
Targeted and complete genomic sequencing of the major histocompatibility complex in haplotypic form of individual heterozygous samples
- Taishan Hu1,5,6,7,
- Timothy L. Mosbruger1,5,
- Nikolaos G. Tairis1,
- Amalia Dinou1,
- Pushkala Jayaraman2,8,
- Mahdi Sarmady2,9,
- Kingham Brewster3,
- Yang Li1,
- Tristan J. Hayeck1,4,
- Jamie L. Duke1 and
- Dimitri S. Monos1,4
- 1Immunogenetics Laboratory, Department of Pathology and Laboratory Medicine, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA;
- 2Division of Genomic Diagnostics, Department of Pathology and Laboratory Medicine, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA;
- 3Sequencing and Genotyping Center, Delaware Biotechnology Institute, University of Delaware, Newark, Delaware 19713, USA;
- 4Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
-
↵5 These authors contributed equally to this work.
Abstract
The human major histocompatibility complex (MHC) is a ∼4 Mb genomic segment on Chromosome 6 that plays a pivotal role in the immune response. Despite its importance in various traits and diseases, its complex nature makes it challenging to accurately characterize on a routine basis. We present a novel approach allowing targeted sequencing and de novo haplotypic assembly of the MHC region in heterozygous samples, using long-read sequencing technologies. Our approach is validated using two reference samples, two family trios, and an African-American sample. We achieved excellent coverage (96.6%–99.9% with at least 30× depth) and high accuracy (99.89%–99.99%) for the different haplotypes. This methodology offers a reliable and cost-effective method for sequencing and fully characterizing the MHC without the need for whole-genome sequencing, facilitating broader studies on this important genomic segment and having significant implications in immunology, genetics, and medicine.
The major histocompatibility complex (MHC), located on Chromosome 6, is a crucial genomic region encompassing over 260 genes, with about half playing critical roles in innate and adaptive immunity (Horton et al. 2004). Importantly, the MHC includes the human leukocyte antigen (HLA) genes critical for transplantation. The MHC also holds the highest density of disease-associated single-nucleotide polymorphisms (SNPs) of any similarly sized segment within the human genome (Clark et al. 2015). However, characterizing the MHC in detail has proven challenging, primarily due to the high degree of sequence variability, structural rearrangements, and extensive linkage disequilibrium throughout the region (Kumánovics et al. 2003; Horton et al. 2004, 2008; Traherne 2008; Barker et al. 2023). Consequently, fine mapping of disease-associated variants within the MHC is difficult. While whole-exome sequencing (WES) can facilitate the identification of trait-associated variants within exons, it cannot accurately characterize long-range haplotype structure and sequence variation within intronic and intergenic regions of the genome. This limitation is significant because 90% of causal autoimmune disease-related variants occur within noncoding regions of the genome (Farh et al. 2015). As a result, characterizing the MHC beyond exons is critical to resolve disease-associated variants accurately.
Acknowledging the importance of complete genomic characterization of the MHC early on, two homozygous B lymphoblastoid cell lines (BLCLs), PGF and COX, were fully sequenced for their MHC (Stewart et al. 2004). Later, with the advent of massively parallel sequencing technologies, another 88 homozygous for Chromosome 6 BLCLs were characterized using a combination of capture array technology targeting the MHC and short-read sequencing via Illumina technology (Norman et al. 2017). However, the MHC sequences of these haploid cell lines were rather fragmented. Most recently an additional six homozygous for Chromosome 6 BLCLs, with partial coverage for their MHC (Horton et al. 2008), were sequenced using a combination of short and long-read technologies (Illumina, Pacific Biosciences [PacBio] and Oxford Nanopore), accomplishing complete coverage of the MHC (Houwaart et al. 2023).
Alternative methods using whole-genome sequencing (WGS) approaches, such as analyzing family trios (Jensen et al. 2017) or individual samples (Chin et al. 2020), successfully produced haploid sequences of heterozygous samples for the MHC, using recently developed de novo assembly approaches. However, due to their reliance on WGS, these approaches are not practical or cost-effective for generating routine haploid MHC sequences for individual heterozygous samples.
Herewith, we propose a novel approach to target and sequence the MHC region of any sample using our region-specific extraction (RSE) method (Dapprich et al. 2016), together with an enriched panel of oligos, whereby the captured MHC is sequenced on the PacBio platform (continuous long read [CLR] or high fidelity [HiFi] reads) and de novo assembled using our strategy and publicly available resources. In the past, we used RSE to show proof-of-concept that targeting and sequencing the MHC of a homozygous sample (PGF) with short-read Illumina technology was feasible. However, the short reads of the Illumina platform and the limited set of oligos designed to capture the MHC of the homozygous PGF cell line, hindered the ability for the haplotypic characterization of the 4 Mb region of the MHCs of random heterozygous samples. A combination of long-read sequencing technologies (PacBio) that enabled phasing and an improved set of oligos designed by AnthOligo (Jayaraman et al. 2020) to target random samples, allowed the haplotypic characterization of the MHCs, as described herein.
Our experimental design includes three approaches to generate haploid sequences and evaluate their accuracy, thereby ensuring the credibility of our overall characterization of a random sample. For this purpose, (1) we have used a mixture of two homozygous BLCLs with known MHC sequences each, to create an artificial heterozygous sample, (2) two family trios (one of European and the other of East-Asian descent), and (3) a single African-American sample. The final haploid sequences generated by our methodology ranged from 96.6% to 99.9% sequence coverage with a depth threshold of at least 30×, with an accuracy of 99.89%–99.99%.
To assess accuracy, we compared (1) the haploid assemblies of COX and PGF to known references, (2) proband haplotypes to independent parental assemblies, and (3) African-American haplotypes to SNP microarray. For all samples, we used HLA typing, construction of the HLA-DR haplotypes of the samples, and the Bionano Genomics platform (referred to as Bionano thereafter) to further assess accuracy. Bionano is an optical imaging technology that provides structural context for most genomic regions (Khan and Toledo 2021) and enables us to confirm the integrity of the assembly and accurately identify structural rearrangements and differences between the two MHC haplotypes.
The approach presented here offers an efficient solution for the credible haplotypic characterization of the MHC of multiple random samples. Accurate and thorough characterization of the MHC haplotypes will enable further studies on the epigenetic and 3D genomic characteristics of this crucial genomic region, ultimately shedding light on the mechanisms affected by variants within both coding and noncoding regions of the MHC that drive human disease risk.
Results
Targeting the MHC sequences using RSE and sequencing on the PacBio platform
Following the strategy outlined in Methods, two homozygous BLCLs, with known MHC sequences, were mixed to generate a single heterozygous sample, and seven blood samples (two family trios and one random sample), were selected to demonstrate the successful haploid characterization of the MHC.
The RSE methodology was used to specifically target the MHC (Dapprich et al. 2016). To achieve MHC specificity (Chr 6: 29,624,000–33,291,000), the AnthOligo software (Jayaraman et al. 2020) was used to design a set of oligos to target and capture this region with RSE. The genomic fragments captured from RSE are then subjected to whole genome amplification (WGA) and sequenced on either the PacBio Sequel or Sequel II platform.
Read selection and editing
Depending on the number of reads obtained for the different samples after sequencing, all or a subset of reads were aligned against the hg38 reference and a panel of MHC sequences (Table 1; Supplemental Fig. S1A; Supplemental Methods: MHC Read Identification). We retain the reads aligned to the MHC (Fig. 1A) and find that the percent of MHC-specific reads aligned ranges from 10.33% to 52.58% (Table 1), yielding 86.09- to 438.13-fold enrichment of the MHC (Table 1). The high accuracy of the Sequel II HiFi reads allowed us to cap read depth to 250× using k-mer abundance (Table 1). Chimeric reads, containing DNA segments from nonconsecutive regions and typically palindromic, are a result of the WGA process (Lasken and Stockwell 2007; Warris et al. 2018; Kiguchi et al. 2021). To mitigate the effects of these WGA artifacts, we apply a bioinformatic process to split these reads at chimeric segment junctions, creating independent subreads (Fig. 1B; Supplemental Fig. S1B; Supplemental Methods: MHC Read Editing by Overlap). These edited reads are then used for the collapsed de novo assembly.
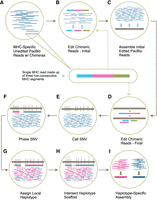
Read editing and haplotype binning detail. (A) Unedited PacBio reads that align to one of the 160 MHC haplotypes in the reference sequence. Inset shows the composition of a single read containing three nonconsecutive or palindromic MHC sequences (chimera) represented by three colors. (B) The purpose of this step is to split each chimeric read into individual segments (initial edited reads). This is done by overlapping chimeric reads and identifying positions along each chimeric read with a buildup of overlap termination sites. (C) Initial edited reads are assembled into haploid mosaic contigs. (D) A final round of chimeric read editing aims at further improving the detection of the chimera boundaries. The chimeric MHC-specific reads from A are aligned to the collapsed assembly, which acts as a reference sequence. The chimeric read is then split based on the reported aligned segments (final edited reads). (E) SNVs are called across the collapsed assembly and (F) SNVs are phased using the final edited read alignments. (G) Final edited reads are assigned a local haplotype if they intersect with variants in a phased block (pink segment = haplotype 1, blue segment = haplotype 2, gray segment = on both haplotypes). (H) Each locally phased block is oriented to the full MHC haplotype by intersecting with the reference-based haplotype scaffold (light blue triangles). (I) Haplotype-specific edited reads are combined with homozygous reads and assembled independently into haplotype-specific assemblies.
MHC capture and sequencing metrics
Collapsed assembly
Edited reads are assembled into contigs representing a mixture of both haplotypes. This assembly acts as a reference sequence used in two contexts: (1) to further edit the chimeric reads and (2) to perform variant calling and phasing across the MHC for later haplotype partitioning. Specifically, edited MHC reads are first assembled into haploid contigs using Canu (Koren et al. 2017), which are then filtered to remove highly overlapping or questionable contigs (Fig. 1C; Supplemental Fig. S1C; Supplemental Methods: Collapsed De Novo Assembly). This first step resulted in 7–41 contigs spanning the MHC, depending on the sample (sample PGF + COX: 7, EAS-P [East-Asian-Chinese proband]: 12, EUR-P [European proband]: 41, AFA [African American]: 11). To address potential issues related to excessive splitting during the reference-free editing process, we performed a reediting of the initial MHC-specific reads. This involved realigning the reads to the generated contigs to enhance the detection of palindromes and other chimeric sequences. Each segment of the chimeric sequence that aligns to separate locations on the assembly is treated as a separate read (Fig. 1D; Supplemental Fig. S1C; Supplemental Methods: MHC Read Editing by Alignment). Table 2 shows the number of MHC-specific reads per sample after editing and Figure 2 shows the size distribution of the MHC-specific reads used for the assembly. The depth of coverage across reference MHC is shown in Figure 3. EUR-P reads are not high enough quality to digitally normalize, resulting in a much wider depth of coverage distribution. The highest scoring aligned segment between each unedited MHC-specific read and contig was retained for variant calling, generating a varying number of heterozygous single-nucleotide variants (SNVs) across each sample (Table 3; Fig. 1E; Supplemental Fig. S1D; Supplemental Methods: SNV Calling and Haplotype Partitioning). All aligned segments were treated as separate edited reads and used to phase heterozygous variant calls into 16–55 phase blocks, depending on the sample. The number of phase blocks depends on the homozygosity of the sample and the accuracy of chimera removal and read correction. Since this phasing is based on the assembly and not alignment to a reference genome (hg38), there are limited phase breaks due to structural variants. A few SNVs (4–10), again depending on the sample, were not part of a specific block and they were not phased (Table 3; Fig. 1F). All other heterozygous SNVs fell within accurately phased blocks, which improved the chance of intersection with the reference-based scaffold and likelihood the SNVs could be used for haplotype partitioning. Finally, reads were assigned a local haplotype (haplotag) based on phased variants within each block (Fig. 1G).
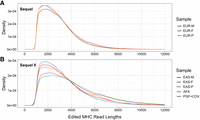
Edited read length distribution. Read length distribution after splitting chimeric reads. (A) Samples sequenced on the PacBio Sequel and (B) Samples sequenced on the PacBio Sequel II. (EAS-F) East-Asian father, (EAS-M) East-Asian mother, (EUR-F) European father, (EUR-M) European mother.
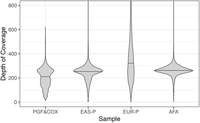
Depth of coverage across the reference MHC sequence after read selection and digital normalization. Digital normalization is not performed on EUR-P because the majority of the reads are not HiFi quality. The median depth of coverage in each sample is represented by a horizontal black bar.
Edited read metrics after chimeric artifact processing
PacBio phasing statistics
Haplotype partitioning and assembly
The next step in phasing the MHC sequences is to connect the locally phased blocks, mentioned above, to a fully phased reference-based haplotype scaffold. This scaffold is created by first calling SNPs across hg38 only at locations on the Infinium Omni2.5-8 array. This restrictive SNP calling was done to avoid false calls within challenging regions of the MHC and to avoid rare variants that could interfere with haplotype estimation. The haplotype scaffold was completed by phasing the genotyped positions directly using the trio and also independently estimated using a prephased reference panel (see Supplemental Methods: Haplotype Scaffold). The proband phasing results derived from the two methods were compared to assess the accuracy of the phase estimation using the reference panel. A minimal number of SNPs were observed as being discrepant: 1 SNP per 1983 SNPs (0.05%) for EAS-P and 1 SNP per 1185 SNPs (0.08%) for the EUR-P sample. For the completion of the MHC haplotypic assembly of the probands, it was the phase estimation and not the trio derivation that was utilized. The phasing of the PGF + COX and the African sample was completed by using the prephased reference panel only.
Moving forward, the PacBio phased blocks are arranged along the haplotype scaffold. If SNVs in the phase block overlap with SNPs in the scaffold, all SNVs in the phase block can be assigned to a haplotype (Fig. 1H; Supplemental Fig. S1D; Methods: SNV Calling and Haplotype Partitioning). We were able to successfully partition 97.30%–99.77% of heterozygous SNV positions to one of the two haplotypes (Table 3). Following the intersecting process mentioned above, 51.98%–85.15% of the total number of reads used were assigned to either of the two haplotypes of each sample (Table 4). Between 14.15% and 47.29% of the reads were homozygous and were not assigned to a particular haplotype. The remaining 0.7%–3.06% of the reads, depending on the sample, could not be assigned to the haplotype scaffold and were assigned to both haplotypes. After the partitioning of the reads to the two haplotypes, each haplotype was assembled independently and the resulting contigs were patched and edited (Fig. 1I; Supplemental Fig. S1E,F; Supplemental Methods: Haplotype-Specific De Novo Assembly, De Novo Assembly Polishing).
Read partitioning statistics
Bionano scaffolding
The consensus Bionano maps were generated for the three samples: EAS-P, EUR-P, and AFA samples. Each sample was characterized by two to four Bionano map segments spanning the MHC. Thereafter, we compare the Bionano maps to the MHC haplotypes using the Hybrid Scaffolding pipeline (part of Bionano Solve, BioNano Genomics) and identify the best matching maps. This approach can be used to estimate the size of the gaps in our assembly, the layout of the RCCX repeats, and identify large-scale insertion and deletion errors, if any, in our assembly. RCCX is a common copy number variation (CNV) in the complement (Class III) region of the human MHC that includes the STK19, C4, CYP21, and TNX genes. The duplicated segment ranges from 26 (short) to 32 (long) kb depending on a human endogenous retrovirus insertion included in the C4 genes (HERVKC4); overall the RCCX region can contain multiple copies of the 26 or 32 kb segments. It should be noted that each of the RCCX repeats has 3 (long) or 2 (short) Bionano direct label enzyme 1 (DLE-1) sites depending on the presence or absence of HERVKC4 (Fig. 4). Therefore, this technology was particularly instructive in identifying the number of repeats in the RCCX region and whether the C4 gene contains the retrotransposon HERV sequence.
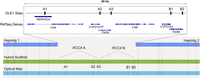
AFA RCCX evaluation using Bionano hybrid assembly. Haplotigs (dark blue) are combined with Bionano optical maps (light blue) to create a hybrid scaffold (green). The gap in between the haplotigs represents the RCCX CNV missing from the assembly. Each RCCX copy has at least two DLE-1 sites in each of the CYP21 and TNX genes, represented (A2, A3, B1, B2); A third DLE-1 site (A1) will be present if there is a HERVKC4 insertion in the C4 intron. Based on the count of the missing DLE-1 sites, the RCCX count and layout can be inferred.
Assessing the accuracy of the MHC haplotypes
For the PGF + COX mixture, even though the phasing was known because the sequences are independently known, the haploid sequences for each of the PGF and COX sequences were generated by using the phase estimation of hg38-based SNPs, restricted to microarray sites, instead of relying on the known sequences. Regarding the trios, while two methods were initially considered, ultimately the haploid sequences of the probands used phase-estimated genotyping data of the proband alone to generate the scaffold for the assembly (i.e., parental haplotypes were not considered). For AFA, trio data were unavailable, so the phase was generated only through estimation. In all cases, the assemblies for the probands (EAS-P and EUR-P), PGF, and COX sequences independently or AFA were assessed for accuracy.
When comparing assemblies, discrepancies are broken down into three different categories: (1) pipeline errors, (2) low coverage discrepancies, and (3) low complexity discrepancies. Pipeline errors can be further divided into two categories, phasing and assembly errors. Phasing errors occur when a variant within a contig, does not overlap with the phased SNPs of the microarray data, and the assignment of this contig to one of the two haplotypes can lead to assembly errors. Assembly errors occur when a read is either not corrected accurately (Sequel data) or when a read with remaining errors is incorporated into the final assembly. Low coverage discrepancies occur when the depth of coverage across a given region is too low for variant calling, read correction, or assembly. Low complexity region discrepancies occur in homopolymers or simple repeats. These regions commonly cause sequencing errors and often have low coverage due to either capture or sequencing challenges.
When comparing our COX or PGF assemblies to the known reference, the accuracy is reflective of just the COX or PGF assembly, however, when comparing family assemblies, the discrepancies come from either the proband or the parental assembly, as there is no true reference sequence. If the source of phasing or assembly discrepancy is from the parental assembly, it is not accounted as discrepant in the final accuracy assessment of the proband. Discrepancies in low coverage and low complexity regions are often difficult to assign to a specific assembly and are, therefore, always included in the accuracy assessment regardless of the assembly source (parental or proband). Overall, all kinds of errors or discrepancies ranged between 0.01% and 0.11% (Table 6).
Evaluation of PGF + COX sample
The PGF assembly generated two haplotigs across the MHC (Fig. 5) for a total coverage of 98.28% (Table 5). The reduced coverage is due to a gap (∼63 kb) located across the RCCX CNV within the Class III region of the MHC. The haplotigs were compared to reference using Quast to determine accuracy, which reported 565 erroneous bp for an overall accuracy of 99.98% (Table 6). Most low coverage regions were found in GC- or AT-rich stretches of the MHC.

Haplotype-resolved assemblies versus PGF. Each haplotype is aligned against PGF for visualization purposes. Each haplotype from the same sample appears in different colors. Black triangles indicate assembly breaks. Breaks in the haplotype alignment without triangles indicate that the sequence in the haplotype is highly divergent from PGF and does not align or the PGF sequence does not exist in the haplotype.
MHC haplotype coverage
MHC haplotype accuracy
The COX assembly generated three haplotigs across the MHC (Fig. 5) for a total coverage of 99.14% (Table 5). The reduced coverage is due to a ∼3.2 kb gap between HLA-B and HLA-C and a ∼24 kb gap located across the RCCX CNV (one copy). Quast reported 4057 bp differences across COX compared to the known COX reference for an overall accuracy of 99.89% (Table 6). The majority of the error comes from a single 3727 bp GC-rich region missing from the COX assembly due to low representation in the capture (subset of 3763 low coverage errors reported for COX in Table 6). The error occurred because this region is not present in PGF, and since there were not enough reads representing this insertion from COX, the pipeline erroneously considered it to be homozygous, adopting the PGF sequence for the region into the final assembly of COX.
Evaluation of proband samples
For the two proband samples, EAS-P and EUR-P, the total coverage was evaluated first. In the EAS-P sample, each of the haplotypes resulted in two haplotigs across the MHC (Fig. 5) totaling 3,466,357 bp for a total coverage of 97.47% for the maternal haplotype (EAS-P-1) and 3,668,815 bp for a total coverage of 97.78% for the paternal haplotype (EAS-P-2) (Table 5). In the EUR-P sample, both haplotypes resulted in three haplotigs across the MHC (Fig. 5), totaling 3,600,868 bp for a total coverage of 98.22% for the maternal haplotype (EUR-P-1) and 3,559,803 bp for a total coverage of 98.24% for the paternal haplotype (EUR-P-2) (Table 5). Both samples had a gap in the RCCX CNV region. Based on the Bionano data, we can predict the arrangement of this region for each haplotype: the EAS-P maternal haplotype is predicted to be a long (∼32 kb)–short (∼26 kb)–long (∼32 kb) arrangement of CNVs (∼89.8 kb), the EAS-P paternal haplotype is predicted to be a long–long–short arrangement (∼83.5 kb), and both haplotypes of the EUR-P sample are predicted to be a long–long arrangement (∼64.7 kb for the maternal haplotype and ∼63.5 kb for the paternal haplotype). Additionally, the EUR-P haplotypes were found to possess a small gap in coverage downstream from the HCG22 gene caused by low coverage across a GGGAGA repeat that is ∼272 bp in the maternal haplotype and 231 bp in the paternal haplotype.
Each assembly was then compared against the parental assembly of the shared haplotype to determine accuracy. For the EAS-P sample, the maternal haplotype differed by 230 bp between the two assemblies, resulting in an identity of 99.99%, and the paternal haplotype differed by 266 bp between the two assemblies, also resulting in an identity of 99.99% (Table 6). For the EUR-P sample, 1030 bp differed between the two assemblies for the maternal haplotype and 1173 bp differed between the two assemblies of the paternal haplotype, resulting in an identity of 99.97% for both the maternal and paternal haplotypes (Table 6).
Evaluation of the independent AFA sample
Parental data are not available for the AFA sample, so the accuracy of the assembly could not be measured in the same way as the previous samples. Instead, we compared the final assemblies of the two haplotypes to (1) the Bionano optical maps, (2) the independently genotyped HLA gene sequences, and (3) the SNP array genotyping data of the sample.
Both haplotype assemblies resulted in two haplotigs spanning the MHC, totaling 3,687,868 and 3,567,034 bp. In both cases, the only break was at the RCCX region. The Bionano consensus map reported 544 (haplotype 1) and 510 (haplotype 2) DLE-1 sites across the MHC. However, in each PacBio assembly across the RCCX CNV, five DLE-1 sites were missing, suggesting one long (three DLE-1 sites) and one short (two DLE-1 sites) CNV for both haplotypes, resulting in ∼58 kb gap in each haplotype (Fig. 4). Aside from the missing DLE-1 sites in the RCCX CNV region, there was only a single discordant DLE-1 site in haplotype 1 and no discordant sites in haplotype 2. Each DLE-1 site is 6 bp long, resulting in 6270 evaluated bp across the MHC. One missing DLE-1 site results in an error rate between 99.90% and 99.98%, depending on the number of errors across the 6 bp of each restriction site.
The 11 classical HLA loci for this sample were genotyped as part of the routine analysis within our clinical laboratory, including intronic sequences, and the corresponding genomic sequences of these HLA alleles were downloaded from the IPD-IMGT/HLA database (Barker et al. 2023). These sequences were compared against the assembly using BLAT to determine the accuracy. In haplotype 1, there was one discrepancy across 42,226 bp for an accuracy of 99.997%. In haplotype 2, there were nine discrepancies across 41,948 bp for an accuracy of 99.979%, where 6/9 errors in haplotype 2 are in the low complexity region downstream from exon 2 in HLA-DRB1 and HLA-DRB3.
The final accuracy assessment compares the microarray SNP genotyping of the sample to the bases in the final consensus sequences. Locations of the 10,352 microarray SNPs spanning the MHC (2430 heterozygous and 7922 homozygous) were found by mapping the probe sequence to each assembled haplotype and identifying the best match. If the probe sequence failed to align entirely or aligned but with more than two mismatches (120 hap1/146 hap2), the probe was excluded from the analysis. Alternatively, if the probe aligned to multiple locations equally well (3 hap1/10 hap2), the SNP was also removed from the analysis, leaving a total of 10,228 and 10,196 positions for the accuracy assessment (see Methods). There were a total of 18 discrepancies across both haplotypes. Each of these discrepancies was manually evaluated by viewing the raw reads at the genomic position of the SNP. The results of this comparison are described below.
Two of the discrepancies were caused by the mismapping of the probe due to indels in the assembly as compared to the probe/reference sequence. In both cases, the placement of the probe was off by a single base pair, and upon manual evaluation, it matched the microarray call.
The remaining 16 discrepancies were true differences between the microarray and assembly data. Seven SNPs were homozygous for the nonreference allele in the microarray with no evidence of the nonreference allele in the assembly or PacBio reads. Furthermore, all samples in this project were called homozygous nonreference in the microarray, including PGF, which is the reference sequence. Based on these two pieces of evidence, we believe the microarray call is incorrect. Additionally, seven SNPs were called homozygous reference while the assembly and raw PacBio reads were both either heterozygous or homozygous for the nonreference allele. The strong presence of the nonreference allele in the raw data is a good indicator that the microarray is incorrect. The final two SNPs were heterozygous in the microarray but appear homozygous in both the assembly and the raw reads. We tend to believe the raw reads and the assembly over the microarray at these two positions.
Therefore, after manual checks, none of the discrepancies appear to be mistakes in the assembly. To determine accuracy, we compared the number of SNPs on the microarray that did not include the 18 discrepancies to the respective positions on the assembled haplotypes. The concordance across the 10,218 and 10,188 evaluated positions of the two haplotypes was 100%.
Gene-centric accuracy assessments
Accuracy of HLA genes
HLA genes are highly polymorphic and arguably the most studied genes in the MHC, making them ideal candidates to further evaluate the accuracy of the assemblies. Each HLA locus was identified in the assembly, genotyped, and compared to traditional Illumina-based HLA typing data at three fields (comparing the nucleotide sequence of the exons for each gene; see https://hla.alleles.org/nomenclature/naming.html for naming conventions). All loci except for HLA-A were concordant at three fields (Supplemental Table S1) (N = 8: HLA-B, HLA-C, HLA-DQB1, HLA-DQA1, HLA-DPB1, HLA-DPA1, HLA-DRB1; N = 4 HLA-DRB3; N = 2 HLA-DRB4; N = 1 HLA-DRB5). The discordant HLA-A locus in the AFA sample appeared to be homozygous in the assembly, but in truth, there is a single synonymous difference between the two haplotypes. The heterozygous position could not be linked to the haplotype scaffold, resulting in the error. Regarding nonclassical HLA genes and HLA pseudogenes, even though we do not have a gold standard for comparison for all samples, we have genotyped these loci within the final assemblies (Supplemental Table S2). For PGF and COX, genotyping information is available (IPD-IMGT/HLA), and it was found that there was concordance for all these genes when compared at three fields. For the remaining samples, the accuracy could not be assessed; however, the majority of the alleles match already described alleles within the IPD-IMGT/HLA database at three fields.
Haplotype structure of the HLA-DR region
The biggest source of complexity across the MHC is the HLA-DR region, which starts with HLA-DRA, ends with HLA-DRB1, and may or may not include the genes: HLA-DRB3, HLA-DRB4, and HLA-DRB5 and the pseudogenes: HLA-DRB2, HLA-DRB5, HLA-DRB6, HLA-DRB7, HLA-DRB8, and HLA-DRB9. There are five major haplotype groups: DR1, DR51, DR52, DR53, and DR8, four of which are represented in our samples. To further test the accuracy of our assemblies, the sequences of each gene and pseudogene were extracted from our assemblies and genotyped. The resulting layout for each assembled haplotype with allele names is shown in Supplemental Figure S2. All our assemblies are consistent with known haplotypes and have realistic genotyping data across the various HLA-DR genes suggesting accurate assembly across this complex region.
Discrepancies and gaps within genic regions
After cataloging discrepancies across each assembly, we decided to identify their locations across the MHC relative to known genes. Each discrepancy was mapped to hg38 and annotated using GENCODE basic transcripts (v45). Discrepancies could have multiple classifications due to alternative transcripts, so the most damaging (coding sequence [CDS] > untranslated region [UTR] > noncoding exon > intergenic/intron) was selected (Table 7). The CDS is estimated to be ∼6% of the MHC, but only contains between 0% and 1.07% of the error, making the CDS accuracy for the three samples >99.99%. Most of the error (94.47%–95.92%) falls within intronic and intergenic regions. This compartment is enriched because most of the observed error is in low complexity regions which mostly fall outside of genes. A final analysis included assessing the gaps in the assembly for gene loss. Gaps across the RCCX CNV region include C4, CYP21, TNX, and the STK19 genes. The three gaps that occur outside of the RCCX CNV region are all intergenic and do not involve known genes.
The number and percentage of discrepancies within various genomic compartments, per haplotype
Discussion
In this report, we present a methodology for haplotypic sequencing of the MHC region of random individual heterozygous samples, which has traditionally been a challenging task despite its biological significance and advances in genomics. However, recent developments in targeted DNA capture, sequencing technologies generating long and accurate reads, and software solutions for de novo assembly, have created an opportunity to address this problem (Jain et al. 2016; Mostovoy et al. 2016; Shin et al. 2019; Wenger et al. 2019; Houwaart et al. 2023). Our approach is characterized by its high coverage, high accuracy, and reasonable cost.
The key features of our method include: (1) targeted capture of the MHC, which avoids the need for massive sequencing of the entire genome, (2) the use of unique bioinformatic tools to address the problem of hybrid reads with palindromes generated after WGA, enabling the editing and identification of most of the MHC sequences after capture, and (3) use of haplotype estimation methods to improve phasing across the MHC. This method has the potential to process multiple samples in a relatively short time.
To ensure the accuracy of the derived MHC sequences, we have designed experiments that secure the proper assessment of haplotypic characterization. We have chosen samples such as a mixture of DNA from two homozygous B cell lines of already known MHC sequences, family trios, and a completely random sample to properly assess accuracy. It is to be noted that the family trios served the purpose of generating MHC haplotypic information for the proband, so the accuracy of the independent derivation of the MHC haplotypes of the proband could be assessed. The parental information from the trio was not used to inform the MHC haplotypic derivation for the probands. It was only a vehicle to assess accuracy. However, the method needs to be further validated with many subjects from different populations to identify the limits of our approach. Most recently, the availability of the HPRC (Human Pangenome Research Consortium) samples (Liao et al. 2023), whereby the MHC has been fully characterized, generates a new opportunity to assess the accuracy of our targeted approach for the MHC. We are using these HPRC samples currently, as we compare long-read sequencing technologies and computational tools for accurate haplotypic characterization of the MHC (work in progress).
Our approach relies heavily on the RSE methodology we have developed for targeting the MHC. This powerful method is complemented by the AnthOligo oligo design software program (Jayaraman et al. 2020), and both have been thoroughly tested and validated using the PGF cell line (Dapprich et al. 2016). In this report, we build on our previous work and demonstrate how the same RSE methodology can be used to characterize the two different MHC haplotypes of random heterozygous samples. Our approach is a promising solution for characterizing other challenging and highly variable genomic regions. In fact, this methodology and software have been successfully used to discover novel HLA alleles (Steiner et al. 2018) and characterize other genomic regions, such as those in zebrafish (Gupta et al. 2010) and RH blood groups (Zhang et al. 2022). While no classical HLA alleles were discovered as part of this study, as the HLA genotype was already known for these genes, we did identify novel allele sequences in the nonclassical HLA genes and HLA pseudogenes (Supplemental Fig. S2; Supplemental Table S2), demonstrating the ability of the method to characterize the variability inherent to the MHC.
One of the key advantages of our RSE method is its elegant design principle. Unlike other methods that depend on multiple overlapping capture oligos, our approach uses oligos every 5–10 kb that hybridize to the region of interest. Through an enzymatic extension process, biotinylated nucleotides are incorporated, which are then used to capture the target fragments (see details in Dapprich et al. [2016] and Jayaraman et al. [2020]). This strategy allows us to capture regions with extensive polymorphisms, regardless of the reference sequence used for the oligo design. The oligos are designed to hybridize to nonpolymorphic regions, so a single set of designed oligos can be used for targeting different samples, without the need for updating the oligos based on the sample. It is conceivable that in some cases we may need to enrich a region with additional oligos if the capture efficiency is low in a particular subregion or entirely absent. This can be easily accomplished by adding more oligos located near the region of interest. As a matter of fact, we used 94 MHC Pangenome haplotypes to assess the performance of this panel of oligos in silico and found that there is an insertion of ∼64 kb that is not targeted by this oligo panel. No other regions were found to not be targeted by this panel in the 94 haplotypes.
The advancement of sequencing technologies and platforms, such as PacBio, has the potential to enable the haplotypic characterization of the MHC. This is primarily due to the technology's capability to produce long reads with a notable level of accuracy. In our work, we have demonstrated that most of the captured fragments used for the assembly of the MHC (as shown in Fig. 2) were reads ranging from 1 to 6 kb in length. The importance of obtaining long reads cannot be overstated, as the MHC region contains structural variation and long repetitive regions that may be missed or ambiguously mapped and aligned using short-read technologies. Therefore, our ongoing efforts are focused on searching for improvements that will enable us to generate even longer reads.
The improvements to the RSE capture efficiency together with the increasing capacity of the sequencing platform allow for multiple samples to be run simultaneously. The on-target percentage of reads (i.e., MHC-specific) varies from 10.33% to 52.58% (86.09- to 438.13-fold enrichment), depending on the quality of DNA used for capture, where we have found that extracting DNA from a fresh sample yields better results. It is not surprising that a good amount of the captured material belongs to nontargeted regions, as nontargeted fragments can be pulled along with the targeted sequences regardless of whether they are biotinylated, simply because they are entangled with the long fragments. The high percentage is not concerning, as they are dispersed among the whole genome with exceedingly low depth of coverage, while the targeted reads are plentiful and generate a high depth of coverage in the targeted region. Additionally, in this work, we have sequenced samples on both the Sequel and Sequel II platforms (Table 1), with a clear difference in throughput. Using the Sequel system, a single sample requires 2–3 flow cells to generate enough reads to characterize the MHC, whereas a single sample run on the Sequel II only needed half of a flow cell. Notably, we utilized only a fraction (600,000/1,912,901 = 31%) (see Table 1) of the HiFi reads obtained for the single sample of EAS-P on Sequel II for the MHC characterization, indicating that a single flow cell can accommodate two to three samples, further improving on the efficiency of the method. While there is a small chance of demultiplexing errors (<0.67%) when multiple samples are run on the same flow cell, none of the reads incorrectly assigned to the wrong sample after demultiplexing caused errors in the assembly. With fresh DNA, one run on the Sequel II, utilizing 8 flow cells, can potentially accommodate MHC sequencing of 16–24 samples. By optimizing the percentage of on-target reads, the number of samples can be further increased and, therefore, reduce cost.
Our approach of using reference-based SNP calling, restricted to microarray sites, to construct the scaffolding that forms the basis for MHC haplotyping, is critical for assembling the MHC when samples are heterozygous. This work has shown that our method of combining physical phasing with estimation (Delaneau et al. 2019) can be successful in generating correct haplotyping without trios, which is especially important for disease association studies where trio information is not always available. It is also possible to validate the reference-based SNP calling using microarray genotyping data. This extra step is a simple way to secure confident genotyping across difficult-to-capture regions. Furthermore, microarray genotyping allows for inexpensive trio phasing when parental samples are available, and assembly is only required for the proband.
It is important to note that any gaps in the phasing of PacBio sequencing blocks during assembly, as shown in Figure 5, do not necessarily disrupt the haplotyping of the MHC sequence as a whole. The continuity of SNPs provided by the haplotype scaffold is sufficient to guide the assignment of PacBio blocks to one of the two haplotypes, even though the scaffold was restricted to 11,500 possible microarray sites across the 3.6 Mb MHC region. The average spacing between SNPs in the scaffold was 321 bp, with the largest gaps near the RCCX CNV (∼32 kb) and DRB5 (∼28 kb). All 142 genes classified as protein coding by Ensembl had microarray SNPs present, with an average count of 60 SNPs per gene plus 5 kb flanking sequences. The genes with the lowest counts were HLA-DRB5 with seven SNPs and C4A with six SNPs. This limited set of SNP sites in the haplotype scaffold contains enough polymorphic sites to secure the assignment of a PacBio block to one of the two haplotypes. The majority of reads obtained were 1–6 kb long, as depicted in Figure 2; however, there are certain challenging regions, such as a segment within the Class III (Complement) region, that remain difficult to assemble. Specifically, the RCCX sequences consist of different numbers of long and short repeats, each being ∼32 and 26 kb, respectively, and cannot be accurately assembled with the size of the sequencing reads obtained using the current protocol. Improving the number of long reads is likely to address this issue. The Bionano data partially address this problem by allowing the determination of the number and order of long and short repeats, thereby determining the overall length of the gap. However, credible sequencing information for each of the long and short repeats is not provided. Despite this challenge, it should be noted that the haplotypic arranged sequences before and after the RCCX gap remain intact. The haplotype scaffold provided the necessary context for the proper assignment of the PacBio phased block on the maternal or paternal haplotype before and after the gap. Therefore, Bionano is a valuable quality assessment technology that allows the identification and characterization of large-scale assembly problems, such as deletions or insertions, in a random sample where trio information is not available.
The successful haplotypic analysis of the MHC region was dependent on the deconvolution of raw reads generated by the sequencing platform, which included hybrid reads containing palindromes. These hybrid reads are primarily generated during the WGA step after capture. The utilization of appropriate bioinformatic tools enabled the editing and identification of most of these hybrid MHC sequences. This bioinformatic intervention facilitated the assembly and formation of the PacBio sequencing blocks, which were then accurately assigned to their respective haplotype scaffold. We recognize that new assemblers, like HifiAsm, have been introduced to resolve haplotypes with PacBio Hifi reads; however, the expectations for reads that feed into HifiAsm are not met with this system, and this tool was unable to produce a usable assembly. We have found that Canu is better for this application because it is highly configurable and able to tolerate errors that may remain after hybrid/palindrome processing. Furthermore, our approach, unlike HifiAsm, is able to incorporate estimation for phasing, which is highly beneficial given the shorter read lengths after processing.
Our experimental design allowed for a credible assessment of the accurate sequencing and haplotypic formation of the selected samples. In summary, the coverage for each haplotype ranged from 97.47% for the EAS-P1 to 99.14% for the COX BLCL. The percent drop in coverage is primarily due to RCCX CNVs in the complement region that for the EAS-P1 sample was the largest (∼90 kb), and, therefore, the comparably less percent coverage for this sample. Accuracy of the same haplotypes was ranging from 99.89% to 99.99%, whereby the primary source of discrepancies was the low coverage of sequencing at particular points or the low complexity of regions with homopolymers. The AFA-haplotypes were characterized by both excellent coverage (98.47% and 98.41% accounting for the ∼57 kb of RCCX CNVs for each haplotype; 100% coverage without the RCCX CNVs) and accuracy at the level of 99.99% and 99.98% for each of the AFA-1 and AFA-2 haplotypes, respectively.
What contributes to both percent coverage and accuracy is the depth of coverage for the MHC region. We have compared the depth of coverage obtained in our results, that is over 200× for all samples (see Fig. 3), to the depth reported by Houwaart et al. (2023), which reports the average depth of coverage for MHC region of six diverse haplotypes using (1) the Illumina-based whole genome sequencing at 15.05× (SD = 2.27); (2) PacBio-based whole genome sequencing at 10.56× (SD = 1.02); and (3) Oxford Nanopore-based at 34.24× (SD = 25.8). Additionally, the HPRC reports an average coverage of 39.7× for 46 samples with PacBio HiFi reads (Liao et al. 2023), although this is not specific to the MHC region. The depth of coverage in our study to some extent has been dictated by the variation observed along the MHC as a result of our capture approach.
Regarding the comparison of our assemblies to similar data in the literature, we evaluated the 35 available 1000 Genomes Project (1KGP) sample assemblies from (Ebert et al. 2021) for continuity across the region captured in our work. For the nine 1KGP samples with both CLR and HiFi reads, we only use the HiFi data for comparison, leaving a total of 21 CLR and 14 HiFi assemblies to compare with our work. Forty-five out of 70 haplotypes were complete across the MHC, the remaining 25 had one or more breaks in the MHC. The most common breaks in the 1KGP samples were across the HLA-DR region (14/70) and the RCCX region (10/70). There were seven breaks outside of the two hotspots, one of which matched with the breaks we observed in the EUR-P sample downstream from HCG22. Breaks in the HLA-DR region for the 1KGP samples led to the loss of HLA-DRB4 (N = 6), HLA-DRB3 (N = 1), breaks within HLA-DRB1 (N = 1), and duplications (N = 2). While breaks were found in both CLR and HiFi samples, gene loss was only in the CLR assemblies. Our assemblies overall are rather comparable; however, our approach across the RCCX CNV region has limitations because of our shorter read lengths, while our assembly of the HLA-DR region is complete in all haplotypes.
It is reasonable to expect that the characterization and collection of a library of complete and accurate MHC sequences from different populations will enable the expedited and accurate analysis of MHC in new samples. This process has already commenced, with initiatives such as the Human Pangenome Reference Consortium's recent efforts to sequence 350 human genomes using long reads and de novo assemblies, contributing to our knowledge and the credible characterization of the whole MHC region. Additionally, the 1000 Genome Project provides MHC sequences, although not very comprehensive. Recently, working with samples from various African populations, we reported 140 new alleles in a population of only 485 individuals (Pagkrati et al. 2023), indicating extensive polymorphism in HLAs and most likely in other parts of the MHC, which has not yet been fully appreciated. As a result, we intend to broaden our studies to include the characterization of the MHC from several populations worldwide, which may require modifications to various parts of the methodology to fully and accurately characterize these samples.
The MHC is a genomic region known to contain genetic variations that may contribute to disease. However, due to extensive linkage disequilibrium within this region, distinguishing disease-causing variants from bystander variants can be challenging. Fortunately, by using detailed and accurate sequencing of the entire MHC in haplotypic form, we can improve our ability to detect disease-related genetic variations. This advancement sets the stage for future studies that could revolutionize our understanding of the relationship between HLA and disease associations, potentially enabling the identification of many polymorphisms and structural variations within the MHC that are linked to different diseases, not just HLAs.
Methods
Sample selection and DNA extraction
A total of nine samples were selected for studying their MHC. First, the BLCL of PGF (obtained from the Coriell Institute GM03107) and COX (obtained from the International Histocompatibility Working Group IHW09022, http://www.ihwg.org/hla/index.html) are both homozygous for Chromosome 6 and, therefore, throughout the MHC region. The MHC sequences of these two BLCL cells are currently used as reference sequences for the human genome (Allcock et al. 2002). Whole blood samples were collected from two family trios (father, mother, and child) and a single individual. One family self-identified as Chinese, the second as European, and the single individual self-identified as African American. All subjects have consented for using their DNA for this project. The PGF and COX cell lines were cultured using RPMI 1640 medium (Gibco, Thermo Fisher Scientific 72400047) containing 15% fetal bovine serum (Sigma-Aldrich F2442) at 37°C and 5% CO2. Genomic DNA (gDNA) was extracted using a Blood and Cell Culture DNA Midi Kit (Qiagen 13343). For the seven whole blood samples, gDNA was isolated on an EZ1 Advanced XL instrument using an EZ1 DNA Blood 350 μL Kit (Qiagen 951054) and EZ1 Advanced XL DNA blood card (Qiagen 9018695).
HLA genotyping
DNA was used to characterize the 11 HLA genes (HLA-A, -B, -C, -DRB1, -DRB3, -DRB4, -DRB5, -DQA1, -DQB1, -DPA1, and -DPB1), using the Holotype HLA kit (Omixon) following the manufacturer's instructions. Sequencing was performed on an Illumina MiSeq instrument using either 2 × 150 or 2 × 250 paired-end sequencing on version 2 flow cells. The HLA genotype was determined using a custom pipeline that combines the output from Omixon's Twin software with GenDx's NGSengine software. Most genes were characterized for the full length of the gene from the 5′ UTR to the 3′ UTR (HLA-A, -B, -C, -DPA1, -DQA1, -DQB1), while other genes were partially characterized (HLA-DPB1 and -DRB5: intron 1 to 3′ UTR; HLA-DRB1 and -DRB3: intron 1 to intron 4; HLA-DRB4: intron 1 to exon 4). All sequences used for comparison purposes were obtained from the IPD-IMGT/HLA database (Barker et al. 2023). The sequences obtained correspond to the amplified region for each gene.
SNP genotyping
All samples were SNP genotyped with the Illumina Infinium Omni2.5-8v3 array and were analyzed with the Illumina GenomeStudio Genotyping Module v2.0.2 at the CHOP Center of Applied Genomics.
RSE oligo design
Oligos for RSE were designed by AnthOligo (http://antholigo.chop.edu), a web-based application developed to optimally automate the process of generation of oligo sequences used to target and capture the continuum of large and complex genomic regions (Jayaraman et al. 2020). A total of 434 oligos were designed to target the MHC Class I and Class III regions (MOG to HLA-DRA, hg38, Chr 6: 29,656,840–32,389,788), based on the PGF cell line, as the reference genome. An additional 290 oligos were designed to target the MHC Class II region (HLA-DRA to DAXX, hg38, Chr 6: 32,389,788–33,275,157) of the standard Chromosome 6 (PGF—HLA-DRB5 positive), but also of the alternative haplotype arrangements of the human genome reference that correspond to the BLCLs COX (HLA-DRB3 positive), QBL (HLA-DRB3 positive), SSTO (HLA-DRB4 positive), and MANN (HLA-DRB4 positive). Oligos targeting the same region in all cell lines were removed. Oligos and supporting information are included in Supplemental Table S3. Oligos were synthesized by IDT (Integrated DNA Technologies), provided in “Lab Ready” format, and prediluted to 100 μM. All 724 oligos were combined in water to an equimolar ratio.
Region-specific extraction
RSEs were performed using the Region Specific Extraction kit (rse100-kit-700) from Generation Biotech. Each 90 μL RSE reaction contained ∼2 μg of gDNA, 4 μL of 100 μM region-specific oligo mixture, H buffer (1×) supplemented with betaine to enhance the capture of GC-rich regions (Henke et al. 1997) and DNase-free water. The RSE mixture was placed on a Veriti thermal cycler (Applied Biosystems, Thermo Fisher Scientific) at 94°C for 5 min to denature the DNA followed by 64°C for 20 min to allow for oligos to bind DNA and for extension to occur. The targeted gDNA was captured by incubating with 90 μL of RSE-B beads for 30 min at room temperature with gentle mixing at 10 min intervals. Beads were collected using a DynaMag-2 Magnet (Thermo Fisher Scientific), washed twice with DNase-free water, and resuspended in 90 μL of R buffer from the RSE kit. Captured DNA was eluted in the supernatant by heating the solution of collected beads at 80°C for 15 min.
WGA
Captured DNA was subjected to WGA using the REPLI-g Midi Kit (Qiagen 150043) using a modified protocol without denaturation and neutralization. Twenty microliters of each RSE sample was mixed with 30 μL master mix in a PCR tube and placed on a Veriti thermal cycler at 30°C for 2–8 h, followed by inactivation of the enzyme at 65°C for 3 min. Residual primers and dNTPs were deactivated with 1 μL of ExoSAP-IT (Affymetrix 78201) according to the manufacturer's protocol. Five micrograms of the final DNA product of each sample was submitted for PacBio sequencing.
Capture efficiency assessment
PrimeTime standard qPCR assays (IDT) were used to assess the capture efficiency of RSE using a standard curve method. The primers and probes were designed using PrimerQuest Tool (IDT, Primer 3 version 2.2.3), targeting the conserved regions across five MHC haplotypes (PGF, COX, QBL, SSTO, and MANN). Six pairs of primers and probes were chosen for use to target different regions of the MHC: M315 and M317 for the MHC Class I region, M321 for the MHC Class III region, and M324, M329, and M332 for the Class II region. β-Actin was used as the negative control. All qPCR primers and probes are contained in Supplemental Table S4. For each qPCR assay, 8 μL of WGA product was combined with 1× Quantitect Probe PCR master mix (Qiagen 204345), 0.5 μM each of forward and reverse primers, and 0.25 μM probe. Six 1:3 serially diluted PGF gDNA standards were run in duplicate for each locus as well as a single background control. The qPCR assay was run on a StepOnePlus Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific) with an initial denaturation at 95°C for 15 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min.
PacBio sequencing
PacBio SMRT bell libraries were prepared using SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences 100-938-900) and sequenced on a PacBio Sequel or Sequel II System after a 5 kb size-selection by the DNA Sequencing and Genotyping Center at the University of Delaware. Samples were sequenced in two sets; the European trio was done earlier using the Sequel platform and the remainder was sequenced on the newer Sequel II platform. Reads generated by the older Sequel platform are not all HiFi quality, so the assembly pipeline differs at a few steps to account for the higher error rate.
Sequencing on the Sequel system required 2–3 flow cells per sample. Subreads from the same SMRTbell molecule were combined into a single consensus read using the PacBio CCS tool (v6.4), requiring a length of at least 1 kb and without filtering on minimum number of passes or read quality (‐‐min-passes 0 –min-rq 0 –min-length 1000). Approximately 80% of the consensus reads were retained, 11%–18% of which were HiFi (≥3 passes, RQ ≥ 0.99).
Sequencing on the Sequel II system accommodated one to two samples per flow cell. Subreads were combined into a single consensus sequence using the PacBio CCS tool, requiring a length of at least 1 kb and retaining only HiFi reads. Reads were demultiplexed with lima (v2.6.0) using default settings. Approximately 40% of the consensus reads were retained per sample.
Structural analysis with Bionano Genomics
Ultra-high molecular weight (UHMW) DNA was extracted from cultured cell lines or whole blood samples using the Bionano Prep SP Blood and Cell Culture DNA Isolation Kit (Bionano Genomics 80042) according to the manufacturer's instructions. The UHMW DNA was digested with DLE-1 and stained with the Bionano Prep DLS Kit (80005) and visualized on the Saphyr System using Saphyr G1.2 chips (20319) according to the manufacturer's instructions. The Saphyr system generated the optical assembly maps using the default parameters for each sample. Bionano de novo assemblies were generated and aligned to the human reference using Bionano Access (v1.6) by the CHOP Center of Applied Genomics. Bionano maps that aligned across the MHC region were extracted and compared to the PacBio de novo assembly using the Bionano hybrid scaffold pipeline (v3.5.1). Scaffolds were checked for large structural variants and to determine the size of the gaps between haplotigs.
Overview of the analysis process and computational pipeline
Read selection for the targeted MHC region was performed by finding reads that aligned to the region using an initial reference panel. The panel of reference MHC sequences was created by using a combination of haplotypes included in the hg38 reference build and haplotype assemblies generated by the HPRC and the Human Genome Structural Variation Consortium (HSGSVC) including a total of 160 haplotypes (see Supplemental Methods: MHC Reference Construction). In settings where the sequence being aligned belongs to a sample that is part of the reference panel, i.e., COX or PGF, the sample was removed from the reference panel for testing purposes. Once the initial filtering process to identify reads within the targeted region was completed, the reference panel was no longer used in order to prevent any potential bias in sequence assembly.
Before assembly, MHC-specific reads (Fig. 1A) were edited to remove potential chimeric segments introduced by the WGA reaction (Fig. 1B). Most of the chimeras are palindromic, meaning the second segment of the chimera is an inverted repeat of the first. Nonpalindromic chimeras are also observed, where the segments are nearby each other in the genome, but are either nonadjacent or inverted with respect to the genome (see Supplemental Methods: MHC Read Editing by Overlap). Edited reads were assembled (Fig. 1C), and contigs that were off target, low depth of coverage, or highly overlapping were removed (see Supplemental Methods: Collapsed De Novo Assembly). The resulting collapsed assembly was used to improve chimera detection in the MHC-specific reads (edited reads) (Fig. 1D; see Supplemental Methods: MHC Read Editing by Alignment).
Edited reads were aligned to the collapsed contigs and the resulting alignment was used for SNV calling (Fig. 1E) and phasing (Fig. 1F). Reads were assigned a local haplotype through a process called haplotagging (Fig. 1G). Hg38-based SNP genotyping, restricted to microarray positions, phased through either trios or computational estimation (see Supplemental Methods: Haplotype Scaffold) was intersected with the SNV calls on the contigs, orienting the local PacBio haplotype blocks to the larger MHC haplotype (Fig. 1H; see Supplemental Methods: SNV Calling and Haplotype Partitioning).
After partitioning the reads into two haplotype pools, a final haplotypic assembly is performed (Fig. 1I; see Supplemental Methods: Haplotype-Specific De Novo Assembly and Haplotype-Specific Patched De Novo Assembly). Further editing was performed to fix breaks, trim palindromic haplotig ends, and merge overlapping haplotigs to produce a final de novo assembly (see Supplemental Methods: De Novo Assembly Polishing).
Accuracy assessment
The sequences across HLA genes were identified in the assembly to (1) check the accuracy of the HLA typing and (2) ensure that the complex HLA-DR region was assembled correctly. First, the final assemblies were aligned to Chr 6 and the HLA-DR regions of Chr6_GL000255v2_alt and Chr6_GL000256v2_alt using minimap2 (Li 2018). Next, sequences that overlap each canonical HLA locus or any HLA-DRB pseudogene were extracted and HLA typed using GenDx's NGSEngine (version 2.31) in PacBio consensus mode with IPD-IMGT/HLA version 3.55. Sequences were also compared using ImmunAnnot (version available on April 9, 2024) with the database version February 2, 2024 using default settings (Zhou et al. 2024).
The assembly HLA typing results for each canonical locus were compared against clinical grade Illumina typing results (see section “HLA Genotyping”) to check for consistency.
The presence or absence of NGSEngine typing results for each HLA-DRB gene and pseudogene was recorded for each haplotype. The map of HLA-DR genes/pseudogenes in the assembly was compared to the five common HLA-DR haplotypes (DR1, DR51, DR53, and DR8) to check for consistency and completeness. Discordant positions in the assembly were mapped to Chr 6 or the HLA-DR region of one of the alternate MHC haplotypes in hg38. Positions were converted to VCF format and annotated using Ensembl's VEP (McLaren et al. 2016) using Ensembl basic annotations and reporting the most severe consequence per variant. Basic annotations prioritize the full-length coding transcripts over partial or noncoding transcripts for the same gene.
Data access
The raw sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA1008148). The code for analysis, microarray data, and assemblies of the haplotypic MHC are available for download at GitHub (https://github.com/tris-10/MHC_RSE_HiFi_Assembly_Pipeline) and as Supplemental Code.
Competing interest statement
D.S.M. is the Chair of the Scientific Advisory Board of Omixon and owns options in Omixon. D.S.M and J.L.D. receive royalties from Omixon. The other authors have no conflicts of interest to disclose.
Acknowledgments
We thank the CHOP Immunogenetics Clinical Laboratory for their work in generating the reference HLA typing for the samples studied. We thank the Center for Applied Genomics at CHOP for running the microarrays and use of Bionano Genomics instrument and server. Finally, we thank the individuals who contributed the biological material that was used for the basis of this study. CHOP institutional funds to D.S.M. were used to support this work.
Author contributions: D.S.M. designed the study; P.J., M.S., T.L.M., and T.H. designed the oligos; T.H., N.G.T., A.D., and Y.L. performed the RSE capture; K.B. prepared the libraries and sequenced the samples on the PacBio; M.S. performed the Bionano Genomic experiments; T.H. prepared the microarray experiments; T.L.M. performed the bioinformatic analysis and haplotype construction; T.H., T.L.M., J.L.D., Y.L., T.J.H., and D.S.M. all wrote the paper. All authors provided critical feedback and approved the final version of the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278588.123.
-
Freely available online through the Genome Research Open Access option.
- Received October 26, 2023.
- Accepted September 19, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








