from the total number of correctly allocated reads divided by the total number of MMRs in the sample. The full script for this calculation, including an example from ENCODE, is available on the Allo GitHub and the Supplemental Code named “read_acc.sh.” Complete accuracy results are shown in Supplemental Table S8.
SmartMap comparison
Read depth percentage error
The subset of testing data sets that used paired-end sequencing were used to compare accuracy with SmartMap (Supplemental Table S2). Paired-end reads were aligned with Bowtie 2 v2.5.0 to hg38 using the arguments “-k 25 ‐‐no-mixed ‐‐no-discordant.” UMRs were extracted from the resulting alignment files using the parser utility in Allo (‐‐parser). These UMRs were then used to get ground-truth peak locations using MACS2 v2.7.1 with the “-f BAMPE” argument. We next artificially shortened these reads from the 3′ end using cutadapt v4.2 with the parameters “-l 30.” The use of Bowtie 2 in this section was owing to its ability to more accurately map paired-end reads even with the pitfall of it being less accurate for short read lengths. We then used the bash join function to extract MMRs from the shortened sample that had a ground truth in the full-length sample. Many of the MMRs in the shortened samples were also MMRs in our ground-truth full-length sample, and we wanted to avoid using these in our final calculations. The resulting alignment files were then analyzed by both SmartMap and Allo. To use an alignment file with SmartMap's prep script, we had to make some modifications. The modified version of this script can be found on the Allo GitHub and the Supplemental Code under the name “sm_prep.sh.” To convert the Allo output to a bedGraph file, we used BEDTools v2.27.1 bamtobed -bedpe and then BEDTools genomecov -bga. We used this same conversion on the ground-truth full-length alignment file.
To get the read depth within peaks, we used BEDTools map with the arguments “-c 4 -o mean -null “0””, in which the counts were generated over the peaks identified by MACS2 in the full-length data sets. The percentage mapping error was then calculated for each peak, and the average was taken for each sample. An example script of this pipeline is available on the Allo GitHub and the Supplemental Code named “smartmap_compare.sh.”
FRiP score calculation
To calculate the FRiP scores, BEDTools v2.27.1 intersect -u was used to find the overlap between peaks called by MACS2 in the sample and the associated alignment files. SAMtools (Li et al. 2009) v1.16.1 view -c was then used to get the number of reads within this overlap. This number of reads was then divided by the total number of reads to calculate the FRiP score for each specific sample.
Performance metrics
CPU usage, execution time, and RAM usage were all tested on Intel Xeon gold 6226R CPUs with a processing speed of 2.90 GHz. We ran Allo using various numbers of processes (one, two, four, six), as well as SmartMap using the simulated data sets from the above section. We normalized values to get the metrics per 1 million reads, making the performance metrics comparable across data sets. The mean was calculated from all simulated data sets and is shown in Supplemental Fig. S2F.
ChIP-seq simulations
Paired-end human ChIP-seq data were simulated using the ChIPOverlapReadSimulator module in ChExMix v0.5 (Yamada et al. 2020). Peak strengths were taken from an ENCODE K562 CTCF data set (experiment ID: ENCSR000EGM). The peak distribution file used was that of CTCF and can be acquired at the following URL: https://lugh.bmb.psu.edu/software/multigps/support/ctcf_chipseq.distrib.txt. BEDTools v2.27.1 random was used to get random locations within the hg38 genome to simulate peaks. To increase the number of MMRs in peaks, we used BEDTools intersect with the hg38 RepeatMasker (Tarailo-Graovac and Chen 2009) annotation to further select peak locations. The arguments used for the simulator were “‐‐c 1 ‐‐r 1 ‐‐a 0.0 ‐‐up 0.0 ‐‐down 0.0 ‐‐frags 5000000 ‐‐reads 10000000 ‐‐paired.” We varied both the read length and noise in the simulation. Following simulation, reads were aligned with Bowtie 2 v2.5.0 to hg38 using the arguments “-k 25 ‐‐no-mixed ‐‐no-discordant.” This allowed us to get alternative locations for all simulated reads. Following this, both Allo and SmartMap were deployed on the resulting alignment file. The procedure to compare median percentage error is identical to that described in the above section “Read depth percentage error.” Simulations were performed 10 times for each noise and read length shown in the text, and the median percentage error was calculated.
Mappability calculations
GenMap v1.3.0 was used to find the uniquely mappable regions in hg38 (Pockrandt et al. 2020). Various k-mer sizes were tested, matching those used in the ChIP-seq simulations (20, 30, 70, 90, 110, and 150 bp). To find the unmappable regions, BEDTools v2.27.1 complement was used on the resulting bedGraph file. The median value for each k-mer length was calculated using the length of each unmappable window in the genome based on the bedGraph output plus the k-mer length.
CTCF analysis
Alignment and peak calling
FASTQ files for CTCF in K562 were downloaded from ENCODE. Samples files were ENCFF000YLW and ENCFF000YLY. The control file used was ENCFF000YRB. Reads were aligned using Bowtie v1.0.0 to hg38 with the arguments “‐‐best ‐‐strata -m 25 -k 25 ‐‐chunkmbs 1024.” Reads were allocated using Allo with default parameters. We extracted the UMRs using grep against the ZA and ZZ tags from Allo. We then called peaks on the UMR alignment file as well as the Allo alignment files using MACS2 v2.7.1 with default parameters. Peaks were called on the replicates separately, and overlapping peaks between the replicates found using BEDTools v2.27.1 intersect -u were used as the peak sets. BEDTools intersect -v was used to identify peaks only discovered via Allo.
Heatmap and profile plot
Both the heatmap and the profile plot were created using deepTools (Ramírez et al. 2016) v3.5.1. The bamCoverage function was used to create bigWigs from each of the alignment files. We used the RPKM normalization argument. To check that this normalization was valid for our specific data sets, we plotted profile plots of random regions using BEDTools v2.27.1 random. We used the computeMatrix function with the arguments “‐‐referencePoint center -a 1000 -b 1000” before plotting.
Motif identification and scanning
Using BEDTools v2.27.1 getFasta with hg38, we extracted sequences for all peaks represented in the UMR sample as well as Allo-only peaks. We then input these sequences into MEME-ChIP v5.3.3 with the arguments “-meme-nmotifs 5 -minw 5 -maxw 20.” The motifs in Figure 5, B and C, were those with the lowest P-values. CentriMo (Bailey and Machanick 2012) in MEME-ChIP was used to create probability plots for the CTCF motif in each region.
TAD overlap
The locations of TADS were downloaded from the 3D Genome Browser for K562, specifically the data set from Rao et al. (2014). We then used these TAD locations to create a BED file of TAD boundaries, which we considered as within 25 kb of either end of each TAD. This file was then intersected with UMR peak calls and Allo-only peak calls using BEDTools v2.27.1 intersect -u. Figures were constructed using pyGenomeTracks (Lopez-Delisle et al. 2021) v3.8.
UMR and MMR count comparison
BEDTools v2.27.1 multicov was used to calculate read counts at Allo-only peaks and UMR peaks. The BAM files used were concatenated files from both CTCF biological replicates. The data were then plotted using ggplot2 with geom_bin2d and a continuous scale with a log2 transformation.
CTCF analysis in mouse and liver samples
ChIP-seq data sets for CTCF in mouse and liver samples were taken from the Schmidt et al. (2012) study under the ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) accession numbers E-MTAB-437 and E-MTAB-424. Bowtie 2 v.2.5.1 was used to align the data sets with the parameter “-k 25.” The human data set was aligned to hg38, and the mouse data set was aligned to mm10. MMRs were allocated with Allo using the default settings. Peaks were called with MACS2 v2.7.1 with default settings. BEDTools v2.27.1 intersect -u was used to find the fraction overlaps between each data set and their corresponding RepeatMasker v4.1.3 annotation file.
ENCODE K562 analysis
Alignment and peak calling
We analyzed the ENCODE public database, focusing specifically on ChIP-seq experiments in K562 cells. From 774 qualifying experiments (retrieval date February 2023), we selected 481 based on the criteria that the data set had at least one replicate, had a control file, had no major audits from ENCODE, and resulted in nonzero peak calls in the UMR sample using our pipeline. We also only analyzed one data set per transcription factor, which was randomly selected from the selection of those available. We retrieved FASTQ files for both single- and paired-end experiments. Replicates were concatenated and aligned using Bowtie 2 v.2.5.1, reporting 25 valid alignments per read with parameters “‐‐no-mixed ‐‐no-discordant” for paired-end reads. The alignments were sorted using SAMtools v1.16.1 collate. We employed Allo on the sorted alignments to obtain rescued reads. For control experiments, reads were randomly assigned during Allo processing (‐‐random). Following alignment and rescue, peaks were subsequently identified using MACS2 v2.7.1, with the argument “-f BAMPE” for paired-end experiments. ENCODE blacklist regions were excluded using BEDTools v2.27.1 intersect -v. A list of all ENCODE data sets used can be found in Supplemental Table S6.
Read depth at peak summits
Peak summit depths were summed across all peaks after MACS2 peak calling on UMR and Allo data. Each dot in Supplemental Figure S5A represents one data set.
Chromosomal locations of Allo-only peaks
The Allo-only peaks were concatenated together for all K562 data sets tested. We removed peaks located in regions outside of the canonical chromosomes. Next, we created a matrix in which the columns corresponded to the chromosomes, and the rows corresponded to bins the size of 1 million bp along the chromosomes. All peaks within these bins across all K562 data sets were added together to get a total value of peaks within each bin. This matrix was normalized to Z-scores using the scale function in R version 4.3.3 (R Core Team 2024). Finally, the bins were plotted as a heatmap using pheatmap (https://cran.r-project.org/web/packages/pheatmap/index.html) v1.0.11, filling in regions outside of chromosomes with NA values.
Profile plots and genome browser images
Fifteen transcription factors were randomly selected as well as five transcription factors with the highest increase in overlap for each annotation we analyzed (TEs, centromeres, and segmentally duplicated genes). Thus, there were 30 data sets total. Data sets in which there were fewer than 50 total peak calls were not included in this analysis. For the randomly selected transcription factors, we plotted read counts at all Allo-only peaks. For the transcription factors that overlapped a specific region type, we only plotted read counts at Allo-only peaks that overlapped those regions. The profile plots were created using deepTools v3.5.1. The bamCoverage function was used to create bigWigs from each of the alignment files. We used the RPKM normalization argument. We used the computeMatrix function with the arguments “‐‐referencePoint center -a 1000 -b 1000” before plotting.
For the genome browser images, we selected two data sets from each annotation group for viewing. We plotted random Allo-only peaks within the enriched annotation using PyGenomeTracks v3.8 and the bigWigs we created using deepTools above.
Overlap with TE classes and families
The RepeatMasker annotation file v4.1.3 was used to define locations of repetitive element classes and families in hg38. Centromere and telomere annotations were extracted from the UCSC Genome Browser hg38 annotations using the tracks “centromeres” and “gap,” respectively (retrieved April 2024). From the “gap” track, telomeres were extracted specifically. The T2T genome (Vollger et al. 2022) annotations were used to define genes that are contained in segmental duplications, and their coordinates in hg38 were found by using their Ensembl IDs against the hg38 GENCODE GFF3 file (v29). BEDTools v2.27.1 intersect -u was used to find the overlaps between each ENCODE peak data set and each repeat type. Overlaps with segmentally duplicated genes were defined as directly overlapping or within 1 kbp of the gene body. The fraction overlaps were then plotted as a box plot using ggplot2.
TE dating
We used data from Simonti et al. (2017), which identified the most recent common ancestor for each TE subfamily. We used the RepeatMasker hg38 file to identify the locations of the TEs. BEDTools v2.27.1 intersect -u was used to find the fraction overlap between Allo-only peaks and UMR-derived peaks for each common ancestor.
TE mappability scores and lengths
A bedGraph file containing UMAP100 (Karimzadeh et al. 2018) uni-read scores for hg38 was downloaded from the Hoffman laboratory website (https://bismap.hoffmanlab.org). To get the average score across each TE insertion, BEDTools v2.27.1 coverage was used. The UMAP K100 bedGraph file contains regions of the genome in which at least one k-mer is uniquely mappable. The fraction of base pairs that intersected the UMAP100 bedGraph file for each insertion site was used as a measure of mappability. Mappability scores were then binned in 0.1 length intervals in order to see the striation of the data better. These mappability bins along with the subfamilies were subsequently plotted using ggplot2. The lengths of the repetitive elements were calculated for each insertion by subtracting the stop coordinate of the RepeatMasker BED file from the start coordinate. These values were grouped by repetitive element family and plotted using ggplot2.
Software availability
Allo is available under an open-source license (MIT license) from GitHub (https://github.com/seqcode/allo). Allo can also be installed from PyPI using “pip install bio-allo” at the command line. Allo is also available as a bioconda package (https://anaconda.org/bioconda/allo). Instructions on usage can be found in the above GitHub repository. The version of Allo used in this study is archived as Supplemental Code.
Data access
Data sets used in this paper were extracted from publicly available sources. Information on the data sets downloaded can be found in Supplemental Tables S1, S2, and S6. MACS2 peak calls (both UMR and Allo pipelines) can be found for all K562 data sets at figshare (https://doi.org/10.6084/m9.figshare.25977160.v1).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This paper is based upon work supported by the National Science Foundation DBI CAREER 2045500 (to S.M.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. The Mahony laboratory is also supported by the National Institutes of Health grant R35GM144135. A.M. gratefully acknowledges funding and training opportunities from the Computation, Bioinformatics, and Statistics (CBIOS) Training Program (T32GM102057). The authors thank the members of the Center for Eukaryotic Gene Regulation at Penn State for helpful feedback and discussions.
Author contributions: A.M. and S.M. conceived Allo's design. A.M. developed Allo and performed accuracy testing including CTCF analysis. D.Q.J. consulted on experimental design. A.M. and J.S. analyzed the K562 data sets. A.M. and S.M. drafted and prepared the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278638.123.
- Received October 16, 2023.
- Accepted June 14, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








