
Genomic origin, fragmentomics, and transcriptional properties of long cell-free DNA molecules in human plasma
- Huiwen Che1,2,3,
- Peiyong Jiang1,2,3,4,
- L.Y. Lois Choy1,2,3,4,
- Suk Hang Cheng1,2,3,
- Wenlei Peng1,2,3,
- Rebecca W.Y. Chan1,2,3,
- Jing Liu1,2,3,
- Qing Zhou1,2,3,
- W.K. Jacky Lam1,2,3,4,
- Stephanie C.Y. Yu1,2,3,
- So Ling Lau5,
- Tak Y. Leung5,
- John Wong6,
- Vincent Wai-Sun Wong7,
- Grace L.H. Wong7,
- Stephen L. Chan4,8,
- K.C. Allen Chan1,2,3,4 and
- Y.M. Dennis Lo1,2,3,4
- 1Centre for Novostics, Hong Kong Science Park, Pak Shek Kok, Hong Kong SAR, China;
- 2Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China;
- 3Department of Chemical Pathology, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China;
- 4State Key Laboratory of Translational Oncology, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China;
- 5Department of Obstetrics and Gynecology, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China;
- 6Department of Surgery, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China;
- 7Department of Medicine and Therapeutics, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China;
- 8Department of Clinical Oncology, Sir Y.K. Pao Centre for Cancer, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China
Abstract
Recent studies have revealed an unexplored population of long cell-free DNA (cfDNA) molecules in human plasma using long-read sequencing technologies. However, the biological properties of long cfDNA molecules (>500 bp) remain largely unknown. To this end, we have investigated the origins of long cfDNA molecules from different genomic elements. Analysis of plasma cfDNA using long-read sequencing reveals an uneven distribution of long molecules from across the genome. Long cfDNA molecules show overrepresentation in euchromatic regions of the genome, in sharp contrast to short DNA molecules. We observe a stronger relationship between the abundance of long molecules and mRNA gene expression levels, compared with short molecules (Pearson's r = 0.71 vs. −0.14). Moreover, long and short molecules show distinct fragmentation patterns surrounding CpG sites. Leveraging the cleavage preferences surrounding CpG sites, the combined cleavage ratios of long and short molecules can differentiate patients with hepatocellular carcinoma (HCC) from non-HCC subjects (AUC = 0.87). We also investigated knockout mice in which selected nuclease genes had been inactivated in comparison with wild-type mice. The proportion of long molecules originating from transcription start sites are lower in Dffb-deficient mice but higher in Dnase1l3-deficient mice compared with that of wild-type mice. This work thus provides new insights into the biological properties and potential clinical applications of long cfDNA molecules.
Fragmentation patterns of cell-free DNA (cfDNA) are shown to be linked to a myriad of biological characteristics, including nuclease activities (Serpas et al. 2019; Han et al. 2020), DNA methylation (Zhou et al. 2022), nucleosome structures (Ivanov et al. 2015; Snyder et al. 2016; Sun et al. 2019), mRNA expression levels (Ulz et al. 2016), and DNA-binding transcription factor activity (Ulz et al. 2019). These characteristics have spurred much research efforts in understanding the underlying biological mechanisms. For example, size analysis of cfDNA molecules reflects characteristic patterns of cfDNA fragmentation. Through short-read sequencing (Illumina), a typical plasma cfDNA fragment size distribution could be depicted with a dominant peak ∼166 bp, with 10-bp periodicities below 150 bp, suggesting a nucleosomal origin (Lo et al. 2010; Ivanov et al. 2015). Thus, cfDNA is generally believed to be a population of short DNA molecules. Moreover, cfDNA fragments from different tissues may bear information tracing to their tissues of origin. As examples, fetal cfDNA molecules in pregnant women and tumoral cfDNA molecules in patients with cancer are generally shorter than background hematopoietic cell–derived cfDNA (Chan et al. 2004; Lo et al. 2010; Jiang et al. 2015). These findings have catalyzed the use of size information of cfDNA for disease detection (Yu et al. 2014; Jiang et al. 2015; Mouliere et al. 2018).
Single-molecule sequencing technologies, including Pacific Biosciences (PacBio) single-molecule real-time (SMRT) sequencing and Oxford Nanopore Technologies (ONT) sequencing, have opened up the possibility of detecting and characterizing long cfDNA molecules. Recent studies using these platforms have uncovered a population of long cfDNA molecules, up to tens of kilobases, in the plasma DNA of healthy, pregnant individuals and patients with cancer (Yu et al. 2021, 2023a,b; Choy et al. 2022). Depending on the sequencing platforms used, long molecules account for about a median of 15% and 5% of the total molecules from SMRT and ONT platforms, respectively (Yu et al. 2023b). Hence, single-molecule, long-read sequencing technologies appear to be capable of sequencing a much wider spectrum of cfDNA molecules.
Studies have investigated the quantity, size distribution, methylation patterns, and end motifs of cfDNA molecules generated by long-read sequencing (Yu et al. 2021; Choy et al. 2022; Katsman et al. 2022; Lau et al. 2023). As single-molecule sequencing technologies are able to measure methylation patterns (Tse et al. 2021; Lau et al. 2023), one can use methylation patterns to explore the tissues of origin of long cfDNA molecules. Additional biological properties of long molecules, however, remain largely unexplored. Hence, in this study, we investigated whether long and short cfDNA molecules might originate from different genomic elements, including euchromatin and heterochromatin. We aggregated cfDNA data generated by SMRT or ONT sequencing to profile the genomic representations and studied the correlation between long cfDNA molecules and gene transcriptional activity. We examined end frequencies of long molecules derived from regulatory regions such as DNase I hypersensitive sites (DHSs) and CCCTC-binding factors (CTCFs). Moreover, we analyzed cleavage profiles surrounding cytosine–phosphate–guanine sites (CpGs) to investigate long cfDNA fragmentomics in the context of cancer detection. We generated additional cfDNA data from various nuclease-knockout mice using SMRT sequencing to gain biological insights regarding the role of nucleases in the generation of long cfDNA molecules.
Results
Long cfDNA molecules originate unevenly across the human genome
A preponderance of molecules with 5′ end-nucleotide of base adenine (A), namely A-end, has been observed in cfDNA molecules >500 bp (Yu et al. 2021). We therefore defined long cfDNA molecules as those of >500 bp. We investigated whether long cfDNA molecules originated evenly across the genome. By pooling sequenced data from previous studies (Yu et al. 2021, 2023b; Choy et al. 2022) using the SMRT and ONT platforms (Supplemental Table S1), respectively, we first compared the genomic representation profiles of long (>500-bp) and short (≤500-bp) molecules, across 100-kb nonoverlapping bins (Methods). Genomic origins of both long and short molecules displayed unevenness along the whole genome. We observed differential genomic representations between long and short molecules in some regions, with Chromosome 10 representation shown as an example (Fig. 1; Supplemental Fig. S1). The differential signals in genomic representation were found in both SMRT (Fig. 1A) and ONT (Fig. 1B) sequencing data, with platform-specific differences. Specifically, we observed that long molecules tended to be overrepresented in regions that overlapped with light bands on Giemsa-stained chromosomes and underrepresented in dark-banded regions shown in ideograms (Fig. 1). The light and dark bands typically correspond to GC-rich euchromatic and AT-rich heterochromatic regions, respectively (Bickmore and Sumner 1989; The BAC Resource Consortium et al. 2001). Thus, long molecules were preferentially derived from euchromatic regions compared with short molecules. This differential representation was particularly prominent on Chromosome 19 (Supplemental Fig. S2), which is well known for high GC-content and is considered as the most gene-rich chromosome (Grimwood et al. 2004).
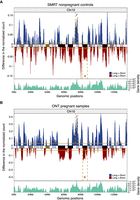
Distribution of long and short molecules from human plasma DNA. (A) Comparison of genomic representation on Chromosome 10 between long and short DNA molecules in 15 nonpregnant controls using SMRT sequencing. The overrepresentation and underrepresentation of long cfDNA molecules with respect to short molecules are indicated in blue and red, respectively. The genomic representation was determined based on 100-kb bins and was further smoothed by a 1-Mb moving average sliding window. The horizontal solid lines indicate normalized median differences between long and short molecules. The dashed rectangular boxes indicate one euchromatic (i) and one heterochromatic (ii) region. The track in between overrepresentation and underrepresentation of long molecules shows the chromosome ideogram. The ideogram band colors correspond to cytogenetic bands in UCSC Genome Browser. Darker bands are AT-rich, and lighter bands are GC-rich. Centromeric regions are indicated in dark green. The bottom track displays gene densities estimated by number of genes in 100-kb windows. (B) Comparison of genomic representation on Chromosome 10 between long and short DNA molecules in 31 pregnant samples using ONT sequencing.
As euchromatin generally tends to be gene-rich, we wondered if the gene density of a genomic region might positively correlate with the difference between long and short cfDNA molecules from that region. Indeed, the difference was significantly correlated to autosomal gene density (Pearson's r = 0.56; P < 2.2 × 10−16), and the gene-rich Chromosome 19 showed a correlation of 0.7 using nonpregnant controls from SMRT sequencing data. The correlation to Chromosome 19 gene density was 0.76 (P < 2.2 × 10−16) for pregnant samples using ONT sequencing data. We further examined the potential relationship between genomic representation of cfDNA molecules and presence of DNA double-strand breaks. Previous studies have found that transcribed genes are hotspots for endogenous double-strand breaks (Crosetto et al. 2013; Lensing et al. 2016; Ballarino et al. 2022). A weak positive correlation (Pearson's r = 0.43; P < 2.2 × 10−16) (Supplemental Fig. S3) between overrepresentation of long molecules and double-strand breaks detected from a lymphoblastoid cell line sample was observed (Methods).
Association between long molecules and gene expression in human samples
Motivated by the observation that long molecules appeared to be enriched in euchromatin, which is associated with active transcription, we assessed the potential relationship between the abundance of such molecules and transcriptional activity. First, we analyzed the abundance of cfDNA molecules originating from unexpressed and housekeeping genes (Methods). Long cfDNA molecules showed higher abundance over housekeeping genes and lower abundance over unexpressed genes in nonpregnant controls and pregnant subjects (Fig. 2A,B). We further examined the abundance of cfDNA molecules originating from gene bodies of differentially expressed genes. Autosomal protein-coding genes were ranked based on their median expression across tissues and grouped into five sets, from a lowly expressed EXP1 to a highly expressed EXP5 set. In line with the enrichment in housekeeping genes, the analysis of nonpregnant controls revealed a stepwise increase of long cfDNA molecule abundance as expression levels increased from EXP1 to EXP5 (P = 0.01374, Mann–Kendall trend test; Pearson's r = 0.78), whereas no such trend was found in short molecules (P = 0.4032, Mann–Kendall trend test; Pearson's r = −0.28) (Supplemental Fig. S4A). We confirmed the observation that long molecule abundance was positively associated with gene expression from SMRT in pregnancies (Supplemental Fig. S4B) and ONT sequencing data (Supplemental Fig. S4C,D). Analyzing the relationship at a higher resolution, we showed that the median abundances of long molecules were positively correlated (Pearson's r = 0.71; P < 2.2 × 10−16) with median transformed expression scores across tissues based on 15,556 expressed genes. In contrast, such positive correlation was not observed in short molecules (Pearson's r = 0.14; P = 0.003) (Fig. 2C). The conclusion was further confirmed by the ONT data (Fig. 2D). Moreover, investigating overall methylation levels revealed lower methylation in long molecules compared with short molecules (Fig. 2E,F).
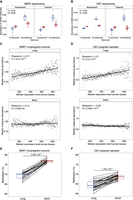
The abundance of long cfDNA molecules shows a positive correlation with transcriptional activity. (A,B) The abundance of long and short molecules on gene bodies of unexpressed and housekeeping genes for nonpregnant controls and pregnant subjects. (A) Data from SMRT sequencing. (B) Data from ONT sequencing. (C,D) The correlation between gene expression and molecule abundance. The median expression level of a gene across tissues was log-transformed and scaled. The median molecule abundance was derived from scaled expression levels. P-values by Pearson's correlation test. (C) Pooled data of 15 nonpregnant controls from SMRT sequencing. (D) Pooled data of 31 pregnant samples from ONT sequencing. (E,F) Comparisons of DNA methylation between long and short molecules. Data points from one sample are connected with a black line. P-values by Wilcoxon rank-sum test. (E) Data from SMRT sequencing. (F) Data from ONT sequencing.
As the abundance of long cfDNA molecules in human plasma showed a relationship with gene expression, we explored whether such a signature could be used to distinguish patients with hepatocellular carcinoma (HCC) from subjects without HCC. Thus, we collected SMRT sequencing data using plasma samples from 20 healthy individuals, 19 hepatitis B virus (HBV) carriers, and 48 patients with HCC (Supplemental Table S2). Median values of 1,021,412, 286,492, and 712,223 high-quality circular consensus sequencing (CCS) reads were obtained for healthy individuals, HBV carriers, and patients with HCC, respectively. Long cfDNA molecules accounted for a median of 21.3%, 19.7%, and 23.7% of total molecules in healthy subjects, HBV carriers, and patients with HCC, respectively. As the number of molecules was relatively low, we attempted to examine a group of genes. The top 5000 expressed genes in HCC tumors from The Cancer Genome Atlas data set (Methods) were identified. We compared the abundance of long and short molecules over these HCC-associated genes. Long molecules showed significantly higher (P = 5.814 × 10−5, Kruskal–Wallis test) abundance in patients with HCC compared with non-HCC groups, whereas short molecules did not show significant differences (P = 0.06504, Kruskal–Wallis test) in this data set (Fig. 3A). A receiver operating characteristic (ROC) curve based on the abundance of long molecules yielded an area under the curve (AUC) of 0.76 to distinguish 39 non-HCC individuals and 48 patients with HCC. When the number of total molecules increased to 1 million for each sample, the AUC improved to 0.9 to distinguish 12 non-HCC individuals and 18 patients with HCC (Fig. 3B).
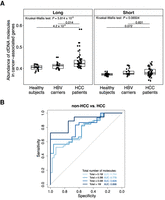
The abundance of long cfDNA molecules for HCC detection. (A) Comparison of the abundance of SMRT sequencing molecules among healthy individuals, HBV carriers, and patients with HCC. The abundance of long and short molecules was measured using the top 5000 expressed genes in HCC tumor tissues. The Kruskal–Wallis test P-value for differences among groups. Post hoc pairwise Wilcoxon rank-sum test P-values with Benjamini–Hochberg adjustment are shown above horizontal lines. (B) ROCs of long molecule abundance measured in A for distinguishing individuals without HCC, including healthy subjects and HBV carriers, and with HCC. Multiple thresholds, including 0.1 million (total > 0.1M), 0.3 million (total > 0.3M), 0.5 million (total > 0.5M), and 1 million (total > 1M) molecules from a sample, were used to include samples for constructing ROCs.
In addition to the abundance of molecules, we assessed molecule ends in specific regions. The end frequencies of cfDNA at transcription start sites (TSSs) and regulatory regions that include DHSs and CTCF binding sites (Methods) were examined. As the expression level rose, the normalized end frequencies of long molecules gradually increased in the vicinity of TSSs (e.g., 50 bp upstream of and downstream from TSSs), rising from 1.01-fold in EXP1 to 1.7-fold in EXP5. Conversely, for short molecules, the normalized end frequencies had a decreasing tendency (Fig. 4A; Supplemental Fig. S4E). Long molecules pooled from the nonpregnant controls displayed higher end frequencies near the peak of tissue-invariant DHSs (Fig. 4B). We observed that the end frequencies of long cfDNA molecules were two times that of short molecules in DHSs (Methods). Analyzing end frequencies near tissue-specific DHSs revealed effects of tissue types on long cfDNA fragmentations. Specifically, at liver-specific and myeloid-specific DHSs, long cfDNA in nonpregnant controls tended to have stronger end preferences (1.4-fold) compared with those of short cfDNA (Supplemental Fig. S4F). As a confirmation, the same pattern around tissue-invariant DHSs was observed in plasma of pregnant women from ONT sequencing (Supplemental Fig. S4G). At CTCF binding sites, long molecules showed higher cfDNA end frequencies as well, in both SMRT and ONT sequencing data (Fig. 4C; Supplemental Fig. S4H). These results have further highlighted that long molecules were preferentially derived from open chromatin regions and that their abundance was correlated with transcriptional activities.
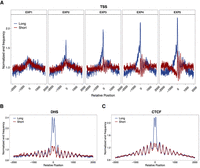
Normalized end frequencies of SMRT sequencing data. (A) Normalized end frequencies of SMRT long and short molecules pooled from plasma of healthy individuals at TSSs of the expression-stratified gene groups EXP1 to EXP5, corresponding to low to high expression. Transcription start positions are denoted as position 0. All transcription start sites were strand-adjusted so that positive positions are in the direction of transcription. (B,C) Normalized end frequencies of SMRT long and short molecules pooled from plasma of healthy individuals at DHSs (B) and CTCF binding sites (C). DHSs or CTCF binding site peaks are denoted as position 0; downstream and upstream 2000 bp is shown.
Differential cleavage profiles between long and short molecules surrounding CpGs
Studies have shown that the cfDNA cleavage preferences occurred at methylated CpG sites (Han et al. 2021; Zhou et al. 2022). Beyond the fragmentation around regulatory regions, we wondered whether fragmentation of long molecules at CpGs would be different from those of short molecules. We analyzed the cleavage patterns at commonly methylated and unmethylated CpGs in pooled healthy data (Methods). The cleavage of short molecules recapitulated earlier findings that nuclease-mediated preferential cleavage occurs at methylated CpGs in healthy subjects analyzed by Illumina sequencing (Zhou et al. 2022). Specifically, the cfDNA cleavage was relatively enriched at the cytosine of methylated CpGs (position 0), followed by a rapid decrease at the nucleotide 1 position immediately preceding methylated CpGs. We found that the cleavage of long molecules did not show such a pattern at methylated cytosines but showed the preferred cutting at positions several nucleotides away from the methylated CpGs (e.g., positions −4, −2, and 4) (Supplemental Fig. S5A).
To explore whether the cleavage profile around CpGs for long molecules would be associated with diseases, we evaluated cleavage patterns using the HCC cohort data mentioned above. Considering the relatively low number of molecules, we evaluated cleavage patterns related to all autosomal CpGs. As the human genome is highly methylated, with 70%–80% of CpGs being methylated (Ziller et al. 2013), the cleavage profiles of short molecules showed a cutting preference at the position 0 cytosine (Fig. 5A). Because of the low number of sequenced molecules, long molecules showed higher variability in cleavage profiles. The cleavages of long molecules, however, consistently showed significant higher proportions (P = 2.6 × 10−6, paired Wilcoxon test, P-values adjusted by Benjamini–Hochberg method) than the adjacent downstream nucleotide at positions −4, −2, 1, and 4 in healthy subjects and HBV carriers (Fig. 5B; Supplemental Fig. S5B). The ratio of 5′ CGN to NCG end motifs (CGN/NCG motif ratio) has been shown to inform methylation level and can be used to distinguish patients with HCC from non-HCC individuals using Illumina sequencing (Zhou et al. 2022). In this SMRT sequencing data set, patients with HCC showed overall lower CGN/NCG motif ratios (P = 2.081 × 10−7, Kruskal–Wallis test) for short molecules. For long molecules, however, CGN/NCG motif ratios of patients with HCC showed no significant difference (P = 0.05137, Kruskal–Wallis test) among the three groups (Fig. 5C).
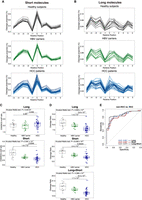
Cleavage profiles of long and short molecules surrounding CpGs. (A,B) Cleavage profiles surrounding all autosomal CpGs for short (A) and long (B) cfDNA molecules. Each line represents one sample. A cleavage window of 11 bases is shown. Positions 0 and 1 indicate cytosine and guanine, respectively. (C) Box plot of CGN/NCG motif ratios for long and short molecules. (D) Box plot of cleavage ratios between aggregating positions −4, −2, 1, and 4 and position −1. (E) AUCs for distinguishing patients with HCC from non-HCC subjects using the cleavage ratios in D. P-values of differences among groups by Kruskal–Wallis tests. Post hoc pairwise P-values by Wilcoxon rank-sum tests with Benjamini–Hochberg adjustment shown above horizontal lines (C,D).
We further attempted to use the ratio between the molecules ending at positions −4, −2, 1, and 4 and those ending at position −1. Using this cleavage ratio, both long and short molecules displayed significant differences (P = 3.893 × 10−8 and P = 8.036 × 10−8, Kruskal–Wallis test) between patients with HCC and non-HCC subjects (Fig. 5D). As a result, long molecules alone achieved an AUC of 0.85 to distinguish 39 non-HCC individuals and 48 patients with HCC. Combining the ratio of long and short molecules, the AUC increased to 0.87 (Fig. 5E).
Investigation of long molecule generation using knockout mice
The amount of cfDNA molecules with 5′ ending in an A or G was reported to increase as the size of cfDNA molecules increased (Yu et al. 2021), and the nuclease DNA fragmentation factor subunit beta (DFFB) showed a preference to cut 5′ to an A or G nucleotide (Han et al. 2020). We hypothesized that long molecules were predominantly generated by DFFB. To gain potential mechanistic insights on generating long cfDNA molecules in human plasma, we studied mouse models in which different nuclease genes had been knocked out. Mice of the C57BL/6N background were used. We sequenced four wild-type (WT), five Dffb−/−, five Dnase1−/−, five Dnase1l3−/−, and five Dnase1−/− and Dnase1l3−/− double-knockout mice samples, with each sample pooled from three to five mice subjects, using SMRT sequencing (Supplemental Table S3). We first analyzed the size profile of plasma cfDNA from these mice by pooling sequenced data of the same genotype. Compared with WT mice, the frequencies of long cfDNA molecules >1 kb in Dffb−/− mice were consistently lower, whereas the corresponding frequencies in Dnase1−/− mice were higher, and such an increasing pattern was further enhanced in Dnase1l3−/− mice and mice with double deletion of Dnase1 and Dnase1l3 (Dnase1−/− & Dnase1l3−/−) (Fig. 6A). For instance, the percentages of cfDNA molecules with a size of >500 bp were 21.7%, 16.8%, 29.3%, 37.2%, and 45% in WT, Dffb−/−, Dnase1−/−, Dnase1l3−/−, and Dnase1−/− & Dnase1l3−/− mice, respectively. The percentages of long molecules were significantly different (P = 0.002053, Kruskal–Wallis test) between different types of mice, with Dnase1l3−/− and Dnase1−/− & Dnase1l3−/− mice higher than WT mice (Supplemental Fig. S6A). Meanwhile, the frequencies of short molecules with a size <250 bp decreased in Dnase1l3−/− and Dnase1−/− & Dnase1l3−/− mice, with a relatively more pronounced reduction in mononucleosomal size (Fig. 6B). The data suggested that DFFB was at least in part responsible for the generation of long cfDNA molecules. The removal of DNASE1L3 and DNASE1 would lead to an enhanced effect in generating long cfDNA molecules, as DFFB might, in the absence of DNASE1L3 and DNASE1, act as a dominant nuclease in contributing cfDNA into circulation. The deletion of Dffb resulted in the reduction of long molecules originating from regions around TSSs, which normally represent open chromatin states (Fig. 6C), further providing evidence that DFFB might be responsible for the patterns related to long cfDNA molecules. The increase of long cfDNA molecules in regions nearby TSSs was elevated in mice in which DFFB functioned prominently by knocking out Dnase1 and Dnase1l3. These patterns could be reproduced in open chromatin regions comprising DHSs and CTCF binding sites (Supplemental Fig. S6B,C). Moreover, the highly expressed genes tended to harbor more long molecules in Dffb-competent mice. However, such enrichment of long molecules was absent in Dffb-deficient mice (Fig. 6D).
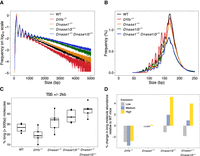
Nuclease-mediated fragmentation in knockout mice. (A) Pooled molecule size distributions of wild-type and nuclease-deficient mice from SMRT sequencing. Visualization of size in the range of 0 to 5000 bp and log10-transformed frequencies were used. (B) Zoom-in plot of A showing size in the range of 0 to 250 bp on the linear scale. (C) Boxplot showing the proportions of long molecules originated from 2000 bp upstream of and downstream from transcription start sites. (D) Percentage of changes in pooled long molecules (>500 bp) abundance relative to wild-type mice on low, medium, and high expression gene groups.
Discussion
This study explores previously unknown properties of long cfDNA molecules in human plasma. First, long cfDNA molecules have been found to be preferentially originated from transcriptionally active regions of the genome, with overall lower methylation and the end frequencies of long cfDNA molecules being more enriched in transcriptional start sites and open chromatin regions. Second, long molecules show a distinctive cleavage profile surrounding CpG sites. The cleavage ratio that makes use of multiple positions surrounding CpG sites can be used to distinguish non-HCC individuals from patients with HCC. In contrast to the previous report that the generation of short cfDNA molecules might be largely attributed to DNASE1L3 (Serpas et al. 2019; Chan et al. 2020), we show that the characteristic distribution of long cfDNA molecules in the genome might at least in part be attributed to the DFFB enzyme activity using genetically modified mice. This observation suggests different nucleases are involved in the fragmentation of long and short cfDNA molecules in human plasma.
By using knockout mice, a plasma DNA fragmentation model based on the end motifs of cfDNA molecules has been proposed, in which DFFB, DNASE1L3, and possibly other nucleases first digest DNA intracellularly, and then extracellular DNASE1L3 and DNASE1 further degrade the plasma DNA (Han et al. 2020). DNASE1L3, DNASE1, and DFFB preferentially cut at C-end, T-end, and A-end nucleotides of cfDNA fragments, respectively (Han et al. 2020). The long molecules uncovered via single-molecule sequencing platforms are likely to be, at least in part, the end product of DFFBs other than DNASE1L3, which is supported by the observation of enrichment of A-end fragments in long cfDNA molecules (Yu et al. 2021). Of note, following knocking out of the Dnase1l3 and/or Dnase1 genes in mice, DFFB appears to take on the leading role in cfDNA fragmentation. Additionally, no clear correlation was found between the total cfDNA concentration and the proportion of long molecules. The higher proportions of long molecules originated from accessible chromatin regions in Dnase1l3−/− and Dnase1−/− & Dnase1l3−/− double-knockout mice were reminiscent of the characteristic of long molecules in human plasma. The long cfDNA molecules in human plasma are therefore likely to be the digestion products of DFFB, which have escaped further or secondary digestion by DNASE1L3. The exact biological mechanisms for the escape of cleavage for long cfDNA remain elusive. Dnase1l3−/− mice have been shown to develop features of systemic lupus erythematosus on both the 129 and B6 genetic backgrounds (Sisirak et al. 2016). DNASE1L3 deficiencies in humans are associated with systemic lupus erythematosus (Al-Mayouf et al. 2011). Long cfDNA that are generated by DFFB might have potential immunomodulatory effects to the innate immune responses.
Prior studies systematically profiled plasma DNA end motifs using mice of different genotypes and found altered end motif frequencies in nuclease-deficient mice (Serpas et al. 2019; Chan et al. 2020). By linking the end motifs and nuclease-deficient genotypes, a nonnegative matrix factorization algorithm-based approach was developed to deconvolute contributions of nucleases. The work has shown that inferring nuclease activities in liquid biopsy is feasible and that DNASE1L3 is a leading contributor to fragmentation in the plasma cfDNA (Zhou et al. 2023). One reason for previous studies focusing on the DNASE1L3 activity is largely because that the Illumina sequencing platform is designed for short-read sequencing, with an upper limit of sequenceable fragments being at ∼600 bp. Long cfDNA molecules investigated in this study are more likely to reflect the DFFB activity. The genome-wide representational analysis of long cfDNA molecules provides another means to dissect the DFFB activity and link the enzyme activity to gene transcriptional activity. Further investigations to elucidate cfDNA fragmentation are warranted. A more comprehensive picture of various nuclease activities could be uncovered when examining the full spectrum of cfDNA molecules in different nuclease-knockout mouse models.
We have uncovered genomic representation difference between long and short cfDNA molecules in human plasma. The enrichment of long molecules in transcriptionally active regions may have important implications. It has been suggested that DFFB first cleaves at nuclear scaffold attachment sites (Nagata et al. 2003) to unfold the chromatin. DFFB induces DNA breaks as a signal for cell fate (Larsen and Sørensen 2017). Although direct evidence of locations of DFFB-induced DNA breaks is lacking, we observed a weak positive correlation between overrepresentation of long cfDNA molecules and DNA double-strand breaks detected in a lymphoblastoid cell line sample. Future examinations at higher resolutions to address the relationship between transcription-associated DNA double-strand breaks and long DNA generation will be needed. Whether DFFB and other nucleases preferentially cleave at those damaged DNA breakpoints and thereafter induce downstream DNA fragmentation remains to be investigated. Additionally, the nuclear morphology at the time of cell death might be another dimension that affects the fragmentation and hence results in overrepresentation of long molecules in euchromatin (Toné et al. 2007). Beyond the mechanism, the enrichment of long molecules in genic regions highlights potential diagnostic value. Retaining the characteristics of chromatin structure, nonrandom fragmentation of cfDNA reflects the transcriptomic and epigenomic status of dying cells (Ivanov et al. 2015; Snyder et al. 2016; Ulz et al. 2016; Lo et al. 2021). A number of studies showed that coverage profiles near TSSs correlate with gene expression and that accessibilities near the transcription factor binding sites reflect the transcription factor activity (Ulz et al. 2016, 2019). Notably, the normalized end frequencies at TSSs in long cfDNA molecules were higher than those of short cfDNA molecules, suggesting a potential higher utility for long cfDNA molecules for inferring gene expression compared with their shorter cfDNA counterparts. Moreover, the abundance of long molecules showed discriminating power in cancer detection.
Compared with sequencing by synthesis, the current throughput of single-molecule sequencing is relatively low, which presents challenges to analyses that require a larger number of molecules. The low number of long cfDNA molecules sequenced creates obstacles for analyzing the association with gene expression at a higher resolution, especially in the context of measuring expression of a single gene. Nevertheless, with technological advances, such as with the recent launch of a higher-throughput version of the SMRT-based sequencer, as well as further optimized protocols, the throughput is likely to improve, potentially facilitating the elucidation of further biological insights and the development of clinical tools. Alternatively, enriching the long molecules of interest may help to adequately harness the advantages for diagnostic purposes. The performance of cancer detection using long molecule abundance and fragmentation patterns can be further enhanced. In addition, a larger cohort that allows validation of these biomarkers is desired. Platform-specific differences were observed from SMRT and ONT sequencing data. Future investigations into the use of a specific platform to maximize the use of long cfDNA are warranted.
In summary, this work expands the current knowledge of fragmentation properties of long cfDNA molecules. The characteristic distribution of long cfDNA molecules along the human genome is correlated with transcriptional activities and disease states, potentially opening up possibilities for biomarker discovery and new clinical applications.
Methods
Human sample collection
The study was approved by the joint Chinese University of Hong Kong (CUHK)–Hospital Authority New Territories East Cluster clinical research ethics committee. PacBio SMRT sequencing data of plasma cfDNA, including healthy individuals (N = 15), pregnancies of different trimesters (N = 28), HBV carriers (N = 13), and patients with HCC (N = 13), were obtained from previous studies (Yu et al. 2021; Choy et al. 2022). Additionally, we collected blood samples from healthy individuals (N = 5), HBV carriers (N = 6), and patients with HCC (N = 35) from the Prince of Wales Hospital, Hong Kong, with written informed consent. Nanopore sequencing data of plasma cfDNA from pregnancies of different trimesters (N = 31) were obtained from a prior study (Yu et al. 2023b). Additionally, plasma cfDNA from nonpregnant healthy individuals (N = 5) was collected and subjected to ONT sequencing. The blood samples were processed by a double centrifugation protocol (1600g for 10 min at 4°C followed by further centrifugation of the plasma at 16,000g for 10 min at 4°C) as previously described (Chiu et al. 2001).
Murine models
Mice with a CRISPR-Cas9-targeted deletion of exon 5 in Dnase1l3 of the C57BL/6N background were generated by The Jackson Laboratory. Mice carrying a targeted allele mutation of Dnase1 (Dnase1tm1.1(KOMP)Vlcg) and mice carrying a targeted allele of Dffb (DffbC57BL/6N-Dffbem1Wtsi), both on the B6 background, were obtained from the Knockout Mouse Project Repository of the University of California, Davis. These mice were obtained under a third-party distribution agreement which stipulated that the animals were nontransferable. Dnase1l3−/− mice were cross-bred with Dnase1−/− mice to produce Dnase1−/− & Dnase1l3−/− mice in the laboratory animal services center of CUHK. Mice including WT B6 were maintained in the same facility. All experimental procedures were approved by the animal experimentation ethics committee of CUHK and performed in compliance with the guide for the care and use of laboratory animals (eight edition) established by the National Institutes of Health.
Murine sample collection
Mice were sacrificed and exsanguinated by cardiac puncture. Blood was transferred into EDTA-containing collection tubes. The blood samples were processed by a double centrifugation protocol (1600g for 10 min at 4°C followed by further centrifugation of the plasma at 16,000g for 10 min at 4°C) as previously described (Chiu et al. 2001). The resulting plasma was collected.
Library preparation and sequencing
Plasma DNA was extracted from 1.2–4 mL of human plasma and 1.1–1.4 mL of pooled mice plasma with the QIAamp circulating nucleic acid kit (Qiagen) according to the manufacturer's instructions. The SMRTbell express template prep kit 2.0 (PacBio) was used for the library preparation of plasma DNA. Briefly, DNA molecules were ligated with hairpin adaptors to form a circularized template. Sequencing primer v4 was annealed to the sequencing template, followed by binding of polymerase to templates using a Sequel II binding kit 2.1 and internal control kit 1.0 (PacBio). SMRT cell 8M was used for sequencing with a Sequel II sequencing 2.0 kit (PacBio), and sequencing movies were collected for 30 h. Nanopore library preparation and sequencing of pregnant samples are as described previously (Yu et al. 2023b). For healthy samples, nanopore libraries were prepared using the native barcoding kit (ONT SQK-NBD114.96) according to the manufacturer's instructions except that a bead-to-sample ratio of three was used in all clean-up steps using AMPure XP beads with prolonged incubation time for DNA repair, end-prep, barcode, and adapter ligation. Short fragment buffer, which retained DNA fragments of all sizes, was used in adapter ligation. Each library was loaded onto a PromethION flow cell R10.4.1 and sequenced on a PromethION device for 72 h.
Sequence alignment
CCS reads that were generated from at least three PacBio subreads were kept. CCS reads from human and mice data were aligned to hg19 and mm10, respectively, using blasr (Chaisson and Tesler 2012). Nanopore-generated sequences were aligned to hg19 with minimap2 (Li 2018) as described previously (Yu et al. 2023b). The size distributions of the raw sequenced molecules before and after alignment were examined. SMRT sequencing data showed highly overlapped size distributions, whereas ONT sequencing data showed deviations comparing before and after alignment (Supplemental Fig. S7). Chimeric reads were filtered out before downstream analyses. For human sequence data, reanalysis of the data using the GRCh38 human reference genome would not affect the results significantly, as the major difference between these two versions of the human reference genome is the sequence representation for centromeres and highly repetitive regions. Similarly, for mouse sequence data, reanalysis of the data using the GRCm39 mouse reference genome would not affect the results significantly because only uniquely aligned reads were included in downstream analyses.
Genomic representation analysis
The human genome was partitioned into nonoverlapping 100-kb bins. Sequences that mapped to blacklist regions (Amemiya et al. 2019) were removed. Long and short molecules were counted in each bin. The bin counts were smoothed by a 1-Mb moving average window and normalized by the median bin count of autosomes. The cytoband information for chromosome ideograms was obtained from UCSC Genome Browser (http://genome.ucsc.edu/). Gene densities were estimated by number of genes in a 100-kb window. The correlations between normalized bin counts and gene densities were calculated using Pearson's method, and smoothed data applying 1-Mb windows were used.
To examine the correlation between the genomic representation of cfDNA molecules and the presence of DNA double-strand breaks, we obtained double-strand breaks detected from a lymphoblastoid cell line sample (Bouwman et al. 2020). DNA double-strand breaks were counted in each 100-kb bin and normalized by the median bin count.
To calculate the proportions of long molecules originated from TSSs, DHSs, and CTCF binding sites in mice, we counted long and short molecules overlapping with these regions; 2000 bp upstream of and downstream from TSSs or peak points of DHSs and CTCF binding sites was regarded as regions of interest. At least 50% size of a molecule needs to be overlapped with these regions of interest to be counted. The proportions of long molecules were computed as the number of long molecules divided by the total number of molecules overlapping with the regions of interest.
Molecule abundance analysis
To quantify molecule abundance over genes, we adopted similar quantification of transcripts using reads per kilobase per million mapped reads. We counted the molecules overlapped with gene bodies and normalized by the total number of molecules and the total length of the genes. For both long and short molecules, we required at least 50% of the size of a molecule to be overlapped with a gene to be counted. To calculate the Pearson's correlation between molecule abundance and gene expression, we used the median molecule abundance of samples and correlated with the median gene expression of each gene set. The Mann–Kendall trend test was performed using the median molecule abundance of samples.
HCC-associated genes were identified using curated HCC expression data from The Cancer Genome Atlas (The Cancer Genome Atlas Research Network et al. 2013). Expression values were averaged across 356 HCC tumor tissue samples and ranked by the averaged expression. The top 5000 expressed genes were selected and used as HCC-associated genes. The abundance of long and short molecules over these 5000 genes was calculated. AUCs were calculated based on the long molecule index.
Single-cell gene expression data were downloaded from The Human Protein Atlas (https://www.proteinatlas.org/) and The Tabula Muris project (The Tabula Muris Consortium 2018) for human and mouse, respectively. In human analysis, housekeeping genes were retrieved from a previous study (Eisenberg and Levanon 2013), and 3510 autosomal housekeeping genes were used. Two thousand one hundred fifty-four genes with a mean of less than one normalized transcript per million across 54 tissues were defined as unexpressed genes. Autosomal protein-coding genes were stratified by median expression across 79 cell types. Gene groups of EXP1–EXP5 correspond to genes with median normalized transcripts per million in the range of (0, 0.001), (0.001, 1), (1, 8), (8, 30), and (30, 11,800), with each containing 4836, 3163, 4187, 4816, and 3088 genes, respectively. To correlate the abundance of molecules with gene expression at relatively high resolution in human samples, the median expression levels of a gene across cell types were log-transformed and scaled to a score ranging from zero to 1000. The median molecule abundance of genes at each scaled expression level was used. Genes with expression scores of lower and upper quantiles (outliers) were discarded from the correlation analysis.
Murine genes were stratified by median expression across 20 tissue types. The low, medium, and high expression gene groups in mice correspond to autosomal protein-coding genes with a median normalized transcript count in the range of (0, 50), (50,180), and (180,450), with each containing 10,987, 3883, and 5195 genes, respectively. The percentage of change in long molecule abundance between the knockout and WT mice was computed using the difference of long molecule abundance between knockout and WT mice, and dividing by the abundance of long molecules in the WT mice.
End frequency analysis
End frequency analysis was performed surrounding the defined region of interests. The regions of interest, including CpGs, DHSs, CTCF binding sites, and TSSs, was extracted as follows.
TSSs were retrieved using NCBI RefSeq data (https://www.ncbi.nlm.nih.gov/refseq/). DHSs and CTCF binding sites were identified from publicly available data. Tissue-invariant DHSs from the study (Meuleman et al. 2020) were identified, and genomic coordinates were converted from hg38 to hg19 using the UCSC liftOver tool. In the analysis, 44,997 tissue-invariant DHSs were used; 157,556 CTCF binding sites from the Encyclopedia of DNA Elements (ENCODE) uniformly processed transcription factor binding site clusters in human were used (Dunham et al. 2012); and 107,227 DHSs (ENCFF855RCO) and 62,461 CTCF binding sites (ENCFF883UPM) from the CH12.LX mouse cell line available via ENCODE were used in the mice analysis.
The peak or midpoint of the region of interests was regarded as position 0. The end frequencies within +/−2000 bp were aggregated and normalized with median count of the region. For regulatory regions (DHS and CTCF) and TSSs, we applied a 10-bp and 20-bp window to smooth the count. The fold change of end frequencies around DHSs was measured as follows. End frequencies within a DHS region (a median size of 211 bp) were aggregated. In parallel, end frequencies of the equivalent region that were 3000 bp away upstream and downstream were aggregated and used as a normalization factor. The fold change of end frequencies around TSSs was measured using a ratio between long and short in human. Normalized end frequencies from position −50 to 50 were aggregated to compute the ratio.
Cleavage profiles surrounding CpGs
For human data analysis, commonly methylated and unmethylated CpG sites were defined as methylation levels >70% and <30%, respectively, in human buffy coat, liver, and placental tissues. Whole-genome bisulfite sequencing subjected to the Illumina platform from the three tissues available from a previous study (Sun et al. 2015) was used to identify 2,422,965 commonly methylated and 558,267 commonly unmethylated CpGs. For analysis of all CpGs, 26,752,699 autosomal CpGs from the human genome hg19 were obtained. The cleavage proportion to measure the cutting frequency was measured as the number of molecule ends at a particular site divided by sequencing depth at the site, as described previously (Zhou et al. 2022). The CGN/NCG motif ratio was calculated using the number of molecule ends at position 0 divided by the number of molecule ends at position −1. The cleavage ratio was calculated using the aggregated number of molecule ends at position −4, −2, 1, and 4 divided by the number of molecule ends at position −1.
Statistical analysis
The Mann–Kendall trend test was used for testing the monotonic trend in molecule abundance over the gene expression groups. The alternative hypothesis that a monotonic increasing trend is present was assumed. The Pearson's correlation test was used to measure relations between the abundance of molecules and gene expression. The Wilcoxon rank-sum test was used to compare two groups at a significance level of 0.05. The Kruskal–Wallis test, or Friedman test in case of dependent groups, was used to compare three or more groups at a significance level of 0.05. Post hoc pairwise Wilcoxon tests were performed with Benjamini–Hochberg adjustment to yield pairwise P-values.
Data access
The sequencing data generated in this study have been submitted to the European Genome-phenome Archive (EGA; https://web2.ega-archive.org/) under accession number EGAS00001005515.
Competing interest statement
K.C.A.C. and Y.M.D.L. hold equities in DRA, Take2, Grail/Illumina, and Insighta. P.J. and W.K.J.L. hold equities in Grail/Illumina. P.J. is a consultant to KingMed Future. W.P. is a consultant to Take2. K.C.A.C. and P.J. are directors of DRA, Take2, KingMed Future, and Insighta. W.K.J.L. is a director of DRA. S.C.Y.Y. received financial support from Oxford Nanopore for attending meetings. H.C., P.J., K.C.A.C., and Y.M.D.L. filed a U.S. patent application (no. 63/544,014) entitled “Genomic origin, fragmentomics, and transcriptional correlation of long cell-free DNA” on October 13, 2023, based on the data in this study. Patent royalties are received from Grail, Illumina, DRA, Take2, and Xcelom.
Acknowledgments
The work was supported by the Innovation and Technology Commission (InnoHK Initiative). Y.M.D.L. received an endowed chair from the Li Ka Shing Foundation. We thank Angel Lai, Chris Kum, Saravanan Ramakrishnan, Xingfu Qin, and Danny Wong for their technical assistance.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278556.123.
-
Freely available online through the Genome Research Open Access option.
- Received September 26, 2023.
- Accepted February 14, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








