
Nanopore strand-specific mismatch enables de novo detection of bacterial DNA modifications
- Xudong Liu1,7,
- Ying Ni2,3,4,7,
- Lianwei Ye1,7,
- Zhihao Guo1,
- Lu Tan1,
- Jun Li1,
- Mengsu Yang2,3,4,5,
- Sheng Chen6 and
- Runsheng Li1,3,4
- 1Department of Infectious Diseases and Public Health, Jockey Club College of Veterinary Medicine and Life Sciences, City University of Hong Kong, Hong Kong 999077, China;
- 2Department of Biomedical Sciences, Jockey Club College of Veterinary Medicine and Life Sciences, City University of Hong Kong, Hong Kong 999077, China;
- 3Department of Precision Diagnostic and Therapeutic Technology, City University of Hong Kong Shenzhen Futian Research Institute, Shenzhen 518000, China;
- 4Tung Biomedical Sciences Centre, City University of Hong Kong, Hong Kong 999077, China;
- 5Key Laboratory of Biochip Technology, Biotech and Health Centre, Shenzhen Research Institute of City University of Hong Kong, Shenzhen 518000, China;
- 6State Key Lab of Chemical Biology and Drug Discovery, Department of Food Science and Nutrition, The Hong Kong Polytechnic University, Hong Kong 999077, China
-
↵7 These authors contributed equally to this work.
Abstract
DNA modifications in bacteria present diverse types and distributions, playing crucial functional roles. Current methods for detecting bacterial DNA modifications via nanopore sequencing typically involve comparing raw current signals to a methylation-free control. In this study, we found that bacterial DNA modification induces errors in nanopore reads. And these errors are found only in one strand but not the other, showing a strand-specific bias. Leveraging this discovery, we developed Hammerhead, a pioneering pipeline designed for de novo methylation discovery that circumvents the necessity of raw signal inference and a methylation-free control. The majority (14 out of 16) of the identified motifs can be validated by raw signal comparison methods or by identifying corresponding methyltransferases in bacteria. Additionally, we included a novel polishing strategy employing duplex reads to correct modification-induced errors in bacterial genome assemblies, achieving a reduction of over 85% in such errors. In summary, Hammerhead enables users to effectively locate bacterial DNA methylation sites from nanopore FASTQ/FASTA reads, thus holds promise as a routine pipeline for a wide range of nanopore sequencing applications, such as genome assembly, metagenomic binning, decontaminating eukaryotic genome assemblies, and functional analysis for DNA modifications.
DNA base modifications are crucial components of epigenetic changes and significantly influence various biological functions. The most extensively studied and understood DNA base modification in vertebrates is 5-methylcytosine (5mC). This type of modification is abundant and can mostly be found in the CpG motif. In prokaryotes, there are three main types of DNA molecule modifications, all of which involve methylation: N6-methyladenine (6mA), N4-methylcytosine (4mC), and 5mC (Beaulaurier et al. 2019). These methylation forms differ in distribution and function in bacteria, but all play pivotal roles in bacterial life processes. Long-read sequencing platforms, including PacBio single-molecule real-time (SMRT) sequencing and Oxford Nanopore Technology (ONT) sequencing, have enabled the direct detection of DNA modifications (Gouil and Keniry 2019; Amarasinghe et al. 2020; Ni et al. 2023a). This is due to the distinct differences in the raw signals produced by modified DNA compared to those produced by canonical DNA sequences. Notably, the detection of 5mC in vertebrate genomes using nanopore sequencing has led to significant advancements over traditional bisulfite sequencing (Liu et al. 2023). However, the challenge of detecting bacterial DNA modifications persists, primarily due to their unique and diverse motifs (Roberts et al. 2023).
Significant advancements have also been made in developing methods for detecting bacterial DNA modifications through nanopore sequencing. The available methods, including Tombo (Stoiber et al. 2017), Dorado (https://github.com/nanoporetech/dorado), Snapper (Konanov et al. 2023), and nanodisco (Tourancheau et al. 2021), rely on interpreting nanopore raw current signals. This process, however, is notably time-consuming and often restricted, as the raw data are typically accessible only to the data producers. Additionally, de novo identification of DNA modification sites necessitates a control sample without modifications, which is generally achieved through whole-genome amplification (WGA). This requirement adds complexity and increases the cost associated with bacterial DNA modification detection in nanopore sequencing. Consequently, de novo detection of bacterial DNA modifications remains a challenging task.
The read accuracy of nanopores may be compromised by basecalling errors, which can be caused by bacterial DNA modifications (Rand et al. 2017; Wick et al. 2019). Bacterial DNA methylation can occur frequently in diverse sequence contexts. In nanopore direct RNA sequencing, mismatches in modified RNA molecules can be used to detect modifications (Liu et al. 2019). However, this feature has not been implemented in bacterial DNA sequencing. A more efficient method for identifying bacterial DNA methylation via nanopore sequencing platforms is needed. A pipeline based on sequence mismatches could serve as a more straightforward and time-efficient alternative.
In this study, we sequenced eight bacterial species, namely, Acinetobacter pittii (Liu et al. 2022), Bacillus cereus (Cui et al. 2016), Enterococcus faecium (Zheng et al. 2007), Escherichia coli (Chen et al. 2017), Klebsiella pneumoniae (Yang et al. 2019), Pseudomonas aeruginosa (Zhao et al. 2023), Salmonella enterica (Chen et al. 2020), and Staphylococcus aureus (Wang et al. 2017). We identified patterns of unbalanced mismatches between forward and reverse strands on these genomes. The sequences of these Gram-negative and Gram-positive strains differ in terms of complexity in chromosomes and plasmids, and these strains can be used as representatives for developing a method for detecting DNA modifications de novo.
We developed Hammerhead to detect bacterial DNA modifications using unbalanced mismatches between forward and reverse strands. Most motifs and sites detected by Hammerhead can be further validated using nanodisco (Tourancheau et al. 2021) and the presence of methyltransferase in different bacterial species. In summary, our results provide insights and solutions for bacterial genome assemblies and identifying modification sites using Nanopore whole-genome reads.
Results
The updated Nanopore sequencing platform shows improvements in read accuracy and assembly quality
Read accuracy is key to successful genome assembly. To evaluate the performance of the R9.4.1 and R10.4.1 flow cells in terms of read quality and genome assembly, we sequenced DNA samples from eight distinct bacterial species under Nanopore R9.4.1, R10.4.1, and high-throughput short-read sequencing (SRS) platforms (see Supplemental Methods). The R9.4.1 platform produced simplex reads only, while the R10.4.1 platform produced both simplex and duplex reads. A total of 9.64 Gb, 8.12 Gb, 15.06 Gb, and 442.4 Mb whole-genome shotgun (WGS) sequencing data were generated for short reads and R9.4.1, R10.4.1 simplex, and duplex long reads, respectively (Supplemental Table S1; Ye et al. 2024). We first assembled the high-quality reference genomes for the eight samples using all R10.4.1 simplex and duplex reads, followed by a polishing phase using both long and short reads (see Methods). Eight circular bacterial chromosomes and 18 circular plasmids were obtained (Supplemental Table S2).
The new R10.4.1 reads had a 99% estimated modal read accuracy, outperforming the 97% in R9.4.1 reads (Supplemental Fig. S1). The median mapping accuracy for R10.4.1 reads was 98%, 2% higher than the R9.4.1 reads (Supplemental Fig. S2). This enhancement was particularly evident in homopolymer regions, where R10.4.1 reads achieved 85% accuracy, surpassing the 74% accuracy of R9.4.1 reads (Supplemental Figs. S2, S3). Nanopore R10.4.1 can produce a small amount (2%–7%) of duplex reads (Supplemental Table S1). These reads were self-corrected between forward and reverse strands and had even higher accuracy than normal R10.4.1 reads, nearly 99.9% (Supplemental Fig. S2).
For quality in bacterial genome assembly, we evaluated the efficacy of using solely R10.4.1 or R9.4.1 reads across a range of coverage from 10- to 120-fold (see Methods). Assemblies from sole R10.4.1 reads outperformed those from R9.4.1 in terms of genome completeness and indels proportion, but these advantages can also be achieved through short-read polishing (Supplemental Fig. S4).
Species-dependent efficacy in mitigating single nucleotide substitutions
Our analyses highlighted distinct patterns of single nucleotide substitutions (SNSs) in bacterial species. For some bacteria, such as B. cereus and S. enterica, additional short-read polishing did not provide further benefits in decreasing the SNS rate. Conversely, in the case of A. pittii, E. faecium, E. coli, and K. pneumoniae, the assemblies based solely on R10.4.1 reads consistently exhibited elevated SNS rates compared to the other assemblies (Fig. 1A). Although one might anticipate that random SNS errors would be rectified with increased long-read coverage, this was not observed for these bacterial assemblies. This trend suggested the presence of systematic bias within the reads. We postulate that the elevated SNS rates observed in these assemblies might be associated with unique genomic features, potentially species-specific k-mer compositions, or DNA modifications.
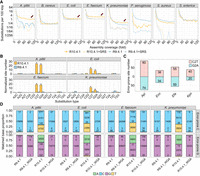
Bacterial DNA modifications influence substitutions in R10.4.1 read–based assemblies. (A) Substitution per 100 kbp of assemblies generated using different coverage of R10.4.1 and R9.4.1 reads, with or without high-quality short-read polishing. The x-axis shows the subsampled read coverage for ONT reads. (B) Normalized per-assembly counts of all 12 SNS types in A. pittii, E. faecium, E. coli, and K. pneumoniae assemblies generated from R10.4.1 (n = 35) or R9.4.1 (n = 35) reads. The R10.4.1 and R9.4.1 assemblies are based on subsampled read coverage from 40- to 100-fold. (*) P-value < 0.01, Student's t-test. (C) Identification of error-prone sites in four bacteria defined by C2T or G2A substitution frequencies exceeding two. (D) Proportions and counts of accurately and inaccurately mapped bases at error-prone C and G sites in four bacterial genomes, respectively. Mapping reads were obtained from R9.4.1, R9.4.1 WGA, R10.4.1, and R10.4.1 WGA. Notably, the C2T and G2A substitutions are more prevalent in R10.4.1 reads. (SRS) short-read sequencing, (Api) Acinetobacter pittii, (Eco) Escherichia coli, (Efa) Enterococcus faecium, (Kpn) Klebsiella pneumoniae.
Substitution types within assemblies exhibit consistent and systematic patterns
To confirm whether SNSs observed in the A. pittii, E. faecium, E. coli, and K. pneumoniae R10.4.1 assemblies were technical errors, we quantified all 12 SNS types to determine whether the errors were randomly distributed within the four bacterial assemblies (see Methods). In particular, the substitutions cytosine to thymine (C2T) and guanine to adenine (G2A) dominated the SNS landscape (Fig. 1B). The frequencies of both C2T and G2A were significantly larger than the simulated distribution (Supplemental Fig. S5). The pronounced prevalence of these specific substitutions, C2T and G2A, suggested that such SNS occurrences are not merely due to random basecalling discrepancies.
To rule out potential biases from the Medaka (V1.8.0) polishing process (https://github.com/nanoporetech/medaka), we additionally polished the assemblies utilizing a different long-read polisher, Racon (V1.5.0) (Vaser et al. 2017). The polishing outcomes still predominantly presented C2T and G2A substitution patterns (Supplemental Fig. S6), indicating that the errors observed cannot be attributed to the polishing process. We can conclude that the bias of substitution type enriched at C2T and G2A in the assemblies originated from technical errors in the R10.4.1 reads. From the SNS frequencies in the 35 assemblies derived from subsampled R10.4.1 reads, we identified the error-prone sites. Specifically, C2T or G2A with more than two occurrences was identified as an error-prone site for C or G, respectively (Fig. 1C; Supplemental Table S4).
Error-prone sites arise from bacterial DNA modifications
Given the distinctive SNS patterns observed in assemblies, we aimed to ascertain whether DNA modifications were the primary cause of the identified SNSs. To this end, we utilized the WGA sequencing approach to produce reads devoid of possible DNA modifications, serving as a control (Ni et al. 2023b).
Analysis of the correct and incorrect mappings at the error-prone C and G sites revealed that the R9.4.1 WGS and WGA reads exhibited similar accuracies, ∼98% (Fig. 1D). However, for the R10.4.1 WGS reads, a significant fraction of the C and G bases were misinterpreted as T (C2T) and A (G2A) (Fig. 1D; Supplemental Fig. S7). In contrast, very few substitutions were detected at the error-prone sites in R10.4.1 WGA sequencing, with 99% of the C and G being accurately basecalled (Fig. 1D). These observations strongly point toward base modifications in these four genomes as the culprits behind the SNS inconsistencies observed in R10.4.1 WGS and WGA reads.
A strand-specific mismatch pattern was observed at error-prone sites
In a deep dive into the distinctions between forward and reverse strands at error-prone sites in the four bacterial species, an intriguing pattern emerged. All the error-prone C sites across the genomes of the two bacteria (E. faecium and K. pneumoniae) exhibited sharp contrasts: forward reads exhibited an impressive mapping accuracy exceeding 99.9%, whereas the reverse reads plummeted below 20% accuracy (Fig. 2A,B). The scenario reversed for error-prone G sites, with low mapping accuracy for forward reads and high mapping accuracy for reverse reads (Fig. 2A,B). A similar result was also observed at the error-prone C and G sites within the A. pittii and E. coli genomes (Supplemental Fig. S8). Considering that forward C is equivalent to reverse G in the genome, as are the SNS patterns of C2T and G2A, error-prone C and G sites may arise from the same type of systemic error.
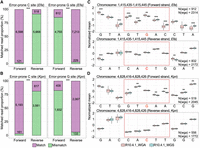
The error-prone sites arising from bacterial DNA modifications show strand bias in R10.4.1 reads. (A,B) Proportions and counts of accurately and inaccurately mapped forward and reverse read at error-prone C and G sites in E. faecium and K. pneumoniae, respectively. (***) P-values <1 × 10−30, Fisher's exact test. (C,D) Illustration of raw current signals at an error-prone G site in E. faecium and K. pneumoniae genome, respectively. Notably, significant differences highlighted between WGS and WGA reads are observed in the forward strand (upper panel), but not in the reverse strand (lower panel). (wgs) whole-genome shotgun, (wga) whole-genome amplification, (Efa) Enterococcus faecium, (Kpn) Klebsiella pneumoniae. (*) P-value < 0.05, (**) P-value < 0.01, and (***) P-value < 0.001, Student's t-test.
To further confirm whether the strand-specific mismatch pattern originated from DNA modification, we investigated the difference between the raw WGS and WGA current signals at the possible modified sites. By checking the signal around a forward error-prone G site in E. faecium, we found that the current signals in the WGS reads were significantly different from those in the WGA reads, while the reverse strand was less different (Fig. 2C). The same example can be found in K. pneumoniae (Fig. 2D). These two sites had a significant proportion of G2A SNSs in the forward reads but not in the reverse ones (Supplemental Fig. S9). From these examples, we believe that the unevenness of the raw signal disturbance between two DNA strands on a modified site is the cause of the strand-specific mismatch pattern. Moreover, the strand-specific mismatch pattern can be used to indicate DNA modification loci in the bacterial genome.
Bacterial DNA modifications can be identified through strand-specific mismatch patterns
With this idea, we developed a pipeline named “Hammerhead” to locate possible bacterial DNA modifications using the mapping accuracy between forward and reverse strands in Nanopore R10.4.1 reads. Briefly, after mapping the reads to the genome, the nucleotide difference indices between forward and reverse reads for each genomic site were calculated (Fig. 3A,B). Theoretically, the difference index for modification-free WGA reads should be zero. However, random errors still occurred in the WGA reads, creating a background-level difference index. To achieve a false discovery rate (FDR) <1 × 10−6 in all four WGA data sets, we obtained an empirical cutoff of 0.35 (Fig. 3C,D; Supplemental Fig. S10). By counting genomic sites with a difference index larger than 0.35 in WGS reads, we obtained 1820 sites in A. pittii, 563 sites in E. faecium, 1860 sites in E. coli, and 1758 sites in K. pneumoniae as potential modification sites. These sites included almost all the error-prone sites identified in the genome assemblies (Supplemental Fig. S11). The motifs linked with these sites in E. faecium and K. pneumoniae were mostly enriched as GATC, which is likely to be a 6mA or 5mC motif in bacterial genomes, confirming that the Hammerhead pipeline can locate DNA modifications using R10.4.1 reads (Fig. 4E,F). Moreover, the motifs of the plasmids inside the two bacteria were similar to those of the chromosome (Fig. 4E,F).
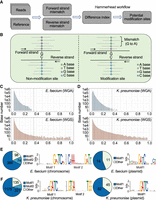
Bacterial DNA modifications can be identified by comparing the mapping accuracy between forward and reverse strands in R10.4.1 reads. (A,B) The workflow and demo of Hammerhead, which is designed to identify the potential modification sites based on the degree of nucleotide difference between forward and reverse reads. (C,D) The distribution of site difference index in WGA and WGS sequencing reads for E. faecium and K. pneumoniae, respectively. The WGA sequencing, representing random read errors, serves as a background filter. High discrepancies between forward and reverse strands in WGS reads suggest potential DNA modifications. A cutoff of 0.35 (FDR < 1 × 10−6 in WGA reads) is used here to identify possible DNA modification sites in WGS reads. (E,F) The motif was enriched from possible DNA modification sites identified by strand accuracy comparison for chromosome and plasmid sequences in E. faecium and K. pneumoniae, respectively. “GATC” is the dominating motif. Note: Only one motif was enriched from E. faecium plasmid (MEME E-value = 1.1 × 10−7). (FDR) false discovery rate.
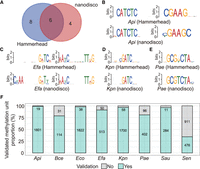
Validation of the potential modification sites from Hammerhead results. (A) The number of shared and unique motifs identified by Hammerhead and nanodisco. (B–E) The comparison of six shared motifs from two pipelines. (F) The proportion of overlap between two groups of modification units identified by Hammerhead and nanodisco. Each modification unit consists of a potential modification site and the surrounding bases (−4 bp to +4 bp). Note: Hammerhead was designed for R10.4.1 reads, while nanodisco was for R9.4.1 reads. (Api) Acinetobacter pittii, (Bce) Bacillus cereus, (Eco) Escherichia coli, (Efa) Enterococcus faecium, (Kpn) Klebsiella pneumoniae, (Pae) Pseudomonas aeruginosa, (Sau) Staphylococcus aureus, (Sen) Salmonella enterica.
To specify that strand-specific errors exist only in the bacterial sequencing reads, we applied Hammerhead to our human R10.4 data (Ni et al. 2023b). The difference index distributions between the R10.4 WGS and R10.4 WGA reads were similar (Supplemental Fig. S12), confirming that this pattern is bacterial-specific.
We then applied the Hammerhead pipeline to the other four bacteria to evaluate its performance. Potential modification sites were also identified in the four other bacteria (Supplemental Fig. S13). These sites in all eight bacterial species were evenly distributed inside the chromosomes or the plasmid (Supplemental Fig. S14). Moreover, the motifs were enriched in sequences near these potential modification sites (−10 bp to +9 bp) with Multiple Em for Motif Elicitation (MEME) E-values < 1 × 10−50 (Fig. 3E,F; Supplemental Figs. S15, S16). The plasmids in bacteria usually share a similar pattern as bacterial chromosomes (Supplemental Fig. S16).
nanodisco validates and supports the identified bacterial DNA modifications from Hammerhead
nanodisco (Tourancheau et al. 2021) is a well-recognized tool for detecting bacterial DNA modifications using the current signal difference between R9.4.1 WGA and WGS reads. Fortunately, we had access to the corresponding R9.4.1 data sets for all the bacterial samples. To validate these potential modification motifs and sites identified from Hammerhead, we utilized nanodisco to perform de novo identification of methylation.
Modifications in bacteria are consistently linked to specific motifs, with more than 95% of modification sequence motifs undergoing methylation (Casadesús and Low 2006; Wion and Casadesús 2006; Beaulaurier et al. 2019). Based on this rationale, the enriched motifs obtained from Hammerhead were compared with those derived from nanodisco to assess whether the identified motifs are indeed indicative of true DNA modifications. A total of 14 motifs were identified by Hammerhead, while 10 were detected by nanodisco. Six motifs were discovered by both approaches (Fig. 4A).
All the shared motifs (n = 6) or their reverse complements (RCs) exhibited current differences (Supplemental Fig. S17). Moreover, four of these motifs were predicted to be involved in 6mA methylation, including C6mATCTC in A. pittii, R6mAYCNNNNNNTTRG and CYA6mANNNNNNGRTY in E. faecium, and G6mATC in K. pneumoniae (Fig. 4B–D; Supplemental Fig. S18). Additionally, one motif, RTAGACGC (the RC of GCGTCTAY), in P. aeruginosa was identified as the 4mC methylation type (Fig. 4E; Supplemental Fig. S18). Although YGAAGC in A. pittii was not characterized as a specific methylation type (4mC, 5mC, or 6mA), we believe that it belongs to the A-base methylation category based on the significant disparity observed in the current signal profile of the first A base (Supplemental Figs. S17, S18).
There are eight motifs uniquely identified by Hammerhead. Among these motifs, GATC in E. coli is a well-known 6mA motif. However, this motif was missed in the nanodisco result. The reason might be attributed to the high frequency of CCWGG motif in E. coli genomes (Breckell and Silander 2022). nanodisco calculates P-values for each base using the Mann–Whitney U test to indicate the significance of the current signal difference between native DNA reads and the negative control. Only the top 2000 peaks of 5 bp smoothed P-values were selected for final motif enrichment analysis (Tourancheau et al. 2021). In the E. coli genome, the 5mC motif CCWGG had smaller P-values than other motifs (Breckell and Silander 2022) thus occupying most of the top 2000 peaks. As a result, the GATC motif could not be enriched using nanodisco using default parameters.
To further confirm whether nanodisco could validate our novel motif finding, we compared the differences in the current signals between WGA and WGS R9.4.1 reads using nanodisco (Supplemental Fig. S19). The differences among all the motifs were confirmed, indicating that these motifs could be identified by nanodisco by fine-tuning its cutoff. Additionally, the detection of six motif-related methyltransferases in their corresponding species strongly supported the occurrence of modifications within the motifs (Supplemental Tables S1, S5).
To further validate all potential modification sites identified from Hammerhead, a comparison was conducted at the site level with nanodisco. Specifically, potential methylation sites identified by Hammerhead with a difference index ≥0.35 were selected. For the nanodisco sites, the RDS files generated from the function of “difference” were used to obtain the site position and mean current differences. Sites with an absolute current difference >2 pA were selected for downstream comparison (Supplemental Fig. S20).
Considering that methylation can impact the current of bases near the modified sites, a methylation unit was employed to assess the overlaps between Hammerhead and nanodisco. The methylation unit consists of a 9-mer base unit centered on the potential methylation sites from Hammerhead or nanodisco. Based on the number of overlaps of these methylation units, the majority of modified sites identified by Hammerhead were confirmed by nanodisco in most bacterial species (>75% intersection), except for S. enterica (∼30% intersection) (Fig. 4F).
The 5mC motif GATC in S. aureus can be detected by decreasing the cutoff
Four motifs were identified by nanodisco but not Hammerhead (Fig. 4A; Supplemental Fig. S21). Among them, the SATSNNSNNSNNS motif seemed to be a false positive result. The bit scores are low for all bases in this motif, indicating low representation and high uncertainty during motif enrichment and typing analysis (Supplemental Fig. S21). Moreover, the difference in the current signal density between the WGA and WGS reads was mild for this motif (Supplemental Fig. S22). The other three motifs, RCCWGGHND, RCCWGGY, and KGATCADYHDNHWR, were predicted to be the 5mC methylation type based on the nanodisco results and the presence of motif-related methyltransferases (Supplemental Fig. S21; Supplemental Table S5).
There are two possible reasons for the false negative results for detecting DNA modification motifs using Hammerhead. The first issue is the cutoff issue. A cutoff difference index >0.35 was used to avoid false positives, with an empirical FDR < 1 × 10−6. The cutoff may be too harsh for low-frequency DNA modifications to be enriched. The other reason is that the motif and modification have been incorporated into training data sets for the current basecalling models. If so, the read accuracy around these motifs should be as high as that of other genomic regions in the WGS reads.
To confirm which of these mutations is the cause of the missing CCWGG and GATC motifs in Hammerhead, we performed a search for the two motifs within the corresponding genomes and examined whether there were any differences in the observed read accuracy. A noticeable decrease in read accuracy was observed for the GATC motif at the G-base and C-base (Supplemental Fig. S23), indicating that this motif could be identified by Hammerhead using the raw basecalling model. The strict cutoff value for the difference index (0.35) might explain the lack of identification of this motif in S. aureus. To validate whether the cutoff made a difference, 3821 sites with a difference index >0.1 (FDR < 1 × 10−3) were selected for downstream motif enrichment analysis. At this time, we identified three motifs, GAT5mC and two other motifs identified earlier, and corresponding methyltransferases were also detected (Supplemental Figs. S15C, S24; Supplemental Table S5).
The 5mC motif CCWGG can be retrieved by fine-tuning the basecalling model
In contrast, the base accuracy within the CCWGG motif in both E. coli and K. pneumoniae was ∼99%, which closely aligns with the expected accuracy for R10.4.1 reads (Supplemental Fig. S23). The basecalling model seems to have incorporated this modified motif into the training data set. To detect modifications within the CCWGG motif using Hammerhead, we needed to revert the model to increase strand-specific errors in the modified CCWGG motif.
The modification system in E. coli has been well studied. C5mCWGG has been associated with the dcm gene, while G6mATC has been associated with the dam gene (Marinus and Løbner-Olesen 2014). We searched for the known dam (NCBI Gene ID: 947893) and dcm (NCBI Gene ID: 946479) genes in our E. coli assembly. The presence of methylation within the CCWGG and GATC motifs was confirmed by the detection of both genes (Supplemental Fig. S25; Supplemental Tables S6–S8). Therefore, we chose E. coli data sets to test whether a new basecalling model could help Hammerhead identify methylation in the CCWGG motif.
We trained a “modification-aware” basecalling model for E. coli, starting from the super accuracy basecalling (SUP) model (dna_r10.4.1_e8.2_400bps_sup@v4.2.0), fine-tuned using methylation-free WGA reads (see Methods). To ensure that the “modification-aware” model does not have an extensive impact on WGS read quality, we first calculated the observed accuracy and mismatch proportion using rebasecalled E. coli WGS reads. The overall read accuracy, mismatch ratio, and homopolymer identification accuracy were consistent with those obtained using the SUP model (Supplemental Figs. S2, S26).
To assess the ability to detect modification sites using the new model, we applied the Hammerhead pipeline to the rebasecalled E. coli WGA and WGS reads. A similar distribution of difference indices was observed for the WGA reads as for the SUP model (Supplemental Fig. S27A). With respect to the WGS reads, a greater number (36,503 vs. 1860) of potential modification sites were identified with the same difference index cutoff of 0.35 (Supplemental Fig. S27A). These sites were selected for downstream motif enrichment analysis. Only two motifs were significantly enriched. One was GATC, and the other was CCWGG (Supplemental Fig. S27B). The significant increase in strand-specific errors around the modification sites enabled the detection of the 5mC modification in CCWGG with Hammerhead, which was missed using the SUP model.
Hammerhead exhibits comparable precision and reliability to ONT official software Dorado in the detection of 6mA methylation
After validating the ability for de novo bacterial methylation finding through comparison with nanodisco using R9.4.1 reads, we further benchmarked Hammerhead against a tool based on R10.4.1 reads to better showcase its performance. Dorado (https://github.com/nanoporetech/dorado) is the official software by ONT for identifying modifications using machine learning. It is important to note that Dorado is not a de novo bacterial methylation detection tool but specifically focuses on identifying 6mA or 5mC methylation.
We needed highly confident DNA methylation sites for benchmarking methylation detection methods using R10.4.1 reads. We used the sites from two 6mA motifs, which have been identified by nanodisco methylation motif typing with R9.4.1 reads. Only the motifs with the modified site prediction percentage higher than 85% were selected. These two motifs were expected to be nearly 100% modified in WGS reads, and 0% modified in WGA reads. The first motif is a type I motif, R6mAYCNNNNNNTTRG, in E. faecium (n = 1063), and the second motif is a type II motif, C6mATCTC, in A. pittii (n = 1318). The genomics sites containing the two motifs could be considered as true positives in WGS reads and true negatives in WGA methylation-free reads.
The distribution of the difference index from Hammerhead and methylation proportion from Dorado demonstrated that both methods performed well in identifying positive and negative sites (Supplemental Fig. S28). Furthermore, the performance of the two methods was illustrated using receiver operating characteristic (ROC) curves and precision-recall (PR) curves (Supplemental Figs. S29, S30). Specifically, the areas under the curve (AUCs) of the two methods for both motifs were >0.99 in the ROC curve (Supplemental Fig. S29). When it comes to the PR curve, Dorado achieved a higher AUCPR value (0.993) compared to Hammerhead (0.980) in the type I motif, while Hammerhead (0.999) outperformed Dorado (0.987) in the type II motif (Supplemental Fig. S30).
In summary, our Hammerhead successfully identified a total of 16 methylation-related motifs across eight representative bacteria using R10.4.1 reads (Table 1). Among them, 14 motifs were further validated either by the results obtained from nanodisco or through the detection of methyltransferases. Meanwhile, Hammerhead exhibited comparable precision and reliability to Dorado in the detection of 6mA methylation. These validations have provided evidence that Hammerhead is effective in accurately locating bacterial DNA modifications.
The summary of validated motifs
A high-accuracy model can be used for modification detection but not a fast model
In the context of converting the current signal into base information, ONT provided three types of basecalling models, namely, fast basecalling (FAST), high-accuracy basecalling (HAC), and SUP, for R10.4.1 reads. We developed the Hammerhead pipeline based on reads basecalled on the SUP model. To test whether the strand-specific error pattern was also present in the FAST and HAC models, the WGA and WGS reads of A. pittii were rebasecalled using the FAST and HAC models, respectively. We first compared the read quality using the basecalled results obtained from three different models. The results revealed that the reads from the SUP model exhibited the highest quality performance compared to those from the other models. On the other hand, reads from the FAST model showed the highest rate of inaccuracy (Supplemental Fig. S31).
We applied the Hammerhead pipeline to both FAST and HAC reads. In WGA reads, the FAST model identified more sites (3561) with a difference index over 0.35 compared to the HAC model, which identified only 57 sites (Supplemental Fig. S32A). The 3561 identified sites were false positives, which could impact the further motif enrichment from the Hammerhead result. The HAC model reads posed a similar WGA/WGS difference index distribution pattern, compared to the reads from the SUP model (Supplemental Figs. S10, S32B). Furthermore, from the potential modification sites detected using HAC model reads, we were able to enrich the same modification motifs as those identified using SUP model reads (Supplemental Fig. S32C). As for FAST model reads, only one motif could be enriched (Supplemental Fig. S33). To quantify the performance of Hammerhead across different models, ROC and PR curves were generated. The results of ROC and PR curves led to the same conclusion that the SUP model had the highest performance, with an AUC of 1.000 and AUCPR of 0.999 (Supplemental Figs. S29B, S30B). On the other hand, the FAST model exhibited the lowest performance, with an AUC of 0.968 and AUCPR of 0.969 (Supplemental Figs. S34, S35). Additionally, the HAC model demonstrated a performance close to the SUP model, with an AUC of 0.999 and AUCPR of 0.996 (Supplemental Figs. S34, S35). Based on these findings, we recommended using HAC reads and SUP reads to detect modifications using Hammerhead.
Duplex polishing resolves nucleotide substitution errors in assemblies
Although Hammerhead can be used to effectively identify modification sites in bacterial assemblies derived from R10.4.1 reads, the issue of substation errors (C2T and G2A) in these assemblies remains a concern when these assemblies are used as references. The “duplex basecalling” technique utilizes both the forward strand and reverse strand of a DNA fragment for sequencing and basecalling (Silvestre-Ryan and Holmes 2021). In such a situation, the Nanopore duplex reads can achieve even greater quality than normal ones (Supplemental Figs. S1–S3). We wanted to investigate whether error-prone sites in assemblies can be corrected by employing duplex reads for polishing.
To this end, we calculated the observed accuracy for each error-prone C and G site. For the R10.4.1 reads, the average observed accuracy at error-prone sites ranged from 54% to 65% (Fig. 5A; Supplemental Fig. S36). In contrast, the average observed accuracies for duplex reads exceeded 88%, except for that for E. faecium, which was ∼78%, suggesting the potential to correct substitution errors in assemblies by polishing with highly accurate duplex reads (Fig. 5A; Supplemental Fig. S36). We then polished the assemblies with duplex reads for E. faecium and K. pneumoniae. The SNS percentages in both bacterial genome assemblies decreased after polishing with duplex reads and were comparable to those polished with short reads (Fig. 5B,C). This result suggested that the SNSs caused by modification could be effectively corrected using duplex reads.
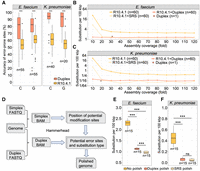
Polishing with duplex reads resolves DNA modification-induced errors in R10.4.1 assembly. (A) Read accuracy comparison for R10.4.1 and duplex reads at error-prone C and G sites. (B,C) Rates of SNS in R10.4.1 assemblies following short-read or duplex polishing in E. faecium and K. pneumoniae, respectively. The assembly only with duplex reads is also shown as a reference. (D) The workflow exclusively focuses on polishing the potential modification sites using duplex reads. (E,F) Comparison of SNS rates in R10.4.1 assemblies following targeted duplex and short-read polishing, focusing on 601 and 369 possible modification sites identified by Hammerhead in E. faecium and K. pneumoniae, respectively. (***) P-value < 1 × 10−8, Student's t-test. (SRS) short-read sequencing.
Considering that most error-prone sites are potential modification sites (Supplemental Fig. S11), we wanted to determine whether focusing solely on polishing those potential modification sites could effectively minimize SNS errors via duplex reads with limited yields. To validate this idea, the Hammerhead method was used to identify potential modification sites. Subsequently, the alignment files of the duplex reads were utilized to polish these sites (Fig. 5D). The duplex-read-polished assemblies exhibited SNS accuracy comparable to that of the short-read (50-fold) polishing, with a coverage of ∼20-fold (Fig. 5E,F). The duplex-read polishing process works well even for bacteria with limited duplex reads, such as A. pittii (10-fold) and E. coli (fourfold), in our study (Supplemental Fig. S37; Supplemental Table S3). The polishing pipeline showed a reliable reduction in SNS density and could be incorporated into the bacterial assembly pipeline using only R10.4.1 reads.
Discussion
In this work, we conducted a benchmark analysis to evaluate the quality of reads and assemblies generated from the most recent ONT R10.4.1 flow cell. Our results demonstrated that R10.4.1 reads were superior in terms of read accuracy and homopolymer detection compared with the R9.4.1 reads. The genome assembly is the consensus of sequenced reads. The quality of raw reads directly impacts genome assembly when using only nanopore reads. In theory, random errors in reads, with a proportion <50%, can be corrected during genome assembly or the self-polishing stage. Consequently, the accuracy of the assembly is usually higher than that of the raw reads. However, some error sites, particularly those with error proportions >50%, which were located within complex repetitive regions or close to other error sites, are difficult to be corrected using only nanopore reads. These errors persist in the final genome assembly. Our analysis revealed that bacterial assemblies obtained using only R10.4.1 reads were comparable to those obtained with short-read polishing in terms of completeness and number of indels. However, in certain R10.4.1 assemblies, the occurrence of base modifications results in a considerable number of error-prone sites with SNSs, particularly C2T and G2A substitutions. We hypothesized that the base methylation is the root cause of the enriched error substitution types. We confirmed this idea by comparing native reads with negative controls at error-prone C and G sites. Based on the novel finding of strand-specific mismatch patterns, we developed a method named Hammerhead to identify bacterial DNA modification. This method was further validated to work effectively and accurately.
The R10.4.1 reads have better sensitivity in bacterial DNA methylation detection than the R9.4.1 reads. The specific strand mismatch pattern was clearly evident in R10.4.1 reads, while not easily observed in R9.4.1 reads at error-prone C and G sites identified from R10.4.1 assemblies (Fig. 1D). The assembly errors from the sole R10.4.1 read have also been identified in different K. pneumonia strains (Lohde et al. 2024). The deviation of Hammerhead's difference index between WGS and WGA from R9.4.1 reads is less pronounced than that of R10.4.1 (Supplemental Figs. S28B, S38). In general, the improvement of read accuracy in R10.4.1 highlighted the technical errors, which are utilized in Hammerhead. The read quality of R10.4.1 is higher than that of R9.4.1 (Supplemental Fig. S2), resulting in lower random errors and reduced background noise, thereby making it easier to detect mismatches caused by methylation. Similarly, we have observed that the FDR for CpG methylation in the human genome is lower in R10.4 compared to R9.4.1, due to the higher read accuracy (Ni et al. 2023b). We could conclude that the difference between R10.4.1 and R9.4.1 reads is the combination effect from the differences of pore protein, motor protein, and basecaller. The library preparation protocols we used for R9.4.1 and R10.4.1 are both “ligation” kits (LSK110 and LSK114). The two kits use different types of motor proteins, which were ligated to DNA molecules during library preparation. The raw current signal is generated when DNA passes through the pore protein, and different protein structures have distinctive signal patterns (Deamer et al. 2016; Peraro and van der Goot 2016; Bhatti et al. 2021; Mayer et al. 2022). The motor protein, however, binds with the pore protein and controls the sequencing speed of the DNA, affecting the sampling frequency and the current pattern. The basecall process, which translates the current signal to nucleotide, is now handled by deep learning-based basecalling models. The model architecture for R9.4.1 and R10.4.1 is similar and embedded in basecallers. However, the models were trained with different data sets, which may also affect the error pattern (Seymour 2019; Xu et al. 2021).
Bacterial DNA methylation differs from that in mammals in both motifs and functions. In mammals, the most abundant form of DNA methylation is 5mC, and cytosine methylation mainly occurs within CpG dinucleotides (Petryk et al. 2020). DNA methylation in mammals is essential for embryonic development and cellular function (Greenberg and Bourc'his 2019; Dahlet et al. 2020; Grosswendt et al. 2020). In bacteria, there are three primary forms of methylation typing: 6mA, 4mC, and 5mC (Casadesús and Low 2006; Beaulaurier et al. 2019). The motifs of certain modified sites vary; for example, 6mA can be detected at GATC and CATCTC sequences. The primary function of bacterial DNA methylation is associated with restriction–modification (RM) systems (Casadesús and Low 2006; Loenen et al. 2013; Roberts et al. 2023). After validating Hammerhead's ability to detect bacterial DNA methylation, we applied our method to human cell line reads to investigate whether technical issues were present in human samples. The similar distribution of the difference index between WGS and WGA reads indicated that this specific-stand error pattern was not observed at human reads (Supplemental Fig. S12). The absence of this pattern may be due to the inclusion of human native DNA reads in the basecall training data sets.
Hammerhead is easy to implement for single-bacterial sequencing or metagenomic sequencing data because it requires only WGS reads, without the need to infer the raw current signal. We set a cutoff of 0.35 for the reference index, which is a 1 × 10−6 FDR based on our R10.4.1 WGA data sets. This cutoff may be too harsh for some species/strains that have a distinct k-mer composition compared to the four strains in our study. The harsh cutoff may result in the absence of some motifs, such as the GATC in S. aureus (Supplemental Fig. S24). We have provided the “cutoff” parameter in Hammerhead, with the default value of 0.35. Users can adjust the cutoff for their own data. Meanwhile, we have added an option to select the top N sites (with a default number of 2000) from Hammerhead. The top sites can be used for downstream motif enrichment analysis, mimicking the behavior of nanodisco, to avoid further cutoff adjustment.
Although the negative controls such as WGA reads are not required by Hammerhead, we have showcased the three usages of negative controls as follows. First, the additional negative controls can help to further validate the results. After the user gets the enriched motifs, a comparison of the difference index distribution of resulting motifs between native reads and methylation-free reads can be conducted to check whether there is a significant difference (Supplemental Fig. S28). Second, a negative control sample could be used to calculate the background noise for the difference index in a specific species. Based on the difference index distribution of control reads, users can adjust the cutoff based on the required FDR. Third, the methylation-free sequencing data can be used to retrain a “modification-aware” model, which can also increase the sensitivity of modification detection. We have demonstrated the process and effect of retraining in the detection of the CCWGG motif in our E. coli strain (Supplemental Figs. S10, S16B, and S27). However, the retraining process takes a longer time and requires GPU resources. For users who do not have WGA reads or GPU resources but still want the high sensitivity from the “modification-aware” basecall model, we have provided our “modification-aware” model trained from our four methylation-free bacterial data sets (https://doi.org/10.6084/m9.figshare.25858072).
Hammerhead was developed based on the stand-specific mismatch pattern. Hammerhead located the potential modifications by selecting genomic sites with a difference index higher than the cutoff. Based on those potential modification sites, the methylation motif could be enriched. However, one of the limitations is the quantitative measurement for individual sites. Hammerhead's difference index cannot directly reflect the proportion of DNA methylation at specific sites. Although the difference index can be used to compare the change of methylation between different samples (Supplemental Fig. S28), it cannot be used to measure the methylation proportion between different motifs. Bacterial DNA methylation is highly motif-driven, with over 95% of nucleotides being modified at methylation motifs, indicating an all-or-none case for a given methylation motif (Casadesús and Low 2006; Beaulaurier et al. 2019). We have demonstrated the all-or-none principle in two 6mA motifs, namely, C6mATCTC and R6mAYCNNNNNNTTRG (Supplemental Fig. S28). Both the difference index from Hammerhead and the methylation proportion from Dorado exhibited a near “one to zero” difference between the native reads and WGA reads (Supplemental Fig. S28), giving an insight that counting methylated or nonmethylated sites in one motif could be an alternative quantitative result from Hammerhead. Another limitation of Hammerhead is the lack of ability to judge the methylation type and the position of the modified site within the motif. To fill this gap, a comprehensive database of bacterial methylation like REBASE (Roberts et al. 2023) is considered to be included in the Hammerhead pipeline in the future.
Hammerhead, as an easy way to detect bacterial DNA methylation, can bring new biological insights and downstream technical applications. Bacterial DNA methylation, primarily mediated through the RM system, governs various cellular functions such as crucial aspects of virulence and metabolism. For instance, the newly identified type I RM system in Pseudomonas syringae has been shown to play pivotal roles in virulence and metabolic pathways, influencing critical processes like the secretion system, biofilm formation, and translational efficiency (Huang et al. 2024). The Hammerhead could quickly detect modification motifs in bacterial genomes, which adds a new layer of information to studying bacterial virulence and horizontal gene transfer (Tisza et al. 2023). Furthermore, there are vast technical applications from the downstream analysis of the Hammerhead result. For instance, in metagenomic sequencing, the contig binning now relies on GC content, k-mer composition, and length. In some complex environment samples, the traditional classification does not work well. We showed that the plasmid generally follows the methylation pattern of chromosomes for certain species (Fig. 3E,F; Supplemental Fig. S16), thus the methylation motif identified from Hammerhead can be further incorporated into the binning processing in metagenomic analysis (Tourancheau et al. 2021). Additionally, according to the outputs of Hammerhead, the strand-specific error pattern is absent in human reads, but present in bacteria reads (Fig. 3C,D; Supplemental Figs. S10, S12). This pattern could be used to distinguish the host and bacterial reads, and help to remove DNA contamination from bacterial DNA, or vice versa.
Hammerhead can de novo identify all potential methylation sites with one command, from FASTQ/FASTA input. The recent development of new deep learning models has enabled Dorado, the basecaller software of ONT, to identify bacterial methylation sites using raw current signal files (FAST5 or POD5). The raw signal files are roughly 5–10 times larger than the FASTQ/FASTA file, typically discarded by users soon after sequencing due to their size. Additionally, Dorado's modification calling requires the use of different deep learning models, separate from the basecalling model. For bacterial DNA modification, users must run three different models: one for basecalling, one for 6mA, and another for 5mC/4mC. These deep learning-based processes also require GPU-based computation, which significantly increases the time and resources needed.
In summary, our study offers valuable insights into methylation detection and solutions for bacterial genome assemblies using only ONT reads. In the R10.4.1 reads, mismatches between the forward and reverse strands were found to be linked with DNA modifications and thus can be used to identify possible modification sites. Building upon the identification of a strand-specific error pattern caused by the base methylation, we developed Hammerhead, the first tool enabling de novo DNA modification calling from ONT basecalled reads, eliminating the need to infer raw ionic currents. Hammerhead demonstrated strong performance in methylation detection, validated by the nanodisco, Dorado, and the presence of methyltransferases. Importantly, Hammerhead holds promise as a routine pipeline for identifying and polishing bacterial DNA methylation sites for a wide range of nanopore sequencing applications, such as genome assembly, metagenomic binning, decontaminating eukaryotic genome assembly, and functional analysis for DNA modifications.
Methods
Read processing
The raw Nanopore data in FAST5 format were subjected to basecalling using Guppy (V6.4.6) (https://community.nanoporetech.com), which employed the basecalling model file dna_r9.4.1_450bps_sup.cfg for R9.4.1 sequencing data and dna_r10.4.1_e8.2_400bps_sup.cfg for R10.4.1 sequencing data. To mitigate the presence of informatic chimeras and concatemeric reads, the split_on_adapter function from the duplex_tools (V0.3.2) (https://github.com/nanoporetech/duplex-tools) was employed with the following arguments: “‐‐allow_multiple_splits ‐‐trim_start 50 ‐‐trim_end 50”.
To acquire duplex reads, we utilized the duplex_tools (V0.3.2) package to initially extract potential paired-read information. Subsequently, we rebasecalled the raw current signal data in FAST5 format by employing the Guppy (V6.4.6) software guppy_basecaller_duplex function, which incorporates paired-read information.
To demultiplex the reads based on the barcoding information (SQK-NBD114-24 for R10.4.1 and EXP-NBD104 for R9.4.1), the guppy_barcoder function of Guppy (V6.4.6) was used with the following arguments: “‐‐enable_trim_barcodes ‐‐num_extra_bases_trim 3”.
To distinguish between R10.4.1 and duplex sequencing reads, we utilized SeqKit (V2.3.0) (Shen et al. 2016) and applied the grep command to filter reads based on their read ID. Finally, any reads with a length <200 bp were removed using SeqKit (V2.3.0) (Shen et al. 2016).
Read quality analysis and homopolymer identification
The reads for each bacterial species were aligned to the high-quality reference using minimap2 (V2.22) (Li 2021) with default arguments. To evaluate the accuracy of the different sequencing libraries, we computed several metrics for
each primary aligned read, including the substitution rate, insertion rate, and deletion rate, and observed read accuracy
using the following equations: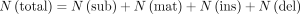
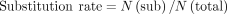
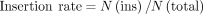
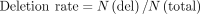
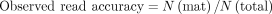 Here, N(sub), N(mat), N(ins), and N(del) are the number of substitutions, matches, insertions, and deletions, respectively, in each read. All the functions could
be achieved by the Giraffe (V0.1.0.14) (Liu et al. 2024).
Here, N(sub), N(mat), N(ins), and N(del) are the number of substitutions, matches, insertions, and deletions, respectively, in each read. All the functions could
be achieved by the Giraffe (V0.1.0.14) (Liu et al. 2024).
To assess the accuracy of homopolymer identification across multiple flow cells, we filtered out homopolymers with a length <3 bp and coverage lower than 3. The remaining homopolymers were analyzed using custom scripts, which calculated the proportion of homopolymers that achieved 100% match accuracy. All the scripts used for read quality benchmarking were packaged as the “observe” function of Giraffe (https://github.com/lrslab/Giraffe_View), and the plotting steps are available at https://github.com/lrslab/Bacteria-Multisequencing/tree/main/code/read_quality.
Genome assembly and read subsampling
To obtain a high-quality reference genome assembly for eight bacterial strains, we employed long-read sequencing data from both the R10.4.1 and duplex data sets as inputs for the different assembly programs: Flye (V2.9.2) (Kolmogorov et al. 2019), Canu (V2.2) (Koren et al. 2017), and Unicycler (V0.5.0) (Wick et al. 2017) (only for S. enterica). The default parameters were used in all the cases. Contigs were selected based on their length, circularity, and synteny across different assembly programs, yielding draft assemblies for each bacterial chromosome and plasmid(s). To correct substitution errors and indels in the draft assemblies, we performed polishing using both long-read and short-read data. Specifically, we used a two-step polishing approach, beginning with Racon (V1.5.0) (Vaser et al. 2017), to polish the drafts in three rounds using ONT R10.4.1 reads, followed by three rounds of polishing using Pilon (V1.24) (Walker et al. 2014) with short reads.
To evaluate genome assembly performance using long-read data from R10.4.1 and R9.4.1, we used 50-fold short-read data, and each long-read data set was subsampled at 10- to 120-fold intervals using Rasusa (V0.3.0) (Hall 2022). Given the potential variability in random subsampling, we performed the subsampling process five times for each long-read data set and coverage level, using seeds ranging from 1 to 5. In brief, we obtained a total of 960 assemblies by performing five repetitions for each long-read data set and coverage level (8 bacterial species × 2 data sets, R10.4.1 and R9.4.1, × 5 repetitions × 12 read coverages). We used Flye (V2.9.2) (Kolmogorov et al. 2019) for genome assembly and polished the resulting drafts with two rounds of medaka (V1.8.0) (https://github.com/nanoporetech/medaka) using the corresponding long-read data set, with or without three rounds of Racon (V1.5.0), with 50-fold short-read data for each bacterium at each read coverage. We compared the assemblies to the high-quality reference, which included the detection of indels and substitutions, using QUAST (V5.2.0) (Gurevich et al. 2013) with arguments of “‐‐min-alignment 1000 ‐‐min-identity 99”. To assess the genome completeness of each bacterium, we used benchmarking universal single-copy orthologs (BUSCO) (V5.4.3) (Manni et al. 2021) with the following databases: pseudomonadales_odb10 for A. pittii and P. aeruginosa; bacillales_odb10 for B. cereus and S. aureus; lactobacillales_odb10 for E. faecium; and enterobacterales_odb10 for E. coli, S. enterica, and K. pneumoniae.
To ensure the accuracy and reliability of our results, we implemented a stringent quality control approach that involved removing the highest and lowest values for each metric at each assembly coverage for each bacterium. We then calculated the average value from the remaining three values to represent the performance of each assembly method for each bacterium. All the codes for processing temperature files are available at https://github.com/lrslab/Bacteria-Multisequencing/tree/main/code/genome_assembly.
Single nucleotide substitution error statistics and current signal comparison
To determine the frequencies of the different substation types, we compared the 35 drafts at the chromosome level assembled from the long-read data (R10.4.1 or R9.4.1) at read coverage ranging from 40- to 100-fold by Quast (V5.2.0) (Gurevich et al. 2013) for four bacteria, namely, A. pittii, E. faecium, E. coli, and K. pneumoniae. The codes are available at https://github.com/lrslab/Bacteria-Multisequencing/tree/main/code/mismatch_fre. Only the sites with coverage over two were considered error-prone.
To count the number of matched base types at those error-prone sites, we utilized minimap2 (V2.22) (Li 2021) to align the four data sets, including R9.4.1, R9.4.1 WGA, R10.4.1, and R10.4.1 WGA, against the reference for K. pneumoniae, E. faecium, A. pittii, and E. coli. The sorted BAM files were subsequently input to the mpileup function from SAMtools (V1.17) (Danecek et al. 2021) with arguments of “‐‐no-output-ends ‐‐no-output-ins ‐‐no-output-ins ‐‐no-output-del ‐‐no-output-del” to calculate the mapped proportion of A, C, G, and T bases at these error-prone sites. The codes can be found at https://github.com/lrslab/Bacteria-Multisequencing/tree/main/code/modification_test.
To directly compare the difference in the current signal between WGS and WGA reads, we used f5c (V1.2) (Gamaarachchi et al. 2020) to resquiggle the reads with the reference and visualized them via nanoCEM (Guo et al. 2024).
Potential modification site identification and motif enrichment with Hammerhead
To determine the sites where the proportion of mapped bases differed between the forward strand and reverse strand, we used
a self-defined index to reflect this difference at a single site and calculated the function via the software Hammerhead (https://github.com/lrslab/Hammerhead).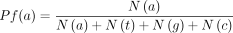
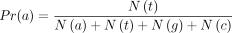
Here, N(a), N(t), N(g), and N(c) represent the number of mapped occurrences of the bases A, T, G, and C, respectively, in the forward strand at a site in
the reference sequence. Given that the DNA strands are RCs, N(a), N(t), N(g), and N(c) represent the number of mapped occurrences of the bases T, A, C, and G in the reverse strand at the same site in the reference.
Pf(a) and Pr(a) represent the proportions of mapped occurrences of base A in the forward strand and reverse strand, respectively. Additionally,
we calculated the proportions of three other bases (T, G, and C) via the same method. Notably, only the sites with a depth
>50 and a depth in forward and reverse strands >25 were retained for downstream analysis. Only the reads with a mapping quality
(mapQ) >30 were used for counting.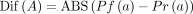 Then, we can calculate the absolute difference in the A bases in the two strands by using the proportion in the forward strand
minus that in the reverse strand. Moreover, we calculated the absolute differences in T, G, and C bases via the same method.
Then, we can calculate the absolute difference in the A bases in the two strands by using the proportion in the forward strand
minus that in the reverse strand. Moreover, we calculated the absolute differences in T, G, and C bases via the same method.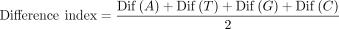
Finally, to calculate the difference index, we sum the absolute differences in A, T, G, and C bases and divide the sum by two.
Considering the presence of random errors during sequencing, a cutoff is needed to filter these background noises. WGA reads, which contain no methylation information, were regarded as negative controls. The differences in expression between the four bacterial WGA data sets were calculated by Hammerhead. To estimate the FDR for the WGA reads, we inferred the distribution of difference indices for the four data sets. The FDR was calculated based on the number of sites over the cutoff divided by the total number of sites. The sites over the cutoff can be considered false positives caused by random errors rather than methylation in WGA reads. To limit the number of false positives to <10 in WGA reads, a cutoff of 0.35 was selected to achieve an FDR < 1 × 10−6. Therefore, the sites with a difference index >0.35 in WGS reads were regarded as potential modification sites.
To identify regular motifs, we selected sequences near the potential modified sites (−10 bp to +9 bp) for motif enrichment. The MEME (V5.5.3) (Bailey et al. 2015) was utilized to identify the motif with the command “meme inputFile.fa -dna -oc . -nostatus -time 14400 -mod zoops -nmotifs 10 -minw 4 -maxw 16 -objfun classic -revcomp -markov_order 0”.
Methylation identification and motif enrichment using nanodisco
nanodisco (V1.0.3) was utilized to identify the modifications in the R9.4.1 WGA and WGS data sets. The functions “process,” “chunk_info,” “difference,” “merge,” “motif,” and “characterize” from nanodisco were used for our eight R9.4.1 bacterial data sets, all with default parameter as the detailed tutorial in the nanodisco documentation (https://nanodisco.readthedocs.io/en/latest/detailed_tutorial.html).
To select the sites with an absolute value of current signal difference >2 pA, the information in the RDS file was processed using the readRDS function of R. These raw RDS files are available at https://doi.org/10.6084/m9.figshare.24298774.
Intersection comparison of methylation units between Hammerhead and nanodisco
Considering that the difference in the current signal and basecalling error may be caused by the nearby modified base (Laszlo et al. 2013; Rand et al. 2017; Wick et al. 2019; Tourancheau et al. 2021), the potential modification site, plus four 5′ and 3′ flanking sites, was considered a modification unit (9 bp). We subsequently conducted an intersection analysis between the modification units identified by Hammerhead and nanodisco using BEDTools (V2.18) with the command “bedtools intersect -wa -a A.bed -b B.bed | sort | uniq -c”.
Methylation metric comparisons between WGS and WGA reads in motifs
To assess the performance of Hammerhead and Dorado in 6mA methylation calling, the metrics between WGS and WGA in two methylation motifs, C6mATCTC and R6mAYCNNNNNNTTRG were used to compare. For Hammerhead, the 15-mer methylation units were chosen, with the center positioned on the 6mA bases and spanning 7 bp upstream and downstream. The maximum value of the difference index was considered as the representative for each unit in WGS reads. Similarly, representatives in WGA reads were selected based on the positions of sites with the maximum values, ensuring that the prediction values were chosen from the same site. For Dorado, the prediction values in both WGS and WGA data were selected based on the positions of the modified 6mA sites.
A methylation-aware basecalling model was retrained using E. coli WGA reads
The new basecalling model was fine-tuned from the original DNA super accuracy model (SUP) using Bonito (V0.7.2) (Seymour 2019) with the following parameters: “‐‐epochs 40 ‐‐lr 5e-4 ‐‐batch 32 ‐‐pretrained dna_r10.4.1_e8.2_400bps_sup@v4.2.0”. To prepare the input SAM file for training, the parameter “save-ctc” must be enabled in the Bonito basecaller.
The fine-tuned methylation-aware model for E. coli can be downloaded from the Hammerhead GitHub repository (https://github.com/lrslab/Hammerhead). The new model was subsequently used for rebasecalling our E. coli WGS and WGA reads.
Genome polishing at potential modification sites with duplex reads
To validate whether polishing only the potential modification sites with duplex reads can reduce the substitution error caused by modification, we used 15 assemblies with 40-, 50-, and 60-fold assembly coverage to compare substitution rates in A. pittii, E. faecium, E. coli, and K. pneumonia, respectively. First, all the potential modification sites with a difference index >0.35 were selected. The BAM file generated from duplex reads mapped against the reference was subsequently used to call the pileup file for these potential modification sites. Ultimately, the decision to rectify or preserve the site would be made based on the proportion accurately depicted in the mapping. The pipeline can be found in the documentation (https://hammerhead-documentation.readthedocs.io/en/latest/#assemblies-polish).
Data access
The whole-genome amplification (WGA) data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA980403. The Hammerhead software, including modification sites finding and duplex-read polish, are available as Supplemental Code and at GitHub (https://github.com/lrslab/Hammerhead). All the processed files and other analysis scripts used in this study are available as Supplemental Material and on GitHub (https://github.com/lrslab/Bacteria-Multisequencing).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Yating Xu, Kaichao Chen, Qiao Hu, and Wai Chi Chan for providing us with valuable bacterial samples. This work was supported by the Early Career Scheme from the Research Grants Council of the Hong Kong Special Administrative Region, China (CityU 21100521); the Hong Kong Health and Medical Research Fund (project number 08194126); the Guangdong General Research Fund (project number 9240054) from the Natural Science Foundation of Guangdong Province; new Research Initiatives support from City University of Hong Kong (project number 9610497) to R.L.; the Theme-based Research Scheme (T11-104/22-R) to S.C.; the Hetao Shenzhen-Hong Kong Science and Technology Innovation Cooperation Zone Shenzhen Park Project (HZQB-KCZYZ-2021017); and the City University of Hong Kong Project (project number 9680217 and number 9678223) to M.Y.
Author contributions: X.L.: Conceptualization, methodology, software, validation, formal analysis, investigation, data curation, writing (original draft, review, and editing); Y.N.: Methodology (whole-genome amplification, nanopore library construction, and sequencing) and writing (review and editing); L.Y.: Methodology (DNA extraction, short-read library construction, and short-read sequencing), writing (review and editing); Z.G.: Writing (review and editing); L.T.: Writing (review and editing); J.L.: Writing (review and editing); M.Y.: Supervision, funding acquisition, writing (review and editing); S.C.: Supervision, funding acquisition, writing (review and editing); R.L.: Conceptualization, methodology, validation, investigation, writing (original draft, review, and editing), supervision, and funding acquisition.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279012.124.
- Received January 30, 2024.
- Accepted September 25, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








