
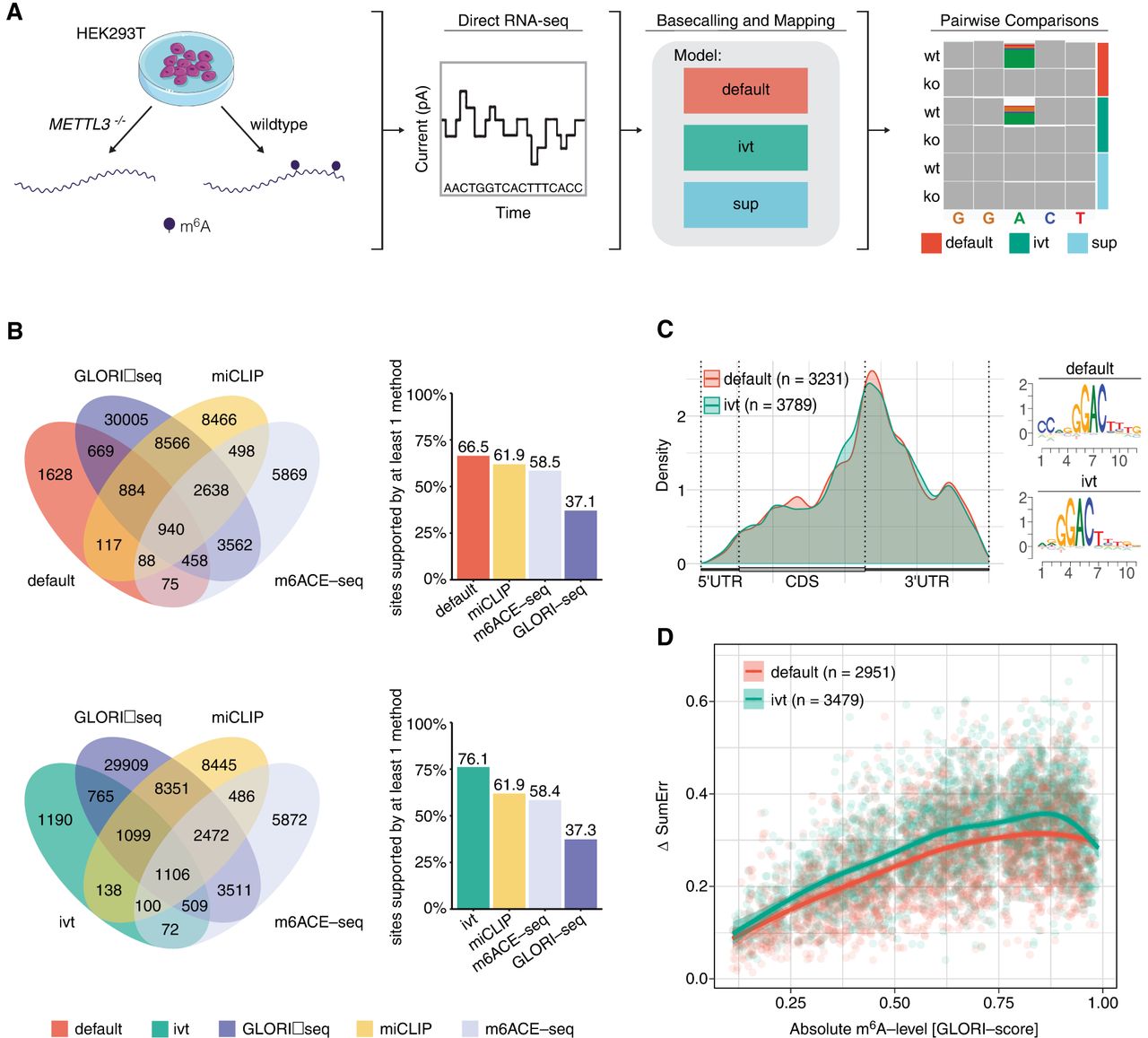
Detection of m6A-modified sites from HEK293T cells using a pairwise comparison approach and validation of identified sites with orthogonal methods. (A) Schematic representation of the workflow to obtain m6A-modified sites using DRS. Samples from wild-type HEK293T and METTL3−/− knockout cells were sequenced using DRS. Raw FAST5 files were basecalled using one of the three models tested (default, IVT, and SUP) and mapped to the human reference genome (hg38). To obtain m6A-modified sites, pairwise comparisons within each model were performed using ELIGOS2 (Jenjaroenpun et al. 2021). (B) Venn diagrams showing the overlap in absolute numbers between the nanopore used with either the default or IVT model and m6ACE-seq, miCLIP, and GLORI-seq (Pratanwanich et al. 2021; Liu et al. 2023). Bar plots show the relative amount of sites supported. (C) Metagene plot of sites identified by either the default or IVT model show the known pattern of m6A distribution with a strong enrichment around the stop codon. Corresponding motifs identified de novo for default (motif AvRec = 0.35) and IVT (motif AvRec = 0.401), using BaMM. (D) Scatterplot depicting the delta summed error (=SumErrWT − SumErrKO) of m6A sites predicted with ELIGOS2 using default and IVT models, and their corresponding absolute m6A-levels as reported by GLORI-seq. Individual points represent single m6A sites. Lines represent models fit using a generalized additive model (GAM).








