
Enhanced detection of RNA modifications and read mapping with high-accuracy nanopore RNA basecalling models
- Gregor Diensthuber1,2,5,
- Leszek P. Pryszcz1,5,
- Laia Llovera1,
- Morghan C. Lucas1,2,
- Anna Delgado-Tejedor1,2,
- Sonia Cruciani1,2,
- Jean-Yves Roignant3,4,
- Oguzhan Begik1 and
- Eva Maria Novoa1,2
- 1Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Barcelona 08003, Spain;
- 2Universitat Pompeu Fabra, Barcelona 08003, Spain;
- 3Center for Integrative Genomics, Faculty of Biology and Medicine, University of Lausanne, 1015 Lausanne, Switzerland;
- 4Institute of Pharmaceutical and Biomedical Sciences, Johannes Gutenberg-University Mainz, 55128 Mainz, Germany
-
↵5 These authors contributed equally to this work.
Abstract
In recent years, nanopore direct RNA sequencing (DRS) became a valuable tool for studying the epitranscriptome, owing to its ability to detect multiple modifications within the same full-length native RNA molecules. Although RNA modifications can be identified in the form of systematic basecalling “errors” in DRS data sets, N6-methyladenosine (m6A) modifications produce relatively low “errors” compared with other RNA modifications, limiting the applicability of this approach to m6A sites that are modified at high stoichiometries. Here, we demonstrate that the use of alternative RNA basecalling models, trained with fully unmodified sequences, increases the “error” signal of m6A, leading to enhanced detection and improved sensitivity even at low stoichiometries. Moreover, we find that high-accuracy alternative RNA basecalling models can show up to 97% median basecalling accuracy, outperforming currently available RNA basecalling models, which show 91% median basecalling accuracy. Notably, the use of high-accuracy basecalling models is accompanied by a significant increase in the number of mapped reads—especially in shorter RNA fractions—and increased basecalling error signatures at pseudouridine (Ψ)- and N1-methylpseudouridine (m1Ψ)-modified sites. Overall, our work demonstrates that alternative RNA basecalling models can be used to improve the detection of RNA modifications, read mappability, and basecalling accuracy in nanopore DRS data sets.
RNA modifications are chemical groups that decorate RNA molecules and are collectively referred to as the epitranscriptome (Saletore et al. 2012). They play a crucial role in a wide variety of molecular processes, including splicing, translation, and RNA stability (Frye et al. 2016; Roundtree et al. 2017; Gilbert and Nachtergaele 2023). Our understanding of the epitranscriptome has dramatically increased in the past years with the development of novel methods that can chart the modification landscape in a transcriptome-wide fashion (Helm and Motorin 2017; Wan et al. 2022; Lucas and Novoa 2023). High-throughput sequencing–based methods were the first to revolutionize the field of RNA modification detection by providing high-accuracy maps for N6-methyladenosine (m6A), 2′-O-methylation (Nm), N4-acetylcytosine (ac4C), and pseudouridine (Ψ), among others (Dominissini et al. 2012; Meyer et al. 2012; Carlile et al. 2014; Schwartz et al. 2014; Marchand et al. 2016, 2018; Sas-Chen et al. 2020). More recently, nanopore direct RNA sequencing (DRS) has emerged as a promising alternative for RNA modification detection owing to its ability to directly detect RNA modifications, without the need for prior chemical treatment, in native full-length RNA reads, thus permitting isoform-specific RNA modification analyses (Novoa et al. 2017; Garalde et al. 2018; Begik et al. 2022; Jain et al. 2022).
In nanopore DRS, single-stranded RNA molecules are threaded through protein pores, embedded within a nonconductive membrane, to which a voltage is applied. This causes the RNA molecules to migrate through the pores, leading to changes in the current intensity signal that are distinct for the four canonical nucleobases. More importantly, systematic changes to the current intensity signal and/or downstream signals are also caused by RNA modifications, providing a basis for modification detection with nanopore sequencing (Garalde et al. 2018; Workman et al. 2019). In the last few years, DRS has been successfully applied to detect diverse RNA modification types, including m6A (Liu et al. 2019; Lorenz et al. 2020; Gao et al. 2021; Leger et al. 2021; Pratanwanich et al. 2021; Hendra et al. 2022), Ψ (Begik et al. 2021; Huang et al. 2021; Tavakoli et al. 2023), Nm (Begik et al. 2021; Stephenson et al. 2022), N7-methylguanosine (m7G) (Smith et al. 2019; Delgado-Tejedor et al. 2023), and inosine (I) (Nguyen et al. 2022), and in a variety of RNA species, such as long noncoding RNAs, viral RNAs, mRNAs, and tRNAs (Liu et al. 2019; Workman et al. 2019; Kim et al. 2020; Leger et al. 2021; Lucas et al. 2024). Identification of modified RNA bases has been typically achieved either (1) by measuring changes in the current signal caused by the presence of modified nucleotides (Stoiber et al. 2017; Leger et al. 2021; Pratanwanich et al. 2021; Acera Mateos et al. 2024) and/or (2) by identifying nonrandom basecalling “errors” that occur at positions where the RNA is modified (Furlan et al. 2021; Jenjaroenpun et al. 2021; Liu et al. 2021; Piechotta et al. 2022).
A central step in nanopore sequencing is the basecalling process, which involves converting the raw electrical signal data generated by the nanopore into a string of nucleotide bases that represent the RNA sequence. To convert the current intensity signal into a nucleotide sequence, the basecalling algorithm requires a basecalling model. These models are typically trained with synthetic and/or in vivo native RNA reads and can predict the four canonical bases (A, C, G, and U in the case of RNA). Current basecalling models, available through the Guppy basecalling software, are not specifically trained to predict RNA modifications. Therefore, when the model encounters a modified RNA nucleotide, it may call the wrong base (mismatch error), not call any base (deletion error), or insert a small piece of sequence (insertion error) at the modified site (Liu et al. 2019; Begik et al. 2021; Jenjaroenpun et al. 2021). Several software exploit this feature and use these basecalling “errors” to identify RNA modifications in DRS data sets (Parker et al. 2020; Jenjaroenpun et al. 2021; Liu et al. 2021; Piechotta et al. 2022; Abebe et al. 2022b). The use of basecalling errors to detect RNA modifications is well suited to detect certain RNA modification types, such as Ψ, which causes a very strong U-to-C mismatch signature at the modified site (Begik et al. 2021; Huang et al. 2021; Tavakoli et al. 2023). In contrast, other RNA modification types, such as m6A, yield relatively modest basecalling “error” signatures (Begik et al. 2021), leading to a relatively high number of false positives and false negatives when predicting them in a transcriptome-wide fashion (Liu et al. 2019).
Basecalling “errors” in nanopore sequencing data can arise because of various factors, such as homopolymeric regions, RNA secondary structures, or helicase missteps (Noakes et al. 2019; Shaw et al. 2023; Liu-Wei et al. 2024). However, a major factor affecting the basecalling “error” patterns is the RNA basecalling model of choice and, more specifically, the data used to train this model. For example, the “default” RNA basecalling model (rna_r9.4.1_70bps_hac) used in Guppy was trained with native mRNA sequences from diverse species and thus contains a significant number of m6A-modified sites in the reads that were used to train the model. Consequently, this basecalling model learned to predict both “m6A” and “A” as “A,” leading to low frequencies in basecalling “errors” when an m6A modification is encountered compared with when the model encounters other RNA modification types that were not included in the training data.
Here, we hypothesized that the detection of RNA modifications could be improved at the stage of basecalling by using alternative basecalling models. We trained novel RNA basecalling models and examined their performance in terms of basecalling accuracy (i.e., sequence identity), read mappability, and RNA modification detection ability. In addition to their applicability for RNA modification detection, these alternative “higher-accuracy” basecalling models may be used for enhanced mRNA vaccine quality control as well as for improved SNP detection, identification of splice sites, and transcript annotation when using DRS data.
Results
RNA basecalling model choice affects both per-read accuracy and RNA modification “errors”
To test whether the use of alternative basecalling models could enhance the detection of RNA modifications using alignment feature-based tools, we trained novel RNA basecalling models using two different approaches (Supplemental Fig. S1A). The first approach consisted in training a model using fully unmodified RNA sequences generated via in vitro transcription (“IVT” model), which we reasoned should lead to increased “error” signatures at modified bases present in mRNAs, compared with the default basecalling model. The second approach consisted in generating a “superaccuracy” RNA basecalling model (“SUP” model) (see Supplemental Table S1), based on the latest CTC-CRF (bonito) DNA model architecture, achieving 96% accuracy for DNA basecalling, which we reasoned should show increased basecalling accuracy for RNA (see Methods) (Supplemental Fig. S1B) and, consequently, improved detection of RNA modifications owing to an improved signal-to-noise ratio. Next, we assessed their effect on both basecalling accuracy and on basecalling “error” patterns, which can be used as proxy for RNA modifications (Fig. 1A).
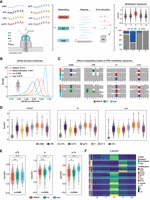
Benchmarking RNA basecalling models and their effect on basecalling accuracy and RNA modification detection. (A) Schematic overview of the in vitro “curlcakes” used in this work, depicting the seven different modification types that were sequenced (m6A, m5C, hm5C, ac4C, Ψ, m1Ψ, and m5U) and the three RNA basecalling models benchmarked (default, IVT, and SUP). To assess the performance of tested RNA basecalling models, error signatures (mismatch, deletion, and insertion frequency) were used. (B) Comparison of basecalling accuracies obtained from each RNA basecalling model on human transcriptomic data (see Methods). The curve represents the density of the per-read identities for each model. Solid lines represent Guppy, and dashed lines represent Dorado as basecalling software. We would like to note that a subset of reads from the HEK293T samples used here was also used for training our “SUP” model (see Methods); however, see also Figure 4A, which shows similar accuracy for SUP model in other species/data sets not included in model training. (C) IGV snapshots showing the effect of using either default, IVT, or SUP models on unmodified (UNM) or modified (m6A, Ψ, and m1Ψ) “curlcake” sequences for two positions each (top and bottom row). Asterisks indicate modified sites. Positions at which the mismatch frequency exceeds 0.2 are colored, and deletions are visualized as drop in coverage. (D) Comparison of the summed errors (mismatch, deletion, and, insertion frequency) for all 5-mers with a central modified base for the three tested basecalling models (default, IVT, and SUP). (E) Box plots showing the delta summed error for 5-mers of selected modifications (m6A, Ψ, and m1Ψ) at the central (modified) position. For equivalent plots of the remaining RNA modifications tested (m5C, hm5C, ac4C, and m5U), see Supplemental Figure S2B. Statistical analysis was performed using a two-sided nonparametric Wilcoxon test with “default” as the reference group. Results were corrected for multiple-hypothesis testing using the Benjamini–Hochberg procedure to obtain adjusted P-values: (ns) P > 0.05, (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001, (****) P ≤ 0.0001. The sample size reported by n represents the number of 5-mers contributing to each box plot per panel. For D and E, the box is limited by the lower-quartile Q1 (bottom) and upper-quartile Q3 (top). Whiskers are defined as 1.5 × IQR with outliers represented as individual dots. (F) Comparison of the median delta summed error (=SumErrMOD − SumErrUNM) for centrally modified (position 0) 5-mers. A Z-score normalization per modification (see Methods) was performed to highlight the effect that different basecalling models have on the obtained delta summed error. Unscaled results are reported in Supplemental Figure S2B.
We compared the basecalling accuracy of both models on human transcriptomic data, finding that the SUP model was the most accurate out of the three models tested, reaching a median read accuracy of 97% on human cells (Fig. 1B), whereas the default and IVT models only showed 91% and 88% accuracy, respectively. Additionally, we included the most accurate model for the newest Oxford Nanopore Technologies (ONT) basecaller Dorado (default model) provided for RNA002 chemistry. The Dorado model reached a median accuracy of 94%, suggesting that our SUP model using Guppy as basecalling software is currently the most accurate basecaller for data generated with RNA002 chemistry (Fig. 1B). Furthermore, we did not observe any significant differences in runtime or CPU usage when comparing the default model to our custom models (Supplemental Fig. S1C,D).
We then examined how basecalling “error” signatures produced by m6A, m5C, hm5C, ac4C, Ψ, m1Ψ, and m5U in synthetic “curlcakes” (Liu et al. 2019) would vary depending on the RNA basecalling model choice (default, IVT, or SUP) (Fig. 1C; see also Supplemental Fig. S1E). Of note, “curlcakes” are synthetic constructs that contain all possible 5-mers, thus allowing the study of the effect in all possible sequence contexts that affect the basecalling process. To this end, we employed per-position mismatch, deletion, and insertion errors, which we consider jointly as summed errors (SumErr; see Methods). Our analyses revealed that SumErr values followed similar trends irrespective of the basecalling model of choice, with pseudouridine modifications (Ψ and m1Ψ) giving the highest error values and cytidine modifications (m5C and hm5C) giving the lowest basecalling errors overall (Fig. 1D). Moreover, we observed that SumErr values in unmodified reads were significantly decreased when basecalled with the SUP model (Supplemental Fig. S2A), in agreement with the increased accuracy of the model.
We then assessed the ability of each model to detect RNA modifications in the form of basecalling errors. For this purpose, we computed the delta summed error (ΔSumErr = SumErrMOD − SumErrUNM) for each position, model, and modification type, which corrects for the different background error rates of each model on a per-5-mer basis. Our results showed that, for most modifications examined—and especially for m5U, Ψ, and m1Ψ—the ΔSumErr was significantly increased when using the SUP model, relative to default (Fig. 1E,F; see also Supplemental Fig. S2B), indicating the signal-to-noise ratio was increased and thus confirming the hypothesis that the use of alternative basecalling models can improve our ability to detect RNA modifications in DRS data. In contrast, the use of the IVT model did not increase the ΔSumErr for most RNA modifications, with the exception of m6A basecalling errors, which were modestly, yet significantly, increased when using the IVT model, compared with the default (Fig. 1E,F), in agreement with our hypothesis.
In addition, we noticed that all RNA modifications, regardless of the basecalling model, showed increased basecalling errors at the modified site (position 0), with the exception of m5C and hm5C, which showed increased errors at position +1, in agreement with previous reports (Begik et al. 2021). Moreover, we observed that for m6A, the increase in “error” using the IVT model compared with the default model was mainly owing to high deletion signatures at the modified site (Supplemental Fig. S2C,D). On the other hand, the increased signal in the SUP model to detect Ψ and m1Ψ modifications was caused by an increase in the global U > C mismatch frequency in SUP, compared with the default (Supplemental Fig. S2C,D; see also Supplemental Fig. S3). Taken together, these results show that the SUP and IVT basecalling models achieve a stronger signal-to-noise ratio by either changing the error signature (IVT model for m6A) or improving previously observed patterns (SUP model for Ψ and m1Ψ).
IVT model outperforms other models in detecting m6A modifications in DRACH contexts
m6A is the most prevalent epitranscriptomic mark in mammalian mRNA molecules, with an average of one to three sites per transcript (Dominissini et al. 2012; Meyer et al. 2012). Thus, the use of native RNA molecules as a training data set for a basecaller (as is the case for both default and SUP models) (see Supplemental Fig. S1) will lead to basecalling models learning to interpret both “m6A” and “A” as “A,” because the training data contain large amounts of m6A-modified residues, whereas its output is limited to the four canonical nucleobases (A, C, G, and U). For this reason, we speculated that the relative basecalling “error” rates in our IVT model should be particularly improved in those sites that naturally contain m6A (DRACH, where D denotes A, G, or U; R denotes A and Gl; and H denotes A, C, or U) and are therefore masked in “default.”
To investigate this, we independently examined the performance of the three models in DRACH and non-DRACH motifs. Indeed, we observed that the modest increase in IVT model performance at m6A-containing sites (Fig. 1E) was significantly stronger when only DRACH sites were taken into account (Fig. 2A), in agreement with our hypothesis. A detailed examination of which individual 5-mers were contributing to this phenomenon revealed that certain DRACH 5-mers showed no or only mild improvement, whereas others were strongly enhanced in their signal-to-noise ratio (Fig. 2B; see also Supplemental Fig. S4A). Notably, the largest improvement in the signal difference was observed for GGACT (Fig. 2B; see also Supplemental Fig. S4B), which is the most prevalent m6A-motif observed in mammalian species (Liu et al. 2020), suggesting that mammalian native RNA was most likely used to train the “default” basecalling model.
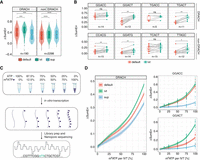
The effect of custom basecalling models on biologically relevant m6A motifs and at different m6A stoichiometries. (A) Comparison of the delta summed errors (ΔSumErr = SumErrMOD − SumErrUNM) at the central modified position for 5-mers that fall within the DRACH motif (D = A, G, or U; R = G or A; H = A, C, or U) and others (non-DRACH). Statistical analysis was performed using a two-sided nonparametric Wilcoxon test with “default” as the reference group. (B) Comparison of the ΔSumErr for individual motifs that are DRACH and non-DRACH. Each dot represents a single observation of that 5-mer within the “curlcake” sequences. To compare the relationship between individual 5-mers, a paired two-sided Wilcoxon test was performed. (C) Schematic representation of the experimental approach to obtain “curlcakes” at different m6A stoichiometries. ATP and m6ATP were mixed at different ratios and used for the in vitro transcription of “curlcakes” with the remaining NTPs, followed by poly(A)-tailing, library preparation, and direct RNA sequencing (DRS). (D) Comparison of error-signature amplitude at different modification stoichiometries between 5-mers that fall within the DRACH motif. The corresponding plot for non-DRACH motifs is reported in Supplemental Figure S5B. Lines were fitted using locally estimated scatterplot smoothing (loess), and lightly shaded areas correspond to the 95% confidence interval. For A,B the box is limited by the lower-quartile Q1 (bottom) and upper-quartile Q3 (top). Whiskers are defined as 1.5 × IQR with outliers represented as individual dots. All statistical tests were corrected for multiple hypothesis testing using the Benjamini–Hochberg correction, and adjusted P-values are displayed: (ns) P > 0.05, (*) P ≤ 0.05, (**) P ≤ 0.01.
Finally, we examined the sensitivity of the different basecalling models with varying modification stoichiometries. To this end, we generated “curlcake” in vitro transcripts at varying modification levels by mixing ATP and m6ATP at different ratios (see also Methods) (Fig. 2C). Our results showed that even at the lowest modification level (12.5%), the IVT model was able to generate a significantly increased error signature at DRACH-bearing 5-mers compared with the default model, which was especially evident in the GGACT context (Fig. 2D; see also Supplemental Fig. S5A–C).
Improved in vivo detection of m6A-modified sites in human samples using the IVT model
Next, we examined whether the IVT model would improve the detection of modified sites in vivo using third-party tools for the detection of differentially modified m6A sites. Therefore, we used publicly available nanopore DRS data from HEK293T wild-type and Mettl3−/− cells, which are incapable of placing the methyl mark (Pratanwanich et al. 2021; Liu et al. 2023). We basecalled the data using our three basecalling models, mapped the reads to the reference genome, and finally used ELIGOS2 (Jenjaroenpun et al. 2021), a tool developed to identify RNA modifications through pairwise comparison of error profiles, to detect m6A sites (Fig. 3A). Comparison of the control and Mettl3−/− samples using ELIGOS2 revealed a similar number of sites detected when basecalling the data using the default (4859 m6A sites) and IVT model (4979 m6A sites) (Supplemental Table S3), showing that the use of the IVT model modestly increased the number of predicted m6A-modified sites. In agreement with our in vitro analyses, we observed a significant increase in the ΔSumErr for sites identified with the IVT model compared with the default when considering m6A sites within the consensus DRACH motif (Supplemental Fig. S6A). Moreover, as previously shown on the “curlcake” data, the largest difference between the IVT and default models was observed for sites falling into the GGm6ACT motif, the most common m6A-containing motif in humans, suggesting that IVT has a higher potential to report true m6A-modified sites than the default model (Supplemental Fig. S6B).
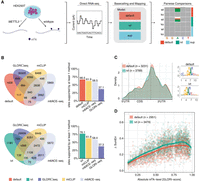
Detection of m6A-modified sites from HEK293T cells using a pairwise comparison approach and validation of identified sites with orthogonal methods. (A) Schematic representation of the workflow to obtain m6A-modified sites using DRS. Samples from wild-type HEK293T and METTL3−/− knockout cells were sequenced using DRS. Raw FAST5 files were basecalled using one of the three models tested (default, IVT, and SUP) and mapped to the human reference genome (hg38). To obtain m6A-modified sites, pairwise comparisons within each model were performed using ELIGOS2 (Jenjaroenpun et al. 2021). (B) Venn diagrams showing the overlap in absolute numbers between the nanopore used with either the default or IVT model and m6ACE-seq, miCLIP, and GLORI-seq (Pratanwanich et al. 2021; Liu et al. 2023). Bar plots show the relative amount of sites supported. (C) Metagene plot of sites identified by either the default or IVT model show the known pattern of m6A distribution with a strong enrichment around the stop codon. Corresponding motifs identified de novo for default (motif AvRec = 0.35) and IVT (motif AvRec = 0.401), using BaMM. (D) Scatterplot depicting the delta summed error (=SumErrWT − SumErrKO) of m6A sites predicted with ELIGOS2 using default and IVT models, and their corresponding absolute m6A-levels as reported by GLORI-seq. Individual points represent single m6A sites. Lines represent models fit using a generalized additive model (GAM).
To further examine this point, we compared the set of ELIGOS2-predicted m6A sites to a list of m6A sites predicted in HEK293T cells using orthogonal methodologies (m6ACE-seq [Pratanwanich et al. 2021], GLORI-seq [Liu et al. 2023], and miCLIP [Cruciani et al. 2023]). This analysis revealed that 67% of the sites reported by ELIGOS2 on reads basecalled with the default model were supported by at least one orthogonal method, whereas 76% of the sites reported by the IVT model had orthogonal support (Fig. 3B; see also Supplemental Fig. S6C–E; Supplemental Table S4). Sites that were supported by at least one orthogonal method demonstrated the typical m6A distribution along the transcript, and motif enrichment of the identified m6A sites matched the mammalian m6A motif, suggesting that these sites are true m6A sites (Fig. 3C). Furthermore, we noted that m6A sites supported by at least one orthogonal method that were predicted with the IVT model captured a higher proportion of GGACT sites compared with those predicted when basecalling with the default model (Supplemental Fig. S6F,G).
Next, we examined the relationship between the reported ΔSumErr and m6A stoichiometry on in vivo sites. To this end, we focused on sites overlapping with those identified by GLORI-seq, which reports absolute quantitative m6A information per site. We found that our IVT model generated higher error-signatures than the default across the entire range of m6A-levels, suggesting that even though it has a higher overall noise, owing to reduced basecalling accuracy, it shows stronger discriminative power than the default model even at lowly modified sites (Fig. 3D). In addition, we found that the IVT model predicted a larger proportion of high-stoichiometry GLORI-seq sites, suggesting that sites predicted with IVT-basecalled data contain a higher proportion of true positives compared with the default (Supplemental Fig. S6H).
These results show that ELIGOS2-reported m6A sites on IVT basecalled data sets are more accurate than m6A sites compared with ELIGOS2-reported m6A sites on default basecalled data sets. In addition, the IVT model was capable of detecting m6A sites that were not reported by any of the orthogonal methods or the other two basecallers tested, but are supported by the most comprehensive m6A database available (Liang et al. 2023), suggesting that IVT can identify a unique subset of sites (Supplemental Fig. S6I). In contrast, the same analysis performed with SUP reported only a very small number of m6A sites (97), which lacked overlap with the ground-truth data sets (Supplemental Fig. S6D,E), suggesting either that SUP is not suited for the detection of m6A using the pairwise-comparison of error-profiles or that ELIGOS2 software, or the default parameters used, are not suited for models that do not generate strong “error” patterns.
A high-accuracy IVT_SUP model does not improve RNA modification detection
We then reasoned that a model trained with IVT data (fully unmodified) and also with the more accurate CTC-CRF architecture might allow for improved RNA modification detection while showing increased accuracy, compared with our “IVT” model (Fig. 1B). We found that this model, which we termed “IVT_SUP,” was able to reach similar basecalling accuracy (97%) to the previously trained “SUP” model (Supplemental Fig. S7A). Subsequently, we assessed its ability to detect m6A, Ψ, and m1Ψ in the form of basecalling errors (Supplemental Fig. S7B,C). We observed a reduced ΔSumErr for pseudouridine modifications (Ψ and m1Ψ) compared with SUP, suggesting that the IVT_SUP model was suboptimal for the detection of these modifications. In contrast, we observed an increase in ΔSumErr at m6A-modified sites compared with the IVT model. A detailed analysis revealed that the increase in overall signal at m6A-modified sites could be largely attributed to an increased signal at non-DRACH-containing motifs, whereas DRACH sites showed a similar ΔSumErr to that observed with the default model (Supplemental Fig. S7D). Altogether, our results show that the IVT_SUP model outperformed neither the IVT model (for m6A) nor the SUP model (for Ψ and m1Ψ) at detecting RNA modifications in the form of basecalling errors, at least when using ΔSumErr as a metric to identify modified sites. For this reason, subsequent analyses were only performed with the “IVT” and “SUP” models.
SUP model demonstrates 97% read accuracy and up to a 50% increase in mapped reads
Nanopore sequencing shows relatively high error rates compared with other sequencing platforms, especially in the case of DRS, with ∼12% per-read error rates (Jain et al. 2022). This high background limits the technology's applicability in several areas such as SNP detection, transcript annotation, and RNA modification detection. In addition to changes in pore chemistry that can mitigate sequencing errors, the introduction of basecalling models with super high accuracy represents an alternative approach to enhancing the technology's accuracy. To this end, we examined the per-read accuracy of the different models trained, across a wide range of model species, using previously published DRS data sets (Parker et al. 2020; Begik et al. 2021; Pratanwanich et al. 2021; Nguyen et al. 2022). Our results revealed a significant increase in median read accuracy of “SUP” compared with the default model, with the SUP model reaching a median accuracy of 97% in human samples (Fig. 4A; see also Supplemental Fig. S8A).
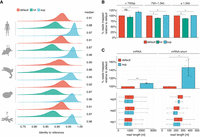
SUP is a super high accuracy model that leads to improved read accuracy and mapping. (A) Cross species comparison of read accuracies obtained from each model. Reads were aligned to the respective reference transcriptome (see Supplemental Table S5). To account for vastly different expression levels that would bias the analysis toward strongly expressed genes, we filtered for the longest reads of each transcript, keeping at most 10 reads per transcript. (B) Comparison of reads mapped to the human reference transcriptome (n = 3) across three bins of read lengths. Error bars indicate ± 1 SD. Statistical significance was determined using a two-sided t-test, corrected for multiple hypothesis testing using the Benjamini–Hochberg procedure: (ns) P ≥ 0.05, (*) P < 0.05). (C) Effect of distinct basecalling models on the proportion of mapped reads obtained from standard poly(A)-selected human samples (median length ≅ 850 bp) and a synthetic RNA library consisting of three polyadenylated in vitro transcripts (median length ≅ 200 nt). Statistical significance was determined using a one-sided t-test, and results were corrected for multiple hypothesis testing using the Benjamini–Hochberg procedure: (ns) P ≥ 0.05, (*) P < 0.05, (**) P < 0.01. Box plots depict the underlying size distribution of the sequenced libraries. The box is limited by the lower-quartile Q1 (bottom) and upper-quartile Q3 (top). Whiskers are defined as 1.5 × IQR with outliers being removed.
We then examined whether the increase in read accuracy would be reflected in the form of improved read mapping during alignment. We hypothesized that improved mapping should particularly impact shorter reads, as errors in those will be penalized more by most mapping algorithms, and may lead to reads remaining unmapped. To test this, we examined the relative proportion of mapped reads compared with the default, binning the reads by their read length. Notably, we found a consistent and significant increase in the number of mapped reads with the SUP model across all bins, with the observed difference to the default model being the largest in the category containing the shortest reads (<750 bp; relative increase compared with default = 117%) (Fig. 4B; see also Supplemental Fig. S8B). We then wondered whether this phenomenon would become even more evident in very short RNAs (mRNA-short, median length ≅ 200 bp), which are frequently observed in highly fragmented samples, such as blood plasma or sperm (Roszkowski and Mansuy 2021; Reggiardo et al. 2023). To test this, we used a library consisting of three polyadenylated in vitro transcripts with an average length of ∼200 bp (see Methods). Indeed, this analysis revealed an even stronger increase in the amount of mapped reads of ∼50% compared with the default model (Fig. 4C; see also Supplemental Fig. S8C).
Taken together, these results demonstrate that our super high accuracy (SUP) model significantly increases the average read accuracy, leading to substantially more reads being mapped, which becomes particularly noticeable in short RNA libraries (<200 bp).
Enhanced U > C mismatch signature and reduced background error in a vaccine model using the SUP model
Recently, the need for an improved quality-control pipeline to characterize mRNA vaccine production has been pointed out (Gunter et al. 2023). In this regard, nanopore DRS offers a unique platform as it can simultaneously provide information about sequence identity, RNA integrity, poly(A)-tail length, and modification status of the RNA molecule. Here, we wondered whether the use of our SUP model, which produced high U > C conversion rates at m1Ψ-modified sites (55%) in the “curlcake” sequences (Supplemental Fig. S9) and demonstrated superior basecalling accuracy (Fig. 4A), would be useful in the quality control of m1Ψ-containing mRNA vaccines, such as those used against COVID-19 (Nance and Meier 2021). To this end, we re-examined publicly available m1Ψ-modified and unmodified matched DRS data, which consisted of a synthetic mRNA vaccine that contained an alpha-globin 5′ UTR, an enhanced green fluorescent protein coding sequence, and an AES-mtRNR1 3′ UTR (Gunter et al. 2023). Our results revealed that all synthetic sequences showed a reduced background error when basecalled with our SUP model, in both the m1Ψ-modified and unmodified variants (Fig. 5A; see also Supplemental Fig. S10A), while simultaneously demonstrating a significant increase in the mismatch errors observed at modified sites (Fig. 5B; see also Supplemental Fig. S10B,C). Indeed, out of a total of 170 m1Ψ-modified sites, we observed an increased U > C error for 151 sites (≈80%) in SUP over the default (Fig. 5C). More importantly, for sites that previously showed a very poor signature in the default, we observed a global improvement in U > C conversion, suggesting that our SUP model is more sensitive toward m1Ψ in this context (Fig. 5D; see also Supplemental Fig. S10D). Overall, our results suggest that our SUP model can be a preferable basecalling model for performing quality control on mRNA vaccines and testing the incorporation of modified nucleotides, owing to its reduced background error and increased signal-to-noise ratio at m1Ψ-modified sites.
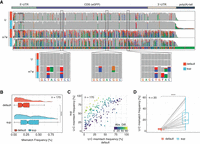
SUP improves the identification of m1Ψ-modified residues in synthetic mRNA vaccines. (A) IGV screenshot showing the synthetic mRNA vaccine containing either unmodified uridine (U) or N1-methylpseudouridine (m1Ψ). The 20,000 longest, uniquely mapped reads were selected for this analysis. Positions at which the mismatch frequency exceeds 0.2 are colored, and those with a mismatch frequency below 0.2 are shown in gray. (B) Comparison of mismatch frequency across all m1Ψ sites (n = 170) found in the synthetic eGPF vaccine. To test for statistical significance, the nonparametric Wilcoxon-test was used, and values were corrected for multiple hypothesis testing using the Benjamini–Hochberg procedure: (ns) P > 0.05, (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001, (****) P ≤ 0.0001. (C) Comparison of U > C mismatch frequency for all 170 m1Ψ sites between the default and SUP. Points located in the upper half of the dotted line represent sites with higher U > C frequency in SUP (n = 151), and points below the dotted line represent sites with higher U > C frequency in the default. (n = 19). Points are colored by the absolute difference between the default and SUP. (D) Comparison of U > C mismatch frequency across the most lowly modified sites in the default (median = 3.45) compared with the SUP (median = 19.43). A paired two-sided Wilcoxon test was performed to test for statistical significance. For B,D, the box is limited by the lower-quartile Q1 (bottom) and upper-quartile Q3 (top). Whiskers are defined as 1.5 × IQR with outliers removed.
Discussion
Recently, nanopore DRS has solidified its position as a promising tool for investigating the epitranscriptome, owing to its unique capability of sequencing native RNAs, thus making it possible to detect multiple modifications within the same native RNA molecule (Garalde et al. 2018; Smith et al. 2019; Begik et al. 2021). Indeed, multiple works have already employed this technology to characterize the epitranscriptomic landscape of mRNAs, rRNAs, and tRNAs (Parker et al. 2020; Delgado-Tejedor et al. 2023; Lucas et al. 2024). Early approaches for the detection of modifications using the nanopore platform have relied on the fact that modified residues will cause an error during the basecalling process (Begik et al. 2022). In this regard, several tools such as EpiNano (Liu et al. 2019), DiffErr (Parker et al. 2020), ELIGOS2 (Jenjaroenpun et al. 2021), DRUMMER (Abebe et al. 2022a), and JACUSA2 (Piechotta et al. 2022) have been developed to exploit this information and perform pairwise comparisons between a modified sample and an unmodified control to identify modified sites (Liu et al. 2019; Parker et al. 2020; Jenjaroenpun et al. 2021; Piechotta et al. 2022; Abebe et al. 2022a). In more recent years, current-based methods such as Tombo (Stoiber et al. 2017), xPore (Pratanwanich et al. 2021), nanoRMS (Begik et al. 2021), Nanocompore (Leger et al. 2021), or m6Anet (Hendra et al. 2022), which interrogate the raw signal space, and modification-aware basecallers such as m6ABasecaller (Cruciani et al. 2023) and Dorado (https://github.com/nanoporetech/dorado), have been developed. These tools show some key advantages compared with error-based methods, such as the possibility of achieving per-read modification resolution (e.g., in the case of m6Anet, m6ABasecaller, and Dorado). Nevertheless, error-based approaches still remain a better approach in specific biological scenarios in which RNA modifications are highly abundant, such as in tRNAs (Lucas et al. 2024). Moreover, training modification-aware basecallers for these RNA species would require the synthesis of RNAs that contain combinations of different modifications, which currently is not feasible. Therefore, error-based approaches provide a valuable alternative to current-based methods and modification-aware basecallers.
A critical component for error-based methods is the basecalling model, which is trained on a specific data set and is responsible for converting the raw current intensity signal that is output by the sequencing machine into a nucleotide sequence. The currently used basecalling model for RNA002 chemistries, which we refer to as the “default” (provided with Guppy 6.0.6 and upward), shows an overall per-read accuracy of 90% using ONT's proprietary basecaller Guppy (Liu-Wei et al. 2024). Previous efforts have been made to develop novel, more-accurate basecalling software, such as RODAN for RNA and Chiron and DeepNano for DNA (Boža et al. 2017; Teng et al. 2018; Neumann et al. 2022), among others. However, the effect of using alternative basecalling models on the detection of RNA modifications has so far not been explored.
Here we present two novel basecalling models, IVT and SUP (Supplemental Table S1), and benchmark their ability to detect a battery of RNA modification types, as well as their basecalling accuracy and effect on read mapping. We find that using a model that was trained on in vitro transcribed RNA (IVT model) significantly improves the detection of m6A modifications (Fig. 1E,F) by generating a strong deletion signature at the modified site (Supplemental Fig. S2C,D), improving the signal-to-noise ratio. This leads to a significant increase in the total number of high-confidence m6A sites recovered in in vivo samples compared with the default model (Fig. 4B). We argue that this improvement is because the default model was trained on native RNA from different organisms containing large amounts of m6A and therefore learned to recognize both unmodified A and m6A as A, especially at naturally modified 5-mers (Fig. 3A). We find evidence for this hypothesis in the fact that the most improved 5-mer coincides with the most highly modified 5-mer in humans GGm6ACT (Fig. 3B; see also Supplemental Fig. S4B).
Our second model, SUP, was trained with the intention of maximizing read accuracy. Therefore, we changed the model architecture from flip-flop, used in both default and IVT, to CTC-CRF (currently used in ONT DNA-seq; see Methods). We found that SUP reaches a per-read accuracy of 97% on human data compared with 91% in the default, which is a substantial improvement and, to our knowledge, the most accurately basecalled DRS run to date (Fig. 4A). We additionally found that this improved accuracy was accompanied by an increased number of mapped reads, becoming particularly apparent in short reads, in which the increase in read mappability reached ∼50% (Fig. 4B,C). Similar performance has been reported regarding ONT's newest direct RNA sequencing chemistry, SQK-RNA004 (96% accuracy). We should note that our improvements in basecalling accuracy were obtained using the SQK-RNA002 chemistry. Furthermore, our SUP model also increased basecalling error signatures at Ψ- and N1-methylpseudouridine (m1Ψ)-modified sites, making it the preferred model for highly modified substrates such as mRNA vaccines (Fig. 5A).
Methods
In vitro transcription
This study used synthetic RNAs known as “curlcakes,” which were designed to include all possible 5-mer sequences while minimizing RNA secondary structures (Liu et al. 2019) and consist of four in vitro transcribed constructs: (1) Curlcake 1, 2244 bp; (2) Curlcake 2, 2459 bp; (3) Curlcake 3, 2595 bp; and (4) Curlcake 4, 2709 bp. To generate “curlcake” synthetic RNAs, the plasmids were digested with BamHI-HF (NEB R3136L) and EcoRV-HF (NEB R3195L) followed by a clean-up using phenol/chloroform/isoamyl-alcohol 25/24/1, v/v (pH 8.05; Sigma-Aldrich P3803). Linearized plasmids were used for in vitro transcription with the AmpliScribe T7-flash transcription kit (Lucigen ASF3507) using 5-methyluridine-5′-triphosphate (5-mUTP, Trilink N-1024-1) instead of UTP, N1-methylpseudouridine-5′-triphosphate (m1meΨTP, Jena Bioscience NU-890S) instead of UTP, or N4-acetyl-cytidine-5′-triphosphate (N4-acetyl-CTP, Jena Bioscience NU-988S) instead of CTP. Products were treated with Turbo DNase (Invitrogen AM2238) to remove the template plasmid and were cleaned up using the RNeasy MinElute kit (Qiagen 74204). All constructs were polyadenylated using Escherichia coli poly(A) polymerase (NEB M0276S) according to the manufacturer's instructions followed by a cleanup using the RNA Clean & Concentrator-5 kit (Zymo Research R1013). Concentrations were determined using the Qubit 2.0 fluorometer. Transcript integrity, as well as polyadenylation success, was determined using an Agilent 4200 TapeStation. For previously sequenced curlcakes used in this study, including unmodified NTPs (UNMs), N6-methyladenosine triphosphate (m6ATP), 5-methylcytosine triphosphate (m5CTP), 5-hydroxymethylcytosine triphosphate (hm5CTP), and pseudouridine triphosphate (ΨTP), publicly available raw FAST5 files were used as input for the analysis (see Supplemental Table S2; Liu et al. 2019; Begik et al. 2021).
DRS of in vitro transcribed RNA
The RNA libraries for DRS of ac4C, m5U, m1Ψ-containing curlcake constructs, and UNM-S (which included Bacillus subtilis guanine riboswitch, B. subtilis lysine riboswitch, and Tetrahymena ribozyme) were prepared following the ONT DRS protocol version DRS_9080_v2_revR_14Aug2019-minion. Briefly, 800 ng of poly(A)-tailed RNA (200 ng per curlcake construct and 250 ng per riboswitch) was ligated to ONT RT adaptor (RTA) using concentrated T4 DNA ligase (NEB M0202T). The optional reverse transcription step was performed using SuperScript III (Thermo Fisher Scientific 18080044) in the case of ac4C, m5U-containing curlcake constructs, and UNM-S and using Superscript IV (Thermo Fisher Scientific 18090010) in the case of m1Ψ. The products were purified using 1.8× Agencourt RNAClean XP beads (Thermo Fisher Scientific NC0068576) and washed with 70% freshly prepared ethanol. RNA adapter (RMX) was ligated onto the RNA:DNA hybrid, and the mix was purified using 1× Agencourt RNAClean XP beads, followed by washing with wash buffer (WSB) twice. The sample was then eluted in elution buffer (ELB) and mixed with RNA running buffer before loading onto a primed R9.4.1 flowcell. The samples were run on a MinION sequencer for 24–72 h. The sequencing runs were performed on independent days and using a different flowcell for each sample.
Training a high-accuracy RNA basecallers (SUP model)
We trained a new RNA basecalling model (SUP) using bonito v0.5.1 (https://github.com/nanoporetech/bonito). The SUP model is based on the latest CTC-CRF (bonito) DNA model architecture, which achieves 96% accuracy for DNA basecalling with chemistry R9.4.1. DNA CTC-CRF models use a window of 19 and stride of five. For our RNA models, we used a window of 31 and stride of 10 (similar to RNA flip-flop models).
Bonito model training requires chunks of fixed length (10,000 samples) and underlying sequences. We kept only chunks that corresponded to 30–270 bases. Chunks from each read were generated by stepping every 50 bases in the sequence space. Training was performed using bonito by keeping 3% of randomly selected chunks for validation. The SUP model was trained using publicly available human data sets containing triplicates of wild-type and Mettl3−/− cells (Pratanwanich et al. 2021). We kept the five longest reads per transcript and ran them in order to balance highly and lowly expressed transcripts. In total, 158,413 reads were used for training. The training was stopped after five epochs. We selected the model with the lowest validation error as the final one from each training, corresponding to epoch 5 for the SUP model. The resulting model was calibrated using the calibrate_qscores_byread.py script from taiyaki and reached 97% median accuracy on native human RNA (Fig. 1B).
Training an RNA basecaller using in vitro sequences (IVT model)
The IVT model was trained using publicly available IVT direct RNA data obtained from CEPH1463 cells (UCSC_Run1_IVT_RNA). RNA model training procedure was performed for 50,000 iterations with following taiyaki hyperparameters: ‐‐size 256 ‐‐stride 10 ‐‐winlen 31 ‐‐chunk_len 4000 ‐‐batch_size 50. We selected up to 100 chunks from every read using a step of 25 bases. IVT model was trained using taiyaki v4.1.0 (https://github.com/nanoporetech/taiyaki).
Demultiplexing DRS
To evaluate the effect of differing m6A-stoichiometries on the sensitivity of each basecalling model, we used publicly available, multiplexed curlcake data (obtained from the NCBI BioProject database [https://www.ncbi.nlm.nih.gov/bioproject/] under accession number PRJEB61874) containing a range of stoichiometries (barcode1 = 12.5%, barcode 4 = 25%, barcode 3 = 50%, and barcode 2 = 75%; m6ATP). Demultiplexing of the barcoded DRS libraries was performed using DeePlexiCon (Smith et al. 2020), keeping reads with demultiplexing confidence scores above 0.80 (-f multi -m resnet20-final.h5 -s 0.8).
Basecalling and mapping of DRS data sets
Publicly available data sets for Homo sapiens (Pratanwanich et al. 2021), Mus musculus (Nguyen et al. 2022), Xenopus laevis (Nguyen et al. 2022), Saccharomyces cerevisiae (Liu et al. 2019), Arabidopsis thaliana (Parker et al. 2020), and the synthetic eGFP mRNA vaccine (Gunter et al. 2023) were downloaded from their respective data repositories reported in Supplemental Table S2. Raw FAST5 files were basecalled and mapped using the MasterOfPores pipeline (version 2.0) (Cozzuto et al. 2023). For basecalling, Guppy (version 6.0.6) was used with either the default, IVT, or SUP model, disabling the filtering step based on read quality (‐‐disable_qscore_filtering) (Supplemental Table S5). For the H. sapiens data set, we additionally examined the basecalling accuracy of Dorado (version 0.6.0) using the highest accuracy model available for the RNA002 chemistry (rna002_70bps_hac@v3) (Fig. 1B). For highly modified curlcake runs and the eGFP mRNA vaccine, basecalled FASTQ files were aligned to the curlcake reference using GraphMap (version 0.5.2) (Sović et al. 2016) with “sensitive” settings (‐‐rebuild-index -v 1 ‐‐double-index ‐‐mapq -1 -x sensitive -z 1 -K fastq ‐‐min-read-len 0 -A 7 -k 5) (Begik et al. 2021). For the in vivo m6A-analysis, basecalled FASTQ files were aligned to the soft-masked human genome (Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa) using minimap2 (v2.17) with “default” settings (-uf -ax splice -k14). Uniquely mapped reads were reported with SAMtools (version 1.15.1) (Li et al. 2009) using samtools view -c -F 260 (Supplemental Table S5).
For the cross-species comparison, reads were mapped to the respective reference transcriptome (Supplemental Table S5) using minimap2 (v2.17) with “transcriptome” settings (-ax map-ont -k14). Aligned reads were first filtered for uniquely mapped reads with a mapQ > 15 using SAMtools (samtools view -q 15 -F 3840 <in.bam> > <out.bam>) (Li et al. 2009). To account for the heterogeneous expression profiles of each transcriptome and to prevent biasing our analysis toward highly expressed genes, we sampled the longest reads per transcript, keeping at most 10 reads per transcript (bam2select.py). Per read identities were calculated using a custom Python script (identity_density_v2.py). All scripts required to reproduce the results in this paper are provided on the project's GitHub page (https://github.com/novoalab/basecalling_models). Silhouettes of the model species were downloaded from PhyloPic (https://www.phylopic.org/).
Comparative analysis of system requirements
To compare the computational requirements of the default model with our custom IVT and SUP models, we subsampled 2 × 100,000 reads from a publicly available mouse brain DRS data set (Nguyen et al. 2022), which was also used in the cross-species comparison (Fig. 4A; Supplemental Table S2). Both replicates (100,000 reads each) were basecalled with Guppy (version 6.0.6), using the Master of Pores pipeline (‐‐granularity 25) on a single GPU (RTX A4000, CUDA11) and a single CPU with 12 GB of memory allocated. Computational requirements (runtime and %CPU usage) were extracted from the nextflow report (‐‐with-report) (Supplemental Fig. S1C,D).
Comparative analysis of basecalled features in curlcakes
To qualitatively assess the difference in basecalling errors introduced by default, IVT, and SUP upon RNA modifications, the sorted and indexed BAM files were visualized using the Integrative Genomics Viewer (IGV, version 2.13.1) (Robinson et al. 2011). Using EpinanoRMS (version 1.1) (Begik et al. 2021), basecalled features (mismatch frequency, deletion frequency, and insertion frequency) were extracted from the alignment files on a per-position basis. To obtain per 5-mer information, this output was converted using epinano_to_kmer.R script, which applies a sliding window of a given size to compile the per k-mer information (R Core Team 2023). These results were further processed to calculate the SumErr (Σ(mismatch frequency, deletion frequency, insertion frequency)) and ΔSumErr(SumErrMOD − SumErrUNM) on a per-position basis and visualized using in-house R scripts (Wickham 2016). To calculate the mismatch directionality, the number of reads corresponding to the correct reference base for that position was divided by the total number of reads obtained for that position. All scripts required to reproduce these results are available under the project's GitHub repository (https://github.com/novoalab/basecalling_models).
Detection of m6A-modified sites in HEK293T using comparative error analysis
To determine whether alternative basecalling models can improve the detection of m6A in vivo, the alignment files of individual replicates for the wild type and Mettl3−/− were merged to obtain greater coverage. To reduce computational load, only protein-coding genes were considered in the comparison. For this reason, lines in which the gene biotype corresponds to protein coding were extracted from the genome annotation file and converted to BED format using bedops gtf2bed (version 2.4.41) (Neph et al. 2012). The resulting BED file was split into individual chromosomes that would serve as regions for testing in the pairwise comparison step. To perform pairwise comparisons, we ran ELIGOS2 (v2.1.0) (Jenjaroenpun et al. 2021) using the singularity image provided on the software's GitLab repository (https://gitlab.com/piroonj/eligos2) with a P-value cut-off of 1 × 10−5, an odds ratio of 2.5, and each of the individual chromosome files as regions for testing. Next, results obtained from individual chromosomes were merged using an in-house shell script and filtered to remove non-A bases as well as homopolymers to identify putative m6A sites (eligos2 filter -sb A ‐‐homopolymer). For comparative analysis of basecalling models, we extracted error-signatures of ELIGOS2-predicted m6A sites using EpiNano (v1.2.4; https://github.com/novoalab/EpiNano).
Metagene analysis and motif identification
To visualize the transcriptome-wide distribution of putative m6A sites identified with distinct basecalling models, we used the GuitaR package (version 2.12.0) (Cui et al. 2016). For de novo motif discovery, sites obtained from ELIGOS2 were converted into the FASTA format, which contained 6 bp upstream of and downstream from the modified site using the table2fa_eligos.mergeextend.sh script. Subsequently, those files were used for de novo motif prediction using BaMM motif (version 1.4.0) (Siebert and Söding 2016) using default settings and a Z-score threshold of five.
Comparison of identified m6A sites against m6ACE-seq, GLORI-seq, and miCLIP-seq
To identify high-confidence m6A sites, we intersected results obtained by each of the three models (default, IVT, and SUP) with data obtained from orthogonal methods. These data sets included m6ACE-seq data that were matched with the nanopore results, as well as GLORI-seq and miCLIP-seq results obtained from the same cell line (Linder et al. 2015; Pratanwanich et al. 2021; Liu et al. 2023). These data sets were filtered to only retain sites for which we had sufficient coverage in the nanopore runs (≥20×) within protein-coding genes (https://github.com/novoalab/basecalling_models). Overlaps between data sets were visualized using the ggvenn R package (version 0.1.9).
For stoichiometric analysis, sites overlapping with GLORI-seq were used as this data set provides per-nucleotide resolution information as well as an estimated stoichiometry for each identified m6A site (Liu et al. 2023). To this end, results obtained by GLORI-seq were first binned based on their reported stoichiometry into sites that were considered either HIGH (≥0.75 modified), MED-HI (≥0.50 and <0.75 modified), MED-LO (≥0.25 and <0.50 modified), or LOW (≤0.25 modified). Only those sites were retained in the final set in which both GLORI-seq replicates were reported within the same bin. This resulted in a total of 47,722 high-confidence sites split between HIGH (n = 8900), MED-HI (n = 7947), MED-LO (n = 12,697), and LOW (n = 18,178).
Processing of eGFP mRNA vaccine data and comparative analysis
Aligned reads were filtered to obtain the 20,000 longest reads and matched between default and SUP (bam2select.py). Mismatch, deletion, and insertion frequencies were calculated and extracted on a per nucleotide basis using EpinanoRMS (Liu et al. 2019). The per-base frequencies were calculated by dividing the number of each of the four canonical nucleobases (A, C, G, or T) for each position by the number of total observations for that position. To determine differences in sensitivity between the two models, we extracted the 20 most lowly U > C-modified sites in default and compared them to the same positions in SUP. All scripts available at GitHub (https://github.com/novoalab/basecalling_models).
Software availability
All code used to build figures shown in this work can be found on GitHub (https://github.com/novoalab/basecalling_models) and as Supplemental Code. All trained basecalling models used in this work are available in OSF (https://osf.io/2xgkp/). Processing of raw FAST5 files was performed using the MasterOfPores (version 2.0) nextflow pipeline (Di Tommaso et al. 2017; Cozzuto et al. 2023; https://github.com/biocorecrg/MOP2).
Data access
FAST5 DRS data sets basecalled with the default, corresponding to “curlcakes” containing ac4C, m5U, and m1Y, as well as UNM-S rep2 and UNM-S rep3, generated in this study have been submitted to the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home), under accession code PRJEB67632. All processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE246151. Further data sets used in this study are specified in Supplemental Table S2.
Competing interest statement
E.M.N. has received travel and accommodation expenses to speak at Oxford Nanopore Technologies (ONT) conferences. G.D. and O.B. have received travel bursaries from ONT to present their work at conferences. E.M.N. is a member of the scientific advisory board of IMMAGINA Biotech. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could otherwise be construed as a conflict of interest.
Acknowledgments
G.D. is part of the ROPES ITN, which received funding from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement no. 956810. O.B. is supported by funds from Merck Innovation 2020. L.P.P. was supported by funding from the European Union's H2020 research and innovation program under Marie Sklodowska-Curie grant agreement no. 754422, and is currently supported by European Research Council funds (ERC-StG-2021 no. 101042103 to E.M.N.). J.-Y.R. is supported by funds from the Swiss National Science Foundation (310030_197906) and the Deutsche Forschungsgemeinschaft (RO 4681/4-2, RO 4681/6-1, and RO 4681/12-1, TRR319 RMaP). This work was supported by funds from the Spanish Ministry of Economy, Industry, and Competitiveness (MEIC; PID2021-128193NB-100 to E.M.N.) and the European Research Council (ERC-StG-2021 no. 101042103 to E.M.N.). We acknowledge the support of the MEIC to the EMBL partnership, Centro de Excelencia Severo Ochoa, and CERCA Programme/Generalitat de Catalunya.
Author contributions: G.D. performed the bioinformatic analyses included in this work, together with O.B., S.C., and A.D.-T. L.P.P. trained the RNA basecalling models used in this work. L.L., M.C.L., and O.B. generated curlcake data sets used in this work. S.C. and A.D.-T. contributed with bioinformatic analyses. O.B. and E.M.N. supervised the project, with the contribution of J.-Y.R. E.M.N. conceived the project and acquired funding. G.D. built the figures. G.D. and E.M.N. wrote the manuscript, with the contribution from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278849.123.
- Received December 12, 2023.
- Accepted September 10, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








