
Characterization of DNA methylation reader proteins in Arabidopsis thaliana
- Jonathan Cahn1,6,
- James P.B. Lloyd1,2,6,
- Ino D. Karemaker3,
- Pascal W.T.C. Jansen3,
- Jahnvi Pflueger1,4,
- Owen Duncan1,
- Jakob Petereit1,
- Ozren Bogdanovic1,
- A. Harvey Millar1,2,
- Michiel Vermeulen3,5 and
- Ryan Lister1,2,4
- 1ARC Centre of Excellence in Plant Energy Biology, School of Molecular Sciences, The University of Western Australia, Crawley, Western Australia 6009, Australia;
- 2ARC Centre of Excellence in Plants for Space, School of Molecular Sciences, The University of Western Australia, Crawley, Western Australia 6009, Australia;
- 3Radboud Institute for Molecular Life Sciences, Radboud University Nijmegen, Nijmegen 6525 GA, The Netherlands;
- 4Harry Perkins Institute of Medical Research, Nedlands, Western Australia 6009, Australia;
- 5Division of Molecular Genetics, The Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands
-
↵6 These authors contributed equally to this work.
Abstract
In plants, cytosine DNA methylation (mC) is largely associated with transcriptional repression of transposable elements, but it can also be found in the body of expressed genes, referred to as gene body methylation (gbM). gbM is correlated with ubiquitously expressed genes; however, its function, or absence thereof, is highly debated. The different outputs that mC can have raise questions as to how it is interpreted—or read—differently in these sequence and genomic contexts. To screen for potential mC-binding proteins, we performed an unbiased DNA affinity pull-down assay combined with quantitative mass spectrometry using methylated DNA probes for each DNA sequence context. All mC readers known to date preferentially bind to the methylated probes, along with a range of new mC-binding protein candidates. Functional characterization of these mC readers, focused on the MBD and SUVH families, was undertaken by ChIP-seq mapping of genome-wide binding sites, their protein interactors, and the impact of high-order mutations on transcriptomic and epigenomic profiles. Together, these results highlight specific context preferences for these proteins, and in particular the ability of MBD2 to bind predominantly to gbM. This comprehensive analysis of Arabidopsis mC readers emphasizes the complexity and interconnectivity between DNA methylation and chromatin remodeling processes in plants.
Epigenomic modifications constitute additional regulatory layers that can alter and/or record transcriptional activity of their underlying genetic sequences (Bird 2007). One such modification is cytosine DNA methylation (mC), the covalent bond of a methyl group (–CH3) to the fifth carbon of cytosine bases. In plants, mC can occur in three DNA sequence contexts: CG, CHG, and CHH (H = A, C, or T). Methylation in each sequence context shows distinct distribution patterns along the genome depending on underlying sequence features and elements (Lloyd and Lister 2022), and is regulated by different enzymes, with DNA methyltransferases responsible for mC deposition (writers), and DNA glycosylases catalyzing DNA demethylation (erasers). Investigation of DNA methyltransferases in Arabidopsis thaliana has revealed sequence context-specific activity for these enzymes: MET1 is responsible for maintaining mCG, being recruited to hemimethylated sites after replication (Finnegan et al. 1996; Ronemus et al. 1996; Shook and Richards 2014); CMT3 and CMT2 maintain mCHG and mCHH, respectively, via positive feedback loops with the histone post-translational modification H3K9me2 (Lindroth et al. 2001; Jackson et al. 2002; Du et al. 2012; Zemach et al. 2013; Stroud et al. 2014); and DRM2 is required for de novo DNA methylation in all three contexts through the RNA-directed DNA methylation (RdDM) pathway (Cao and Jacobsen 2002; Matzke and Mosher 2014; Lloyd and Lister 2022). DNA demethylases appear to exhibit less sequence context specificity, where DME is involved in all demethylation events in the companion cells of the plant gametes but primarily in the CG context, and ROS1, DML2, and DML3 are partially redundant in vegetative tissues for all mC contexts (Gong et al. 2002; Gehring et al. 2006; Penterman et al. 2007; Hsieh et al. 2009; Ibarra et al. 2012).
In Arabidopsis, heterochromatin is marked by high DNA methylation levels in all three contexts and is associated with transcriptional silencing of transposable elements (TEs) (Cokus et al. 2008; Lister et al. 2008; Lloyd and Lister 2022). In addition to an important role in silencing TEs in plants, notably in germline cells, mC is also involved in other aspects of plant development, such as imprinting by regulating MEDEA, a histone methyltransferase of the polycomb repressive complex 2 (PRC2) (Satyaki and Gehring 2017), in recombination patterns during meiosis, where mCHG limits heterochromatin rearrangements (Underwood et al. 2018), or in controlling alternative splicing in male sex cells of meiosis factor MPS1 (AT5G57880) (Walker et al. 2018). Many genes (10%–20%) (Zhang et al. 2020) in euchromatic arms also harbor DNA methylation but only in the CG context, located in the gene body with a modest bias toward the 3′ end, called gene body methylation (gbM) (Zhang et al. 2006; Cokus et al. 2008; Lister et al. 2008). In contrast with heterochromatic mC, gbM is associated with ubiquitously and moderately expressed genes in plants (Zhang et al. 2006; Coleman-Derr and Zilberman 2012; Takuno and Gaut 2012). Despite being present in almost all land plants, whether gbM has a molecular function has been highly debated since some plant species do not harbor gbM (Bewick et al. 2016; Niederhuth et al. 2016; Zilberman 2017). A current hypothesis proposes that gbM is a coproduct of stable mC maintenance, and the lack of a described mechanism that specifically regulates gbM to date would currently support this as the most parsimonious explanation (Bewick et al. 2016, 2017; Bewick and Schmitz 2017; Wendte et al. 2019).
An important factor in understanding the molecular roles of mC in different sequence and chromatin contexts is the discovery and characterization of the proteins that can bind to DNA methylation (mC readers). Two protein domains were identified to confer the ability to bind to DNA methylation: methyl-binding domain (MBD), first identified in methyl-CpG-binding protein 2 (MeCP2) (Nan et al. 1993), and the SET and RING-associated (SRA) domain (Baumbusch et al. 2001; Johnson et al. 2007). By sequence homology with these founder proteins, three families were identified in Arabidopsis, two containing a SET-domain—three variant in methylation (VIM) proteins and 10 suppression of variegation 3–9 homolog (SUVH) proteins—and 13 members in the MBD family (Baumbusch et al. 2001; Berg et al. 2003; Springer et al. 2003; Zemach and Grafi 2003; Springer and Kaeppler 2005). The VIM proteins can bind to mCG and mCHG in vitro but preferentially bind to hemimethylated DNA in the CG sequence context (Woo et al. 2007, 2008), and are responsible for recruiting MET1 for mCG maintenance after replication (Stroud et al. 2013; Shook and Richards 2014). Several members of the SUVH family have been well characterized by their involvement in mC deposition: SUVH4/KYP, SUVH5, and SUVH6 are able to bind to mC in all contexts, but preferentially bind mCHG and mCHH (Johnson et al. 2007; Rajakumara et al. 2011; Du et al. 2014). They contain an active histone methyltransferase domain that catalyzes H3K9me2, thus forming a feed-forward loop with CMT3 and CMT2 that bind to H3K9me2 and in turn deposit mCHG and mCHH, respectively (Jackson et al. 2002; Ebbs et al. 2005; Ebbs and Bender 2006; Stroud et al. 2013, 2014; Zemach et al. 2013). SUVH2 and SUVH9 bind all methylated contexts but preferentially to mCG and mCHH, respectively (Johnson et al. 2008). They are partially redundant in the RdDM pathway, recruiting PolV for the generation of long noncoding RNAs that—in concert with other factors—recruit DRM2 for de novo DNA methylation (Kuhlmann and Mette 2012; Johnson et al. 2014; Liu et al. 2014). SUVH1 and SUVH3 have also been identified as mC readers, and were proposed to have a role in promoting the transcription of genes with hypermethylated promoters (Li et al. 2016; Harris et al. 2018; Zhao et al. 2019).
The functions and binding abilities of MBD proteins are less well understood. Like their animal counterparts, MBD proteins seem to preferentially bind to mCG, despite some reports of their binding to non-CG context methylation too (Ito et al. 2003; Scebba et al. 2003; Zemach and Grafi 2003). MBD5, MBD6, and MBD7 are the most studied members (Li et al. 2017; Ichino et al. 2021, 2022), and have been shown to be enriched at chromocenters by GFP-fusion microscopy analysis, a localization that is dependent on the chromatin remodeler DDM1 (Zemach et al. 2005). They can interact with each other and with alpha-crystallin domain (ACD) proteins, but their molecular function remains unclear due to conflicting results regarding their involvement in chromatin remodeling or DNA demethylation (Zemach et al. 2008; Li et al. 2015, 2017). MBD8, MBD9, MBD10, and MBD11 are involved in diverse developmental processes, but based on biochemical characterization, they are unlikely to bind to mC (Berg et al. 2003; Peng et al. 2006; Preuss et al. 2008; Stangeland et al. 2009; Yaish et al. 2009). It is unclear whether MBD1, MBD2, and MBD4 can bind to DNA methylation and their molecular functions are still unknown (Zemach and Grafi 2007), but they have recently been found in complex with histone deacetylases and may have a role in controlling flowering timing (Zhou et al. 2021).
The mC-binding ability of MBD proteins had been exclusively investigated by electrophoretic mobility shift assay, leading to some conflicting results (Ito et al. 2003; Scebba et al. 2003; Zemach and Grafi 2003). A different approach was taken more recently, by performing a DNA affinity pull-down followed by mass spectrometry analysis (Harris et al. 2018). They used probes containing endogenous sequences of highly methylated promoters, which might have biased the discovery of proteins binding to these specific loci. In this study, we performed a DNA affinity pull-down experiment followed by mass spectrometry using DNA probes reflecting context-specific DNA methylation states in the Arabidopsis genome, resulting in the identification of all known mC readers as well as a variety of new candidate DNA methylation-binding proteins. We then focused on the characterization of their genome-wide binding pattern in vivo, and of high-order mutant plants lacking these proteins to interrogate the direct role of DNA methylation in Arabidopsis.
Results
Identification of Arabidopsis mC readers
To identify potential mC readers in Arabidopsis, we performed a DNA pull-down affinity assay of nuclear proteins using methylated DNA oligonucleotides. For each methylated sequence context (CG, CHG, CHH), DNA probes were designed to maximize the relevance of the binding proteins by searching for a representative 5 bp motif frequently found in methylated sequences of the Arabidopsis genome (Supplemental Fig. S1; see Methods). After combining probes with Arabidopsis nuclear extracts and performing affinity enrichment, mass spectrometry analysis of the pulled-down proteins using a statistical analysis that compares protein enrichment for methylated and unmethylated probes of each context identified 36 proteins enriched in at least one methylated cytosine context (Fig. 1; Supplemental Table S1). Among these proteins, all but one known mC readers were found to be enriched in binding to the methylated probes, and often in the contexts relevant to their functions (Supplemental Table S1). For example, the three VIM proteins were found enriched in symmetrical contexts; SUVH2 and SUVH5, as well as SUVH1 and SUVH3, were enriched in all contexts; SUVH4/KYP and SUVH6 were enriched in mCHG; and MBD5 and MBD6 were enriched only in mCG. Only MBD7 was excluded from our follow-up analyses (below), since it was recovered in only two out of three mCG probes. We also found expected proteins enriched for binding to unmethylated probes (Supplemental Table S1). For example, 13 WRKY transcription factors were enriched in the CHH probe (Fig. 1B; Supplemental Table S2), which have previously been found to prefer binding to unmethylated DNA by DNA affinity purification sequencing (DAP-seq) (O'Malley et al. 2016). The CHH probe sequence contains their known target site sequence (TGAC), likely underlying their binding to this probe (O'Malley et al. 2016). This screen also identified new mC reader candidates, most notably MBD1, MBD2, and MBD4, which while related to other mC readers, have had mixed results regarding their affinity for methylated DNA in prior studies (Ito et al. 2003; Scebba et al. 2003; Zemach and Grafi 2003; Harris et al. 2018).
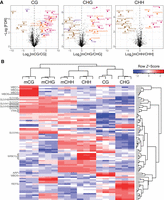
Identification of mC reader proteins in Arabidopsis from affinity pull-downs. (A) Volcano plots of the proteins statistically enriched for binding to methylated probes (known mC binders are pink, previously unknown mC binders are orange) or unmethylated probes (brown) in each context (CG, CHG, CHH). Statistically enriched proteins, often identified in mass spectrometry experiments and thus listed as contaminants in Van Leene et al. (2015), are colored purple. (B) Hierarchical clustering of the proteins (rows) statistically significantly enriched in at least one set of probes (columns, each representing a replicate). The names of proteins of interest were highlighted, the rest of the 83 proteins identified can be found in Supplemental Table S2.
Among new candidate mC readers we found proteins with DNA-binding domains, including the uncharacterized AP2/B3-like transcription factor AT5G25475 enriched in all three sequence contexts; HOMEOBOX-1 (AT1G28420, also called RINGLET 1, RLT1) enriched in mCG, which interacts with the Imitation Switch (ISWI) chromatin remodelers CHR11 and CHR17 (Li et al. 2012), and with chromatin remodelers BRAHMA (BRM), SWI3B and SWI3C (Efroni et al. 2013); and HOMEODOMAIN GLABROUS 11 and 12 (HDG11 and HDG12) enriched in the mCHG probes, known to be involved in cell differentiation and meristem development, yet previously thought to bind to A/T rich sequence (Nakamura et al. 2006; Xu et al. 2014; Horstman et al. 2015). Other proteins could be indirectly bound to DNA methylation through interactions with other readers. For example, the RING/U-box protein BMI1C, part of the polycomb repressive complex 1 (PRC1) (Sanchez-Pulido et al. 2008), was enriched in mCG and mCHH sequence contexts, potentially through contacts with MBD1 that was enriched in the same contexts and with which potential direct interaction was previously identified (Arabidopsis Interactome Mapping Consortium 2011).
Binding preferences of mC readers
To investigate the genomic-binding sites of seven selected mC readers (MBD1, MBD2, MBD4, MBD5, MBD6, SUVH1, and SUVH3), mutant lines of each reader (Supplemental Table S3) were isolated and then complemented with an epitope (2xStrep-HA-6xHis) tagged version of the corresponding protein driven by its predicted endogenous promoter, as estimated by DNase I hypersensitivity (see Methods). Given the lack of a physiologically observable phenotype in the mC reader single mutants, we selected lines that restored gene expression to close or above endogenous levels (Supplemental Table S3). Chromatin immunoprecipitation sequencing (ChIP-seq) against the HA epitope was performed on two independent insertion lines in T2 populations (Supplemental Table S3). Initial ChIP-seq peak calling analysis showed similar profiles in each replicate (Supplemental Fig. S2). However, the line with higher protein expression had higher enrichment signals (i.e., more peaks called), and merging the replicates enabled the identification of additional peaks that would have been missed if independent replicates were used (Supplemental Fig. S2). Thus, merged samples were used for further analyses. Peak analysis of merged replicates showed specific genome-binding profiles for each protein (Fig. 2A). MBD5, MBD6, SUVH1, and SUVH3 exhibited preferential localization to heterochromatic peri-centromeric regions, whereas the other MBD proteins studied were mostly localized along euchromatic chromosome arms (Fig. 2B). MBD1, MBD2, and MBD4 were mostly associated with genes, whereas MBD5, MBD6, SUVH1, and SUVH3 were primarily associated with TEs. The regions bound by MBD5, MBD6, SUVH1, and SUVH3 were highly methylated in all sequence contexts, whereas MBD2 peaks were mostly methylated in the CG context, and MBD1 peaks were unexpectedly lowly methylated (Fig. 2C). Sequence motif analysis (Supplemental Fig. S3), demonstrated that no clear motif was found for any of the candidates, indicating that their ability to bind DNA was not dependent on the genetic sequence but rather on the epigenomic state of the genomic region (Supplemental Fig. S3). However, the most strongly enriched motifs for MBD5 and MBD6 harbor a central CG dinucleotide, which would support their preference for methylation in the CG context. The preferential binding of these proteins was also assessed in vitro by DNA affinity pull-down sequencing (Bartlett et al. 2017). We incubated recombinant mC reader proteins with WT Arabidopsis fragmented gDNA libraries before (DAP-seq) and after PCR amplification (ampDAP-seq), where the amplification depletes the library of DNA methylation. MBD5 and MBD6 showed sequence enrichment, but only for DAP-seq, where DNA methylation was present, and with binding site profiles that closely match those identified by ChIP-seq (Fig. 2D). In contrast, (amp)DAP-seq experiments on MBD1, MBD2, MBD4, SUVH1, and SUVH3 did not show any signal, suggesting that additional requirements are necessary for their DNA-binding in vivo (Supplemental Fig. S4). Similarly, HDG11, enriched in mCHG probes, seems to prefer binding to unmethylated DNA in (amp)DAP-seq experiment (Supplemental Fig. S4H). These results support the hypothesis that MBD5- and MBD6-binding abilities are largely or solely dictated by DNA methylation in the CG context, whereas the other mC readers tested might require other cofactors, such as protein complexes to interact with or specific chromatin states.
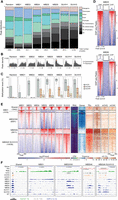
The diverse binding profiles of mC readers across the Arabidopsis genome. (A) Distribution of ChIP-seq peaks in genomic features. The total numbers of peaks for each sample are indicated at the bottom. Original annotations for TEs are indicated in parentheses. Promoters include [−1 kb; +100 bp] around the transcription start site; Terminators include [−100 bp; +1 kb] around the transcription termination site; Genes include both exons and introns. MBD1 peaks were shuffled in the genome to generate a random set of peaks. (B) The density of ChIP-seq peak positions along the Arabidopsis chromosomes, relative to their distance from the centromere (C, on the left) to the telomere of the chromosome arm they are on (T, on the right). (C) Average DNA methylation levels in WT Arabidopsis seedlings under protein-binding sites. Boxplots show the mean and quartiles of all peaks for each sample. For each context, only peaks containing at least three cytosines and a sequencing coverage ≥3 (average on all cytosines in the peak) are presented. (D) DNA affinity purification sequencing for MBD5 (top) and MBD6 (bottom) recombinant proteins on methylated (DAP) and unmethylated (ampDAP) genomic DNA libraries. Heatmaps show enrichment levels at all DAP-seq peaks, and the correlation with in vivo binding assessed by ChIP-seq. (E) Clustering of all mC reader ChIP-seq peaks based on their colocalization. Five clusters were identified, which are bound by different subsets of mC readers, and show different chromatin environments as highlighted by DNA methylation levels in each context, the transcript abundance, and the presence of annotated gene and TE features. (F) Browser displays a representative locus for each cluster defined in (E). ChIP-seq enrichment (log2 Fold Change [IP/Input]) is shown for each mC reader as well as a WT control (where the IP was performed on a WT population, with no tagged protein), along with DNA methylation in each sequence context and RNA expression in WT seedlings (log2 CPM).
To more closely examine the similarities and differences of where these seven selected mC reader proteins were binding in the genome, we merged the peaks from each of the proteins into a nonoverlapping set of regions, which were then clustered based on their ChIP-seq signal for each of the mC reader proteins (Fig. 2E). Five major clusters were defined, with the first cluster corresponding to regions methylated in all sequence contexts, often intersecting annotated TEs, and bound by all seven mC reader proteins (Fig. 2E,F). The second cluster comprises expressed, unmethylated genes, which are not bound by any mC reader, except for MBD1 (Fig. 2E,F). The regions in the third cluster are bound by MBD2, MBD5, and MBD6, with these peaks corresponding to expressed genes that are methylated in the CG sequence context, reflective of gbM (Fig. 2E,F). Looking specifically at all genes with gbM (Supplemental Fig. S5A) further confirmed this observation. The fourth cluster is only bound by MBD5 and MBD6, and is TE-rich and gene-depleted, highly methylated, and transcriptionally silenced (Fig. 2E,F). Finally, the fifth cluster, bound by MBD5, MBD6, SUVH1, and SUVH3 is also TE-rich but surrounded by genes, potentially representative of shorter euchromatic TEs (Fig. 2E,F). Taken together, this reveals that different mC readers have vastly different binding preferences, which are not only specific to the DNA methylation levels and sequence contexts, but to the more general chromatin state as well.
To further investigate the binding preferences of the mC readers for protein-coding genes or TEs, and to alleviate some of the limitations of peak calling algorithms, we centered the analyses on the protein-coding (genic) and TE regions that overlapped at least one mC reader-binding site (Fig. 3). This showed that MBD1 is enriched in genic regions (Fig. 3A), but is largely absent from TE sites (Fig. 3B). Despite our initial probe-based approach identifying MBD1 as enriched in binding to methylated DNA (Fig. 1), the ChIP-seq profiling of MBD1 reveals that, in vivo, MBD1 binds mostly to regions without mCG methylation, corresponding to unmethylated genes largely absent from the binding of other mC readers profiled here, and a relatively small number of highly methylated regions that appear to attract all examined mC readers studied here (Fig. 3A). In contrast, MBD5 and MBD6 generally bind to the same regions as one another, which are defined by the presence of mCG, irrespective of whether it is a protein-coding gene or TE, and of whether it is expressed or not (Fig. 3). MBD5 and MBD6 bound regions are often cobound by MBD2 (78%), but only in protein-coding regions (Fig. 3A). Genes with body methylation are the main targets of MBD2 identified in this study (Fig. 3; Supplemental Fig. S3A). MBD4 binds to the fewest sites in the genome, to methylated sites bound by other mC readers (mainly MBD1/2/5/6) (Fig. 3), as well as to unmethylated promoters (Fig. 2A). SUVH1 and SUVH3 were previously shown to bind to RdDM targets (Harris et al. 2018; Zhao et al. 2019). We confirmed this preference, and showed that SUVH1/3 targets are short euchromatic TEs and borders of larger heterochromatic ones (Fig. 3B), located in the promoter of protein-coding genes (Fig. 3A), with high levels of mCHH (Fig. 2C,E).
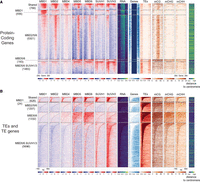
mC readers differ in their binding preferences across genes and TEs. (A) Heatmaps of ChIP-seq signal enrichment of mC readers in protein-coding genes (TAIR10). For each cluster from Figure 2, all unique protein-coding genes intersecting at least one peak were selected, and within each cluster, they were sorted by descending means of the total read number in all bins of the region. For each locus, the methylation level in each context and the RNA expression level (normalized by RPKM) were calculated from the Col-0 WT seedlings sample. For the methylation data, bins with no WGBS coverage are displayed in gray. ChIP-seq signal enrichments were calculated by comparing each ChIP sample to its corresponding input DNA control, scaled in log2 Fold Change (FC) from −1.5 to +1.5. The number of loci in each cluster is reported on the left-hand side of the heatmaps. The presence of an annotated gene or TE in each bin is shown in blue and red, respectively (0 if absent, 1 if present). Genes were scaled to 2 kb, with 1 kb upstream of the TSS and downstream from the TES, plotted in 100 bp windows for DNA methylation tracks and 20 bp windows for the others. (B) Heatmaps of ChIP-seq signal enrichment of mC readers in TEs (TAIR10). For each cluster from Figure 2, all unique TEs and TE genes intersecting at least one of the peaks were selected, and within each cluster, they were sorted by descending means of the total read number in all bins of the region. Heatmap details as in A, except that TEs were aligned on their 5′ end, plotting 1 kb upstream and 6 kb downstream.
To quantitatively analyze the binding profiles of these proteins, we next investigated the chromatin states to which these proteins bind. To do so, we generated a chromatin-state model with ChromHMM (Ernst and Kellis 2017), using the mC reader ChIP-seq, DNA methylation, and histone modification ChIP-seq data (Supplemental Fig. S5B). While the majority of states were unbound or only marginally bound by mC readers (states 1, 2, 4, 5, 6, 7, 8, 9, 11, 13, and 19; 75% of the genome), we noted several states associated with mC reader binding, as follows: state 3 (4330 loci; 2.3% of the genome), unmethylated genes marked by H2AKub, H3K27ac, H3K4me2, and H3K4me3, representing 25% of MBD1 targets; state 10 (3335 loci; 1.8% of the genome), genes with gbM, representing ∼50% of MBD2-binding sites; states 12, 17, 18, and 20 (26,768 loci; 12.3% of the genome), short TEs with little H3K9me2 but high DNA methylation (except state 20, which is enriched in transcriptional start sites), corresponding to >80% of SUVH1 and SUVH3-binding sites; states 14 and 15 (5034 loci; 7.8% of the genome), long TEs enriched in DNA methylation and H3K9me2, contributing to 25% of MBD5 and MBD6 targets, which also bind states 10 and 17; and finally, state 16 (1802 loci; 1% of the genome), which is defined by all mC readers and most histone marks, with repeats and, notably, ribosomal RNA, and corresponding to the shared cluster from Figure 2E (Supplemental Fig. S5C). Genome-wide correlation of ChIP-seq signal with DNA methylation in each context, and with gbM specifically, further confirms these association: MBD1 is not correlated with mCG (Pearson's = 0.05 for mCG, Spearman's = 0.15 for gbM); MBD2 is more correlated with gbM than with mCG alone (Spearman's = 0.45 vs. Pearson's = 0.16); MBD4, MBD5, and MBD6 are highly correlated with mCG (Pearson's = 0.42, 0.59, 0.72, respectively); while SUVH1 and SUVH3 are more highly correlated with mCHH (Pearson's = 0.48 and 0.55, respectively) (Supplemental Fig. S6A). Similar enrichments were found for the ChIP-seq of the same readers generated from other tissues in previous studies (Harris et al. 2018; Ichino et al. 2021; Wang et al. 2024), and when comparing to previously established chromatin states of the Arabidopsis genome (Supplemental Fig. S6A–C; Sequeira-Mendes et al. 2014; Jamge et al. 2023). Together, these analyses reveal that mC readers have specific binding specificities in vivo: MBD1 binds mostly unmethylated genic regions; MBD2 binds almost exclusively to genes with body methylation; MBD5 and MBD6 bind to all mCG without discrimination, including in gbM-genes, short euchromatic TEs, and long heterochromatic TEs; and SUVH1 and SUVH3 bind to short euchromatic TEs (RdDM targets) that are highly enriched in non-CG methylation. Furthermore, these results suggest that the binding of different combinations of mC readers to a single genomic site may form its own code that might define or represent the epigenomic context of the region.
Assessing the effects of the absence of multiple mC readers
Single, double, and triple mutants of mC readers have previously been reported, exhibiting only mild effects on the transcriptome and epigenome (Harris et al. 2018; Feng et al. 2021; Ichino et al. 2021, 2022; Zhou et al. 2021). Given the cobinding of multiple mC readers to the same genomic sites within the Arabidopsis genome, we generated different combinations of mutants in two of the seven profiled mC readers by CRISPR–Cas9 editing (Supplemental Table S4). After confirming the presence of frame-shifting indels, these mutants were then crossed to produce higher-order mutants, including the triple mutants mbd2 mbd5 mbd6 (referred to as mbd2;5;6), mbd1 mbd2 mbd4 (mbd1;2;4), and the quadruple mutant mbd1 mbd2 mbd5 mbd6 (mbd1;2;5;6). We also generated a new line of the suvh1 suvh3 (suvh1;3) double mutant. No morphological impact of these higher-order mutants could be observed, so we explored their molecular phenotypes by performing RNA sequencing (RNA-seq) and whole genome bisulfite sequencing (WGBS) (Supplemental Fig. S7). In totality, very few changes were found in either DNA methylation (Supplemental Fig. S7A) or gene expression (Supplemental Fig. S7B), the only exception being the suvh1;3 double mutant, in which we detected over 2000 differentially expressed genes (DEGs) (Supplemental Fig. S7B). Previously, SUVH1 binding in the promoter region immediately proximal to the TSS of genes was found to slightly enhance their expression (Harris et al. 2018). With the data generated here, we looked to see if loss of SUVH1 and SUVH3 (suvh1;3) (Supplemental Table S4) led to decreased expression of protein-coding genes with SUVH1 binding in their promoter regions, but detected no genome-wide relationship between SUVH1 binding close to the TSS and changes in gene expression (Fig. 4A). We confirmed that our suvh1;3 double mutant was exhibiting the expected molecular phenotype by verifying that known targets of SUVH1/3 enhancement were downregulated: ROS1, ESM1, ESP, and MBP1 (Figs. 3D, 4B; Li et al. 2016; Harris et al. 2018; Zhao et al. 2019). We did detect a small but significant reduction in the expression of genes where SUVH1 was bound immediately after the transcriptional end site (TES) (Fig. 4A). These data suggest that this genome-wide effect is modest or experimental-condition specific, and that many of the DEGs in suvh1;3 may be the results of changes in direct target dysregulation, such as of ROS1 and others to be characterized. Indeed, we see a significant overlap between DEGs in suvh1;3 and publicly available RNA-seq data (Kim et al. 2019) for ros1 mutant (Hypergeometric test: P-value = 4.89 × 10−21) (Supplemental Fig. S7C).
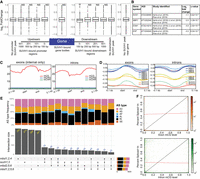
Transcriptomic changes of higher-order mC reader mutants. (A) Expression of protein-coding genes with and without SUVH1-binding sites at various locations relative to the TSS and TES. SUVH1-bound gene bodies were compared to genes with no binding in the gene body or within 1 kb of the gene with regards to changes in expression in the suvh1;3 mutant. The effect of binding within windows downstream from the gene on expression in the suvh1;3 mutant was compared to genes without binding in the downstream region. The same was performed for those binding upstream. (NS) adjusted P-value > 0.05, (**) adjusted P-value < 0.01, after Benjamini–Hochberg correction of Mann–Whitney U test/Wilcoxon rank-sum test. (B) Known targets of SUVH1/3 gene expression changes (log2FC and q-values) in our suvh1;3 double mutant. (C) Metaplots of DNA methylation levels in each sequence context over exons and introns for the de novo transcriptome generated in this study. Exon/intron lengths were normalized to 100 bp using deepTools. (D) Metaplots of mC reader ChIP-seq (log2FC [IP/Input]) over exons and introns similarly to (C). (E) Upset plot showing the number of differential alternative splicing (DAS) events shared between the different higher-order mutants studied. DAS types are skipped exon (SE), retained intron (RI), alternative last exon (AL), alternative first exon (AF), alternative 5′ splice site (A5), and alternative 3′ splice site (A3). (F) Histogram 2D plot of the correlation between exon (top; orange) or intron (bottom: green) mCG level and inclusion rate of the exon (or intron) within the final transcript, as measured by percent spliced-in (PSI). The black line represents a linear relationship between methylation of the exon (or intron) and inclusion of the region in the final transcript and the red line shows the regression from a linear model of their relationship. The Spearman's correlation coefficient for exon mCG and PSI is 0.02 (P-value = 0.4) and the Spearman's correlation coefficient for intron mCG and PSI is −0.001 (P-value = 0.9).
To investigate whether the 4446 CG-differentially methylated regions (DMRs) that we did identify between WT and higher-order mutants (suvh1;3, mbd1;2;4, mbd2;5;6, mbd1;2;5;6) were potentially linked to the loss of an mC reader, we overlapped the CG-DMRs with the mC reader ChIP-seq peaks, revealing little overlap between them (Supplemental Fig. S7D), in agreement with previous work (Li et al. 2016; Harris et al. 2018; Zhao et al. 2019), and no overlap between CG-DMRs and DEGs (Supplemental Fig. S7E,F). To further investigate the potential role for an mC reader in directly controlling transcription and further investigate the potential role of gbM, we performed nuclear global run-on sequencing (GRO-seq) and 5′ GRO-seq (Hetzel et al. 2016) on the mbd2 mutant, since MBD2 was specifically binding to gbM-genes. GRO-seq allows us to look beyond steady-state RNA levels and to look at transcription initiation rates. We found few differences between WT and mbd2 plants in terms of transcriptional activity as measured by GRO-seq and 5′ GRO-seq (Supplemental Fig. S7G), and almost no overlap between the statistically different regions of transcription and MBD2 ChIP-seq peaks (Supplemental Fig. S7H), suggesting that any differences were not resulting from the loss of a localized direct effect of MBD2 at these loci. Finally, considering the histone marks correlating with the mC reader binding (Supplemental Figs. S5, S6), we considered potential cross talk between mC readers and histone modifications. Thus, we performed ChIP-seq in WT and mutant plants to examine a range of histone post-translational modifications associated with active (H3K27ac, H3K4me1, H3K4me2, H3K4me3, H3K36me3) and silenced (H3K9me2, and polycomb-deposited H3K27me3, H2AKub) chromatin states. Few differentially marked peaks were identified (Supplemental Fig. S6A), and the binding profiles of most histone marks were highly similar between WT and each mutant examined, including the width of the peaks (Supplemental Fig. S6B). Taken together, these data suggest that the loss of even multiple mC readers concurrently do not have profound effects on the DNA methylome, the several histone modifications tested here, or the transcriptome in Arabidopsis seedlings.
In many organisms, including flowering plants, mCG is enriched in exons relative to introns and has been linked to exon definition during RNA splicing (Fig. 4C; Feng et al. 2010; Zemach et al. 2010). We hypothesized that mC readers may act as adaptor proteins to recruit splicing factors to the chromatin (Luco et al. 2010; Pradeepa et al. 2012). We found that all mC readers examined except SUVH1 and SUVH3 were also enriched in exons compared to introns (Fig. 4D), matching the enrichment of mCG levels, thus potentially supporting the notion that mC readers could recruit splicing factors to the chromatin during transcription. Further investigation, however, identified few (∼600) differentially alternatively spliced (DAS) events in the higher-order mutants (Fig. 4E), and there was very little overlap between higher-order mutants with overlapping mutations (Fig. 4E), suggesting that the few DAS events detected were likely background noise. Given the proposed role of DNA methylation within gene bodies in exon definition, we looked at inclusion levels of differentially spliced exons and introns in WT seedlings. If mCG was a mark for a region to be included in the final transcript, we would predict a correlation between exon (or intron) mCG level and inclusion rate of the exon (or intron) within the final transcript, as measured by the percent spliced-in (PSI). Therefore, we correlated the WT mCG levels with the PSI values for all skipped exons and retained introns, but again found no correlation (Fig. 4F). These data indicate that while mCG and many mC readers are enriched in exons, mCG does not appear to direct splicing inclusion of events in Arabidopsis seedlings, and mC readers do not direct alternative splicing.
Identification of mC reader protein interactions
To gain insights into molecular mechanisms that the mC reader proteins may be involved in, protein interactors were identified by (tandem) affinity purification coupled with mass spectrometry, (T)AP–MS, of the same tagged mC readers used for ChIP-seq. To ensure stringent identification of protein–protein interactions with mC reader proteins, one TAP–MS experiment was performed in one replicate (Set 1) and another AP–MS experiment was performed in triplicate (Set 2). TAP–MS for Set 1 was performed using the Strep and His tags on MBD1, MBD2, MBD6, and SUVH1 (Supplemental Tables S3, S5). Set 2 consisted of these same baits as well as MBD4, MBD5, and SUVH3, isolated using the Strep tag (Supplemental Tables S3, S5). To identify only the most robust interactions, we applied the following stringent rules: high-confident interactors that appear in the (T)AP–MS from both sets (Fig. 5A), high confidence in the replicates from Set 2 experiment (Supplemental Table S5), or overlap with previously published interactions (Supplemental Table S6). Correlation of the peptide enrichment of putative interactors in each replicate revealed several protein interactors (Fig. 5A). Notably, MBD1 pull-downs in both sets of experiments revealed interactions with chromatin regulators UBN1, HAT3.1, and HUA2 (Fig. 5). UBN1 is a component of the HIRA complex, responsible for histone variant H3.3 deposition (Nie et al. 2014). Other HIRA complex members (HIRA, UBN2, and H3.3) were also identified in Set 1 (Supplemental Table S6), and while not passing our stringent criteria, still suggest that MBD1 interacts with the HIRA complex. MBD1 also interacted with the transcription factors HAT3.1 and HUA2 (Fig. 5B). HAT3.1 and HUA2 were also found to interact with MBD4 in Set 2. Another high-confidence interaction that we identified was between MBD6 and ACD protein ACD21.4 (AT1G54850, formerly IDL2) (Fig. 5). This interaction has been reported previously (Ichino et al. 2021), as has the direct interaction of SUVH1 with SUVH3 that we identified (Fig. 5; Harris et al. 2018). A number of interactors that did not pass our initial stringent criteria (Supplemental Table S5) have been previously reported as mC reader interactors (Supplemental Table S6), thus they have been validated by another independent study and were included here in the network of mC reader interactors (Fig. 5B). Such interactors include DNAJ proteins (SDJ2/DNAJ2, SDJ1/DNAJ1, and SDJ3) with SUVH1, and DNAJ proteins SLN and SDJ2/DNAJ2 with MBD6 (Fig. 5B; Supplemental Table S6). We also found evidence for the interaction of MBD6 with ACD protein ACD15.5 (AT1G76440, formerly IDL3) and the demethylation factor IDM3 (AT1G20870, ACD51.9) (Fig. 5B; Supplemental Table S6). Previous studies showed that MBD1, MBD2, and MBD4 interact with a histone deacetylase complex (Feng et al. 2021; Zhou et al. 2021). We found evidence for both MBD1 and MBD2 interacting with HDA6 and other proteins from this complex: SANT3 (AT2G47820) and ATHIP2 (AT3G17880) (Fig. 5B; Supplemental Tables S5, S6), suggesting that MBD1 can take part in multiple complexes, one for regulating histone acetylation, and another for H3 variant deposition. Taken together, these data show that not only do the different mC readers have different binding profiles throughout the genome, but also form multiple different complexes, indicating a subfunctionalization of these factors.
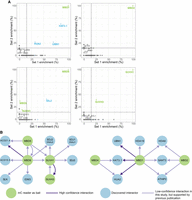
Protein interactors with mC readers. (A) Scatter plots showing the correlation between Set 1 and Set 2 experiments for MBD1, MBD2, MBD6, and SUVH1. The gray lines represent the thresholds to reduce false positive proteins being selected for the interaction network. A 15% threshold for each replicate was selected. mC reader proteins that have been used as baits in this study are green. Interactors identified within this study are blue. (B) A conservative interaction network of proteins in this study. Interactions (edges) identified in both Set 1 and Set 2 experiments are indicated in dark purple. Those edges that were present in our data sets but did not meet our strict criteria, yet that have been identified in another study are indicated in light purple. Nodes (proteins) are colored as in A.
Discussion
Here, we used an unbiased screening approach to identify DNA methylation-binding proteins in Arabidopsis and characterized the binding profiles of seven of these: MBD1, MBD2, MBD4, MBD5, MBD6, SUVH1, and SUVH3. While some of these mC readers had overlapping binding sites within the genome, clear differences between them could be identified. MBD2 predominantly binds to gbM, while MBD5/6 bound mCG sites within genes and TEs, often overlapping with SUVH1/3-binding sites, which largely bound non-CG methylation TE sites. Despite being identified within our screen as an mC reader, MBD1 is largely bound to unmethylated sites within gene bodies, which raises some interesting questions regarding what determines it's in vitro versus in vivo-binding profiles. It is worth noting that our in vitro pull-downs were performed using the Ler ecotype but our subsequent in vivo data were collected from the Col-0 ecotype. However, there are no protein variants in MBD1 between these two ecotypes, so we expect the differences to reflect the molecular complexes formed by MBD1 in vivo rather than something intrinsic to the protein differing between the two strains of A. thaliana used here.
Despite the main focus of this study to be on mC readers, the methylated-probe pull-down screen performed has also identified proteins which preferentially bind unmethylated DNA (Fig. 1). MBD9 has never been shown to bind DNA but instead histone H4 (Zemach and Grafi 2007; Yaish et al. 2009), and we found that MBD9 was strongly enriched with unmethylated DNA (Fig. 1). The only protein enriched in two unmethylated contexts (CG and CHH) (Fig. 1) was the AP endonuclease ARP (AT2G41460), known to be involved with DNA repair after active demethylation by DME or ROS1 (Córdoba-Cañero et al. 2011; Lee et al. 2014; Akishev et al. 2016), which along with the detection of the DNA repair protein RAD4 (AT5G16630) (Kunz et al. 2005; Liang et al. 2006), suggests that methylation sensitivity may play a role in this part of DNA repair.
Two Jumonji-C (JmjC) domain-containing proteins, JMJ24 and JMJ28, were found enriched with unmethylated CHG (Fig. 1). JMJ24 acts to reduce H3K9me2 levels indirectly, through ubiquitin E3 ligase activity via its RING domains that target CMT3 for proteasomal degradation (Deng et al. 2015, 2016; Kabelitz et al. 2016). JMJ28 is a homolog of JMJ24 that also contains a JmjC and a RING domain (Lu et al. 2008). Recently, Arabidopsis JMJ28 was identified as guiding H3K4me3 deposition (Xie et al. 2023), supporting a role in the formation of euchromatin free of mCHG. Therefore, our results, and those of others, support an association between JmjC-domain proteins and unmethylated DNA, especially in the CHG context, and might suggest a role in preventing heterochromatin marks from spreading into euchromatin like in other species, such as JmjC-domain proteins DMM-1 in Neurospora crassa and Epe1 in Schizosaccharomyces pombe (Tamaru 2010).
Like MBD5/6, MBD2 binds mCG in gene bodies (∼50% of MBD2-binding sites), but unlike MBD5/6, MBD2 rarely binds TEs (∼15% of MBD2-binding sites) (Fig. 2, Supplemental Figs. S5, S6). Our binding profiles of MBD5/6 agree with the previously reported localization of GFP-fusion proteins at chromocenters (Zemach 2005). In contrast, MBD2-GFP localization was diffused throughout the nucleus, unless DNA patterns were disrupted in ddm1 or met1 mutants (Zemach 2005), supporting our finding that in WT plants MBD2 is preferentially bound to methylated genes and is not enriched in heterochromatin. MBD2 may interact with a range of chromatin remodelers, notably with BRAHMA (BRM) (Supplemental Table S5). BRM has been shown to prevent deposition of H3K27me3 (Li et al. 2015), which may aid in preventing the repression of genes with gbM, helping to explain their constitutively expressed pattern (Coleman-Derr and Zilberman 2012). The recruitment of BRM to gbM would occur in addition to its recruitment by the H3K27 demethylase REF6. Given that REF6 preferentially binds unmethylated mCHG sites (Fig. 1A,B), we propose a synergistic effect of REF6 and MBD2 might thus recruit BRM at genes with mCG but unmethylated CHG sites. Contrasting with this hypothesis, we do not see an increase in H3K27me3 in mbd2 mutants, potentially due to further redundancy, the need for simultaneous REF6 disruption, or a cell-type-specific role for this recruitment. Since gbM is absent from some plant species (Bewick et al. 2016), it would be interesting to know whether MBD2 presence and/or function is also conserved in these species, or if its involvement downstream from gbM has become moot.
Given the enrichment of many mC readers in exons relative to introns (Fig. 4D), mirroring the reported patterns of CG methylation (Fig. 4C; Feng et al. 2010; Zemach et al. 2010), we explored whether alternative splicing could be directed by mC readers. Previously, histone modifications have been shown to direct alternative splicing in mammalian cells via adaptor proteins (Luco et al. 2010; Pradeepa et al. 2012). While an example of non-CG methylation in male sex cells in Arabidopsis is related to alternative splicing patterns (Walker et al. 2018; Lloyd and Lister 2022), our results suggest that gbM does not direct splicing patterns (Fig. 4). This is supported by previous work in plants that compared DAS in WT to the met1 derived epiRILs (Bewick et al. 2016). Other work has raised questions over whether exonic enrichment of DNA methylation is relevant to alternative splicing in insects (Patalano et al. 2015; Harris et al. 2019). Given the lack of an obvious effect on the loss of DNA methylation and mC readers on alternative splicing, it is likely that in many species, exonic DNA methylation is involved in a different mechanism, potentially as a by-product of CMT3 activity in flowering plants, as previously suggested (Bewick et al. 2016, 2017; Wendte et al. 2019), and that the MBDs examined here do not act in the “chromatin–adaptor complex” model of the regulation of alternative splicing (Luco et al. 2011).
The lack of obvious phenotypes within mC reader mutants, including higher-order mutants, suggests that their role in normal plant growth is complex and masked from conventional approaches to study them. This is reminiscent of work in mammals in which disruption of all MBD-encoding genes did not derepress TEs (Kaluscha et al. 2022). Indeed, methylation-sensitive DNA-binding transcription factors played a much larger role in regulating the expression of methylation-responsive genes than MBD proteins did (Kaluscha et al. 2022). It is possible that further redundancy between mC readers exists and that even higher-order mutants would need to be generated to eliminate this possibility. For example, MBD7 may be redundant to MBD5/6 as they partially colocalize to chromocenters (Zemach et al. 2008), but as MBD7 was not identified in all three replicates of our in vitro screen, likely due to the structural constraints of the three MBD domains, it was not a focus of this study (Fig. 1). The lack of phenotype identified here with our higher-order mutants is in contrast to Ichino et al. (2021), which found that the mbd5;6 had defects in silencing (Ichino et al. 2021). However, the use of floral tissue for their RNA-seq work would indicate a cell-type-specific function for MBD5/6 (Ichino et al. 2022). The role in repression by MBD5/6 only appears to be detectable when normal chromatin compaction is lost, either naturally in pollen vegetative cells or via a mutation in the H1 encoding genes (Ichino et al. 2021, 2022). A similar situation is true for MBD2 and its recently published role in TE repression in the vegetative cell of mature pollen (Wang et al. 2024). Considering the specialized function and regulation of DNA methylation in some tissues such as pollen vegetative cell and endosperm, it is likely that the loss of mC readers would only create phenotypes in these specific cell types, or during restricted developmental stages. The ChIP-seq profiles of the mC readers generated in seedlings for this study are very similar to previously published data sets generated in floral tissues (Supplemental Fig. S6), suggesting that the binding of these proteins is consistent across tissues and thus that other changes—at the chromatin level or via the presence of tissue-specific cofactors—are necessary to reveal their functions. Alternatively, these proteins may have a more constitutive yet unknown role, and the expression levels of the tagged proteins might explain the small differences in binding profiles seen between studies.
We found that loss of suvh1;3 led to more transcriptomic changes than for any other higher-order mutant studied here (Supplemental Fig. S7) and found that previously reported targets of SUVH1/3-mediated expression enhancement, including ROS1 (Harris et al. 2018; Zhao et al. 2019), were downregulated in the suvh1;3 double mutant (Fig. 3D). Given that a similar number of genes were up- and downregulated in suvh1;3 plants, it is likely that many of these dysregulated genes are indirect targets of SUVH1/3 and might be the consequences of the loss of ROS1 and other targets. We showed here with both ChIP-seq and DAP-seq that MBD5 and MBD6 bind methylated CG in all genomic contexts (Figs. 2, 3; Supplemental Fig. S3). Also, no direct interactions with DDM1, MET1, or H1 were found in our TAP–MS (Fig. 5; Supplemental Table S5) nor previously published data sets (Li et al. 2015, 2017; Ichino et al. 2021). The mislocalization of MBD5/6 in ddm1 mutants (Zemach et al. 2005) must then be due to the redistribution of mCG. MBD5 and MBD6 have also been shown to contribute to rDNA silencing and nucleolar dominance (Preuss et al. 2008; Ren et al. 2024). The role of these proteins might thus be to simply prevent transcription by steric hindrance, as the increase in accessibility in upregulated genes in pollen grains might suggest (Ichino et al. 2022). Analysis of DNA accessibility on the chromatin state corresponding to genes with gbM (chromatin state 7) (Sequeira-Mendes et al. 2014), showed low DNA accessibility (Leduque et al. 2024), which could suggest a role for MBD2/5/6 proteins in limiting accessibility independently of transcription. Analyzing the impact on DNA accessibility and genome-wide distribution of mC readers in the absence of these MBD2/5/6 proteins could provide an additional understanding of their function.
Our protein–protein interaction network for mC readers (Fig. 5B) may indicate some mechanistic roles for these mC readers. The chromatin landscape in Arabidopsis is a complex interconnection of mechanisms that control gene expression, while maintaining TE silencing. We found that mC readers in Arabidopsis likely participate in this intricate regulation, binding to methylated sequences in specific chromatin contexts. It has been shown previously that MBD5 and MBD6 participate in gene silencing with SILENZIO (Ichino et al. 2021, 2022). However, they also showed that MBD5 and MBD6 can interact with SUVH1 and SUVH3, proteins that participate in gene activation in Arabidopsis (Harris et al. 2018; Zhao et al. 2019). We have shown here that MBD5, MBD6, SUVH1, and SUVH3 are often colocalized in their genomic-binding sites in seedlings, with the main difference being the preference for SUVH1/3 for non-CG methylation in contrast to CG methylation for MBD5/6. However, why some loci are bound by both the reportedly activating SUVH1/3 mC readers (Harris et al. 2018; Zhao et al. 2019) and the repressive MBD5/6 mC readers (Ichino et al. 2021, 2022) is unclear, especially given that the apparently repressive MBD5/6 proteins also interact with components of an activation complex containing IDM3 and SDJ2/DNAJ2 (Fig. 5; Li et al. 2017). It is conceivable that MBD5/6 might moonlight with both activator and repressor roles, depending on the exact chromatin context, such as whether they are in complex with SUVH1/3 or not (Figs. 2, 3). While IDM3 and SDJ1/2/3 have been reported to control DNA methylation levels (Miao et al. 2021), we did not see evidence for MBD5/6 or SUVH1/3 playing a role in this DNA methylation maintenance role (Supplemental Fig. S7), which is consistent with previous reports of stable methylomes in mC reader mutants (Harris et al. 2018; Zhao et al. 2019; Ichino et al. 2021).
We found that MBD1 and MBD4 interacted with transcription factors HAT3.1 and HAU2, suggesting that these transcription factors may recruit MBD1/4 to specific sites in the genome, or that MBD1/4 may aid in providing specificity to HAT3.1/HAU2 with regards to the epigenomic context. The HIRA complex is responsible for the deposition of the H3.3 histone variant (Nie et al. 2014). In addition to identifying HIRA complex component UBN1 as a high-confidence interactor of MBD1 (Fig. 5), we also found HIRA, UBN2, and H3.3 as putative MBD1 interactors (Supplemental Table S5), suggesting that MBD1 may play a role in the recruitment of the HIRA complex. Previous reports have shown that MBD1/2/4 interact with a deacetylation complex composed of HDA6 and SANT-domain proteins, and the TE-derived HHP1/HARB (Feng et al. 2021; Zhou et al. 2021). Feng et al. (2021) found that loss of MBD1, MBD2, and MBD4 lead to a small change in H3 acetylation by ChIP-seq (Feng et al. 2021), and while we did observe a slight decrease in H3K27ac in mbd1 and mbd1;4, we did not examine the triple mbd1;2;4 mutant as they did (Supplemental Fig. S8). Also, we used an antibody to a specific acetylation mark (H3K27ac) rather than a general antibody to any H3 acetylation, as performed in Feng et al. (2021).
In contrast with the ability of MBD1 to bind DNA methylation in vitro (Fig. 1), MBD1 binding in vivo is mostly at unmethylated loci (Fig. 2; Supplemental Figs. S5B,C, S6A,B). Our data suggest that, in vivo, MBD1 is recruited by multiple protein complexes that it interacts with: the HIRA complex (Fig. 5), the histone deacetylation complex (Fig. 5; Feng et al. 2021; Zhou et al. 2021), and binding with transcription factors HAT3.1 and HUA2 (Fig. 5). While we did not identify BMI1C in our (T)AP–MS data sets, previous interactions between MBD1 and BMI1C were identified (Arabidopsis Interactome Mapping Consortium 2011), and BMI1C was identified in our in vitro analysis in the same contexts as MBD1 (Fig. 1). The relationship between MBD1 and H2AKub loci might also highlight such interactions (Supplemental Fig. S5B,C). MBD2 preferentially binds CG methylated gene bodies (Fig. 2). The cobinding of MBD1 and MBD2 to the HDA6 complex (Fig. 5; Feng et al. 2021; Zhou et al. 2021) may indicate that this complex interacts with the fraction of MBD1 bound to methylated regions of the genome, whereas MBD1-specific complexes, notably the HIRA complex (Fig. 5), may be recruited to unmethylated regions of the genome. It has been recently shown that the chromatin remodeler DDM1 is only able to facilitate histone H3.1 deposition in nucleosomes containing unacetylated histone H4, thus allowing for DNA methylation to take place (Lee et al. 2023). MBD1, MBD2, and MBD4 might then participate in this interplay between DNA methylation, histone acetylation, and histone variant deposition, crucial for epigenetic inheritance (Lee et al. 2023). The lack of a clear phenotype, even in the higher-order mutants, raises questions regarding the function of mC readers in plants. The gbM-binding profile, exemplified by MBD2, could indicate a protective role for this protein against DNA damage. DNA methylation is mutagenic, but genes with gbM have a lower rate of evolution (Takuno and Gaut 2012), suggesting that a mechanism exists that prevents mutations of these cytosines. Confirming this hypothesis, or showing that negative selection of these mutations is alone responsible for a lower evolution rate, would require a much longer time frame and many generations to see the consequences of mutations.
Taken together, we have shown that Arabidopsis mC readers form multiple distinct protein complexes, potentially influencing their genome-wide binding profiles and suggesting several subfunctions. These results support the existence of independent regulatory mechanisms downstream from DNA methylation that function in a chromatin context-dependent manner. Further investigation, in specific cell types or in plants deficient in epistatic mechanisms, could be key to address the apparent redundancy and to understand the precise function of these proteins.
Methods
DNA affinity pull-down assay
Nuclear proteins were extracted from A. thaliana Landsberg erecta (Ler) ecotype cell suspensions derived from root callus, by protoplasting and isolating nuclei (Supplemental Methods). DNA affinity pull-down assays were then performed following an adapted version of Spruijt et al. (2013), using probes designed for each sequence context following the model of Spruijt et al. (2013) (Supplemental Methods). Enriched proteins were measured by LC–MS/MS (Supplemental Methods), peptides were searched against the UniProt A. thaliana proteome (version 2014-09-03) with MaxQuant version 1.5.1.0 (Cox and Mann 2008), and results were analyzed with Perseus version 1.4.0.0.
DNA affinity purification sequencing
DAP-seq has been performed according to the original protocol (Bartlett et al. 2017), using genomic DNA libraries created from 2-week-old Col-0 seedlings. Plasmids containing the proteins of interest in the pIX-HALO backbone were ordered from the Arabidopsis Biological Resource Center (ABRC) (Supplemental Methods).
Plant material
All plants were A. thaliana accession Col-0, grown on soil at 22°C in 16 h light/8 h dark cycles. Details about single and higher-order mutant lines, complemented lines, and genotyping can be found in Supplemental Methods, while primers used in this study are listed in Supplemental Table S7.
Chromatin immunoprecipitation sequencing
Of 14-day-old seedlings, 3–8 g was used for chromatin extraction (Supplemental Methods). Immunoprecipitation was performed using the following primary antibodies (αHA antibody: Biolegend 901502; αH2A.Z antibody: Abcam ab4174; αH2AK121ub antibody: Cell Signaling Technology 8240S; αH3K4me1 antibody: Abcam ab8895; αH3K4me2 antibody: Abcam ab32356; αH3K4me3 antibody: Abcam ab8580; αH3K36me3 antibody: Abcam ab9050; αH3K27me3 antibody: Abcam ab6002; αH3K27ac antibody: Abcam ab4729; αH3K9me2 antibody: Abcam ab1220; αH3 antibody: Abcam ab1791). Libraries were created according to Rubicon thruPLEX DNA-seq kit and sequenced on a HiSeq 1500 or NextSeq 500 instrument (Illumina) (Supplemental Methods).
Affinity purification coupled with mass spectrometry
Of 14-day-old seedlings, 3–8 was used for nuclear protein isolation (Supplemental Methods). For the experimental Set1, tandem affinity purification was performed using IBA MagStrep “type3” XT beads (Fisher Biotech) and Dynabeads His-Tag Isolation and Pulldown (Thermo Fisher Scientific). For the experimental Set2, a single strep IP protein pull-down was performed (Supplemental Methods). Purified protein complexes were digested and measured by LC–MS/MS (Supplemental Methods).
Whole genome bisulfite sequencing
WGBS was performed by MethylC-seq (Lister et al. 2008). Genomic DNA was extracted from 2-week-old seedlings with DNeasy Plant Mini Kit (Qiagen) and further purified with Isolate II PCR and Gel Kit (Bioline). Libraries were then generated from fragmented DNA (Covaris, 250 bp) with NxSeq AmpFREE Low DNA Library Kits (Lucigen, now LGC Biosearch Technologies). After size selection by AMPureXP beads, bisulfite conversion was performed with the EZ DNA Methylation-Gold Kit (Zymo Research). Libraries were amplified with Kapa HiFi HotStart Uracil+ ReadyMix (Roche) and purified with AMPureXP beads. MethylC-seq libraries were sequenced on the HiSeq 1500 platform (Illumina) using single-end 100 bp format, according to the manufacturer's instructions.
High-throughput RNA sequencing
Three biological replicates were performed in parallel for all genotypes from populations of 2-week-old seedlings grown on half strength Murashige and Skoog media supplemented with 1% sucrose. Total RNA was extracted with RNeasy Plant Mini Kit (Qiagen) and treated with RQ1 DNase (Promega). Libraries were then generated with TruSeq Stranded Total RNA Library Prep Kit (Illumina), after depletion of ribosomal RNAs with Ribo-Zero rRNA Removal Kit Plant (Illumina). RNA-seq libraries were sequenced on a NextSeq 500 (Illumina) using paired-end 42 bp format, according to the manufacturer's instructions.
Global run-on sequencing
GRO-seq and 5′-GRO-seq were performed on 10–20 g of 2-week-old seedlings (∼10 plates), according to the published protocol (Hetzel et al. 2016).
Data analysis
Details about ChIP-seq, DAP-seq, WGBS, RNA-seq, and GRO-seq data analysis can be found in Supplemental Methods. Read mapping statistics can be found in Supplemental Table S8.
All annotated genes in TAIR10 were split into three categories based on their DNA methylation levels: genes with >2% mCHG and mCHH were classified as TE-like methylation; in the remaining list, genes with >5% mCG were labeled as gbM-genes; and the rest are unmethylated genes (Supplemental Table S9).
Chromatin states were generated with ChromHMM (Ernst and Kellis 2017) as described in Supplemental Methods. Details about the parameters, the model, and the final genome segmentation are available in Supplemental Table S10.
Affinity purification–MS analysis is detailed in Supplemental Methods. Of note, we filtered out the contaminant proteins listed in (Van Leene et al. 2015), since they are common contaminating proteins from TAP–MS experiments in Arabidopsis, and the rules for inclusion in our network of mC reader protein–protein interactions were:
-
If above threshold (15%) in Set1 experiment and Set2 combined replicates.
-
If above a stringent threshold (35%) in combined Set2 replicates (and over 30% in at least 2/3 individual replicates).
-
If found in other published studies as an interactor and peptides were identified in either Set1 experiment or Set2 experiment.
Data access
All of the raw read files generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1065964. The proteomic IP-MS data generated in this study have been submitted to the ProteomeXchange database (https://www.proteomexchange.org/) under data set identifier PXD050777. ChIP-seq peaks of mC readers, the de novo transcriptome assembly, and the associated scripts used to analyze the data can be found at Zenodo (https://doi.org/10.5281/zenodo.10828673). Associated scripts can also be found as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Dmitri Nusinow for the SHH-tag plasmid used for cloning and advice on the affinity purification experiments, and all members of the Lister lab for their insightful discussions. J.C. was supported by a UWA International Postgraduate Research Scholarship. The Vermeulen lab is part of the Oncode Institute, which is partly funded by the Dutch Cancer Society (KWF). This work was supported by the following grants to R.L.: Australian Research Council (ARC) Centre of Excellence in Plant Energy Biology (CE140100008), ARC DP210103954, NHMRC Investigator Grant GNT1178460, Silvia and Charles Viertel Senior Medical Research Fellowship, and Howard Hughes Medical Institute International Research Scholarship.
Author contributions: J.C. and R.L. conceived of the project and designed the experiments. J.C. performed the ChIP-seq, DAP-seq, RNA-seq, and GRO-seq experiments. J.Pf. conducted the sequencing. J.C. and O.B. performed the affinity pull-down experiment. I.D.K. performed the corresponding mass spectrometry and its analysis. J.C., J.P.B.L., P.W.T.C.J., O.D., A.H.M., and J.Pe. performed the (T)AP–MS experiments and/or their analysis. J.C., J.P.B.L., and R.L. analyzed the data and/or its significance. A.H.M., M.V., and R.L. acquired funding. J.C., J.P.B.L., and R.L. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279379.124.
- Received March 19, 2024.
- Accepted October 17, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








