
Comprehensive isoform-level analysis reveals the contribution of alternative isoforms to venom evolution and repertoire diversity
- Xinhai Ye1,2,3,
- Chun He1,
- Yi Yang1,
- Yu H. Sun4,
- Shijiao Xiong1,
- Kevin C. Chan2,
- Yuxuan Si3,
- Shan Xiao1,
- Xianxin Zhao1,
- Haiwei Lin1,
- Yang Mei1,
- Yufeng Yao5,
- Gongyin Ye1,
- Fei Wu2,3 and
- Qi Fang1
- 1State Key Laboratory of Rice Biology and Breeding & Ministry of Agricultural and Rural Affairs Key Laboratory of Molecular Biology of Crop Pathogens and Insects, Institute of Insect Sciences, Zhejiang University, Hangzhou 310058, China;
- 2Shanghai Institute for Advanced Study, Zhejiang University, Shanghai 201203, China;
- 3College of Computer Science and Technology, Zhejiang University, Hangzhou 310027, China;
- 4Department of Biology, University of Rochester, Rochester, New York 14627, USA;
- 5Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou 310020, China
Abstract
Animal venom systems have emerged as valuable models for investigating how novel polygenic phenotypes may arise from gene evolution by varying molecular mechanisms. However, a significant portion of venom genes produce alternative mRNA isoforms that have not been extensively characterized, hindering a comprehensive understanding of venom biology. In this study, we present a full-length isoform-level profiling workflow integrating multiple RNA sequencing technologies, allowing us to reconstruct a high-resolution transcriptome landscape of venom genes in the parasitoid wasp Pteromalus puparum. Our findings demonstrate that more than half of the venom genes generate multiple isoforms within the venom gland. Through mass spectrometry analysis, we confirm that alternative splicing contributes to the diversity of venom proteins, acting as a mechanism for expanding the venom repertoire. Notably, we identified seven venom genes that exhibit distinct isoform usages between the venom gland and other tissues. Furthermore, evolutionary analyses of venom serpin3 and orcokinin further reveal that the co-option of an ancient isoform and a newly evolved isoform, respectively, contributes to venom recruitment, providing valuable insights into the genetic mechanisms driving venom evolution in parasitoid wasps. Overall, our study presents a comprehensive investigation of venom genes at the isoform level, significantly advancing our understanding of alternative isoforms in venom diversity and evolution and setting the stage for further in-depth research on venoms.
Venom undergoes independent evolution across various animal phyla, such as snakes, platypuses, spiders, wasps, and cone snails. It serves as a crucial adaptive trait in these venomous species, offering a remarkable model for comprehending the molecular mechanisms that drive evolutionary innovations (Arbuckle 2020; Zancolli and Casewell 2020). Moreover, venom presents excellent opportunities for investigating the emergence of new gene functions and the evolution of novel regulatory networks. Different venomous animals have developed diverse venom genes for distinct purposes (Casewell et al. 2013; Reyes-Velasco et al. 2015; Schield et al. 2019, 2022; Giorgianni et al. 2020; Almeida et al. 2021; Barua and Mikheyev 2021; Mason et al. 2022; Perry et al. 2022). Although extensive studies on venom genes have provided profound insights into venom diversity, origins, turnover, regulatory mechanisms, and biological functions, the majority of these studies have focused on the gene level (Casewell et al. 2013; Suryamohan et al. 2020; Almeida et al. 2021). For instance, the transcript encoding the longest protein is often used as the representative transcript for subsequent analyses. However, the lack of resolution at the isoform level can impede the precise understanding of venom biology. This limitation can be attributed to the use of short-read sequencing technology during transcriptome assembly. Although Illumina short reads, in combination with assembly software like StringTie (Pertea et al. 2015) and Trinity (Grabherr et al. 2011), can be used to assemble isoforms, defining genes with complex alternative splicing remains challenging. Consequently, only partial RNA transcripts are covered, and the transcriptome assembly fails to distinguish alternative RNA splicing landscapes. Eukaryotes have been widely reported to possess variant transcripts resulting from diverse alternative splicing events, as well as alternative transcription start sites and alternative polyadenylation sites (Merkin et al. 2012; Mazin et al. 2021; Wright et al. 2022). In a few investigations on a snake and a spider, multiple isoforms of venom genes have been discovered (Cousin et al. 1998; Haney et al. 2019; Ogawa et al. 2019). A study focusing on the Protobothrops snake, using long-read RNA sequencing, identified three venom gene families with extensive alternative splicing, which likely contribute to the production of divergent venom proteins (Ogawa et al. 2019). However, it remains unclear whether this phenomenon occurs in other venomous species and, if so, whether a high proportion of venom genes possess multiple isoforms. Furthermore, several fundamental aspects of alternative isoforms underlying the rapid evolution of venom composition remain poorly understood, including the contribution of these isoforms to venom functional diversity, the evolution of venom genes with multiple isoforms, the role of alternative isoforms in venom recruitment, and the regulatory processes governing these isoforms.
Parasitoid wasps encompass a diverse group of venomous species. Their venoms fulfill multiple functions in regulating gene expression, immune systems, and metabolism in their invertebrate hosts, which are crucial for parasitism (Asgari and Rivers 2011; Martinson et al. 2014; Yang et al. 2021). Notably, significant turnover of venom repertoires at the gene level has been observed among parasitoid wasp species, even within the same genus, likely driven by the necessity to adapt to changing host ranges (Martinson et al. 2017; Ye et al. 2022). As a result, parasitoid wasps are emerging as a rapidly evolving model for studying venom evolution and the emergence of new gene functions. However, our understanding of parasitoid venoms is primarily limited to the gene level, with only a few studies identifying venom isoforms of individual genes using computational predictions or relying solely on short-read RNA sequencing, which may yield incomplete or inaccurate results (Colinet et al. 2014; Yan et al. 2016; Teng et al. 2017). Additionally, previous research has demonstrated that the recruitment of existing genes to venom through expression shifts (i.e., gene co-option) plays a dominant role in the rapid turnover of venom in parasitoid wasps, representing a fast mechanism for the emergence of new gene functions (Martinson et al. 2017; Ye et al. 2022). And most venom genes of parasitoid wasps are not organized as tandem arrays as in most other venomous systems (Ye et al. 2020). However, the contribution of alternative isoforms, another avenue for rapid evolutionary innovation, to the venom evolution of parasitoid wasps has not been thoroughly assessed.
In this study, our objective was to investigate the diversity of isoforms within genes encoding venom repertoires. To achieve this, we used a comprehensive isoform profiling workflow based on long-read sequencing. Our focus was on Pteromalus puparum, a representative parasitoid wasp species with a global distribution. By analyzing the isoform landscape of venom genes, along with gene/isoform expression and venom proteome data, we aimed to elucidate the role of alternative isoforms in the diversification of venom proteins. Additionally, we aimed to trace the origins of venom genes with alternative isoforms, providing novel insights into the genetic mechanisms responsible for the emergence of venom genes.
Results
Generating a high-resolution RNA landscape through a hybrid-sequencing workflow
Our approach combined multiple sequencing technologies with distinct technical strengths, along with a series of computational methods for complete transcriptome reconstruction of P. puparum (Fig. 1A). Firstly, Pacific Biosciences (PacBio) HiFi long-read sequencing (PacBio Iso-Seq) and strand-specific Illumina RNA-seq, coupled with isoform pruning and refining, were used to capture high-quality isoform models. Subsequently, short-read sequencing was performed to obtain the 5′ ends containing a 5′ cap (cap analysis of gene expression; CAGE) and the 3′ ends preceding the poly(A) tail (polyadenylation site sequencing; PAS-Seq). This allowed for the identification of precise transcription start sites (TSSs) and transcription end sites (TESs), and the obtained information was integrated into isoform end corrections (Fig. 1A; Supplemental Figs. S1, S2).
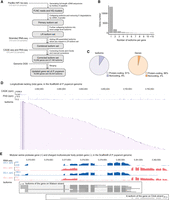
Overview of the high-resolution RNA isoform annotation in adult female P. puparum. (A) The workflow for sequencing and analysis, which includes multiple sequencing strategies such as PacBio HiFi Iso-Seq, stranded RNA-seq, CAGE-Seq, and PAS-Seq, as well as bioinformatic analyses for full-length transcriptome reconstruction and error corrections. (B) Distribution of the number of isoforms per gene. (C) Pie charts illustrating the composition of coding and noncoding isoforms/genes. (D) An example gene (longitudinals lacking gene, lola) with 266 isoforms. The top panel shows CAGE reads, the middle panel shows PAS reads, and the bottom panel shows a subset of isoforms (for complete set, refer to Supplemental Fig. S4). (rpm) reads per million. (E) An example demonstrating the precise annotation of two overlapping genes from different strands. The top panel shows strand-specific RNA-seq (blue represents Watson strand mapping reads; red represents Crick strand mapping reads), and the bottom panel shows isoforms. (VG) venom gland, (CA) carcass, (rpm) reads per million.
The developed workflow was applied to the venom gland tissue and carcass (i.e., female adult tissues excluding the venom gland) of the adult female parasitoid wasp P. puparum. Detailed statistics of the sequencing libraries can be found in Supplemental Tables S1–S3. Combining the results, a total of 27,160 isoforms were obtained, corresponding to 10,292 gene loci. Among these, 1067 isoforms represented novel gene loci that were not included in the official gene set (OGS 2.0) previously generated in our genome sequencing project of P. puparum (Ye et al. 2020; Mei et al. 2022). Consequently, an updated gene set of P. puparum (OGS 2.1) was established, comprising 18,560 genes and 35,434 isoforms (Supplemental Dataset S1). Notably, 96% of the genes (17,901) and 91% of the isoforms (32,212) were predicted as protein-coding genes and isoforms, respectively (Fig. 1C; Supplemental Table S4). Analysis of the splice sites indicated that the majority (98.82%) followed the canonical GU-AG pattern (Supplemental Fig. S3). Subsequent studies were conducted based on this version of genome annotation.
Our sequencing and correction efforts have resulted in a comprehensive RNA isoform annotation for adult female P. puparum. We identified a total of 4970 genes with two or more isoforms (a total of 21,942 isoforms, of which 19,125 were protein-coding isoforms), enabling in-depth exploration of splicing diversity (Fig. 1B). We discovered 76 genes that produced over 10 isoforms. One notable example is the longitudinals lacking (lola) gene, which exhibited 266 isoforms, making it the gene with the highest number of identified isoforms in adult female P. puparum (Fig. 1D; Supplemental Fig. S4). The RNA-binding protein lark gene ranked second with 80 isoforms (Supplemental Table S5). By leveraging the CAGE and PAS data, we were able to overcome the potential limitations associated with determining the precise 5′- and 3′-boundaries of isoforms using PacBio Iso-Seq alone (Wang et al. 2019; Sun et al. 2021). Additionally, we identified 2672 gene overlapping loci in P. puparum (Fig. 1E). Overall, our findings provide a high-resolution and accurate transcriptome for adult female P. puparum.
Diverse alternative RNA processing events
We conducted a comprehensive analysis of alternative splicing (AS) characteristics, based on our updated annotation. A total of 13,195 AS events were identified, involving 3220 genes (Fig. 2A; Supplemental Table S6). Gene Ontology (GO) enrichment analysis revealed that these AS genes are significantly enriched in multiple biological regulation processes (P < 0.05, false discovery rate adjusted, FDR-adjusted; Supplemental Table S7). Furthermore, 1405 genes exhibited co-occurrence of two or more types of AS events, indicating the complexity of AS in P. puparum. Within the venom gland, we identified 4442 AS events from 1673 genes, whereas the carcass exhibited 8899 AS events from 2613 genes. Comparative analysis demonstrated that intron retention (IR) accounted for the majority (27.7%) of AS events in the venom gland, whereas it ranked fourth (15.0%) in the carcass (Fig. 2A). This suggests that AS event patterns in the venom gland and carcass have largely diverged, likely because of their distinct functions.
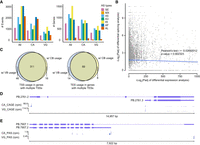
Differential alternative RNA processing events between the venom gland and carcass. (A) Statistical analysis of alternative splicing (AS) events and AS genes in three gene sets: all genes (All), genes expressed in the venom gland (VG), and genes expressed in the carcass (CA). Expressed genes were defined as those with TPM values higher than 1 in at least one replicate of the corresponding sample. The chart displays seven types of AS events: skipping exon (SE), mutually exclusive exons (MX), alternative 5′/3′ splice sites (A5/A3), retained intron (RI), and alternative first/last exons (AF/AL). (B) Scatter plot illustrating the relationship between differentially spliced genes (DSG) and differentially expressed genes (DEG). Pearson correlation analysis revealed no significant association between the DSGs and DEGs. The lines represent linear regressions, and the shaded areas depict 95% confidence intervals around the mean predictions. (C) Alternative usage of transcription start sites (TSS) in genes with multiple TSSs and alternative usage of transcription end site (TES) in genes with multiple TESs. We identified 311 genes with at least one venom gland-biased TSS and one carcass-biased TSS, as well as 89 genes with at least one venom gland-biased TES and one carcass-biased TES. (D) An example of a gene exhibiting distinct TSS usage between the venom gland and carcass, resulting in two different isoforms. The panels show isoforms, CAGE-Seq in the carcass, and CAGE-Seq in the venom gland. (E) An example of a gene with distinct TES usage between the venom gland and carcass. The panels display isoforms, PAS-Seq in the carcass, and PAS-Seq in the venom gland. (rpm) reads per million.
Using percent spliced-in (PSI) values calculated by SUPPA2 (Trincado et al. 2018), we identified 874 genes with significantly differential AS events between the venom gland and carcass (P < 0.05; Supplemental Table S6). Notably, 25.1% of these differentially spliced genes (219 DSGs) were also differentially expressed genes (DEGs) between venom gland and carcass (log2FC > 2 or log2FC < −2, p-adjusted < 0.05; Supplemental Table S8). We observed a lack of significant positive correlation between DSGs and DEGs (r = −0.027, Pearson's correlation test; Fig. 2B). This led us to speculate that, in the venom gland, DEGs and DSGs may play complementary roles in venom-related functions. Given that the parasitoid wasp venom gland expresses a relatively small set of genes at high levels (specifically, 7636 genes were expressed in the venom gland with mean transcripts per million (TPM) > 1, of which 487, accounting for 2.6% of all genes, contributed to 90% of RNA-seq reads), we further correlated the information related to DEGs and DSGs with their gene expression levels. We identified 50 highly reliable venom gland expressed genes (with TPM values higher than the N90 value of the venom gland) that exhibited differential splicing and significant up-regulation in the venom gland (Supplemental Table S9). These genes serve as potential candidates for exploring the functions of differential AS underlying venom diversity. GO analysis revealed their association with various biological processes, including small molecule metabolism, defense response, and immune system processes (Supplemental Table S10).
To investigate alternative transcription initiation and termination events, we used independent 5′ and 3′ end profiling approaches, enabling us to accurately determine isoform ends. Our analysis revealed intriguing insights into the diversity of transcription start sites (TSSs) and transcription end sites (TESs). Specifically, we identified 1934 genes with at least two TSSs and 2811 genes with multiple TESs based on the mapped CAGE and PAS reads, respectively. We then assessed the TSS usages of each gene, quantifying the fraction of all TSS reads originating from a specific TSS in both the venom gland and carcass. Notably, we observed tissue-biased TSS usage patterns in 373 genes, where the difference in TSS fraction exceeded 0.4 (Fig. 2C). Among these genes, 311 displayed both venom gland-biased and carcass-biased TSSs, illustrating tissue-based TSS switches (Fig. 2D provides an example, Supplemental Table S11). Similarly, we applied the same criteria to define tissue-biased TES usage and identified 89 genes with both venom gland-biased and carcass-biased TESs (Fig. 2E showcases a representative example; Supplemental Table S12). These findings underscore the complexity of RNA isoforms in P. puparum, complementing our gene-level characterization. Notably, a significant number of genes exhibited venom gland-biased alternative RNA processing events, thereby enhancing our genomic resources and facilitating a comprehensive understanding of the function of venom gland genes in parasitoid wasps. It is essential to note that although the venom gland is recognized as a highly specialized organ with a limited number of highly expressed genes (Martinson et al. 2017), our comparative analysis between the venom gland (a well-defined tissue) and carcass (a mixed tissue) may introduce potential biases when interpreting the expression differences of certain genes. To gain a more precise understanding of gene expression patterns, further exploration through intensive sampling of different tissues and single-cell profiling will be necessary.
Comprehensive identification of venom genes in P. puparum
Based on our updated annotation, we used a comprehensive approach combining transcriptomic (transcription-level) and proteomic (translation-level) evidence to systematically characterize venom genes and identify splicing isoforms that contribute to protein products. In this study, we adopted two key criteria for identifying venom genes in P. puparum, which have been widely used in venom gene identification studies in various parasitoid wasp systems (Martinson et al. 2017; Ye et al. 2022): (1) The genes must exhibit highly reliable expression in the venom gland, indicated by TPM values higher than the N90 value of the venom gland; (2) proteins encoded by these genes must be supported by proteomic profiling of secreted venom (see Methods). In this classification of venom genes, we did not strictly require the presence of a signal peptide, as previous evidence has shown that some RNA-seq-supported proteins lacking signal peptides can still be detected in venom proteomes (Martinson et al. 2017; Yang et al. 2021; Ye et al. 2022). In total, we identified 179 venom genes in P. puparum, of which 89 genes did not contain a signal peptide (Supplemental Table S13).
Numerous venom genes produce multiple isoforms and diverse proteins within the venom gland
Subsequently, we conducted an analysis to determine the presence of multiple isoforms among these venom genes. According to our updated annotation, 82.5% (148/179) of venom genes were found to have multiple isoforms, whereas only 31 genes produced a single isoform. We then focused on identifying the number of venom genes expressing multiple isoforms specifically in the venom gland tissue. By considering isoforms with expression levels above the mean TPM value of 1 in the venom gland, we identified 10 venom genes that had multiple isoforms but only one isoform was expressed. Thus, within the venom gland, we observed that 138 venom genes produced multiple isoforms, ranging from two to 24 isoforms (Fig. 3A; Supplemental Table S14). Furthermore, we investigated whether these multi-isoform venom genes generated diverse proteins in the venom gland. We identified 95 genes that encoded distinct proteins (Fig. 3A; Supplemental Table S13; Supplemental Fig. S5). To gain insights into the utilization rates of these multiple isoforms, we calculated the isoform fraction value (referred to as isoform usage, see Methods) for each isoform of every individual gene in the venom gland using strand-specific RNA-seq data. Our analysis revealed that 77 genes surpassed our threshold, indicating that the expression percentage of the second most frequently used isoform was greater than 10%. This provides robust evidence that numerous venom genes generated multiple isoforms with notable utilization within the venom gland.
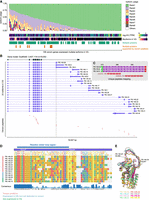
Diverse isoforms of venom genes in P. puparum. (A) Isoform usage of venom genes. Only isoforms expressed in the venom gland are included, and isoforms with TPM values higher than 1 in at least one replicate of the venom gland sample are considered expressed. The usages of isoforms within each gene are ranked (Rank1 represents the most used isoform in a gene) and depicted in different colors. Venom genes with multiple isoforms encoding different proteins are indicated, with emphasis on genes supported by mass spectrometry-based proteomics. This figure showcases venom genes that express multiple isoforms in the venom gland. Please refer to Supplemental Figure S5 for the isoform usage landscape of all venom genes. (B) PpSerpin1 exhibits the expression of 23 isoforms in the venom gland, encoding a total of 12 proteins, with eight of them supported by venom proteome evidence. (C) A zoomed view of the alignments between a protein encoded by isoforms (PB.148.3/.9/.29) and venom peptides. Additional supporting evidence for seven other venom serpin1 proteins can be found in Supplemental Figures S16 and S17. (D) Local alignment of amino acid sequences encoded by PpSerpin1 isoforms starting with Valine343. The putative reactive center loop region is indicated. Isoform PB.148.34 is not included in the alignment because of the absence of the serpin domain. (E) Comparison of 3D protein models predicted using AlphaFold2, highlighting variations in the reactive center loop among the eight venom serpin protein isoforms produced by PpSerpin1 through alternative splicing. The highlighted residues in the PB.148.30 protein structure represent the predicted start and end of the reactive center loop.
Evidence of a single venom gene producing diverse venom proteins by alternative splicing
To validate the generation of diverse venom proteins through alternative splicing, we conducted a manual inspection of the mapped peptides supported by mass spectrometry. We defined a protein as a venom protein if it was encoded by a venom gene and had at least one completely matched peptide. Through this analysis, we identified eight cases where alternative splicing contributed to venom protein diversity. These cases included the serine protease gene, venom allergen 3 gene, alpha-amylase 2 gene, venom acid phosphatase Acph-1 gene, pancreatic lipase-related protein gene, serpin 1 gene, CCHC-type zinc finger protein gene, and a lipase-like gene (Fig. 3A; Supplemental Figs. S6–S17). This finding provides further evidence that alternative splicing plays a role in expanding the diversity of venom proteins in parasitoid wasps.
A representative example demonstrating the contribution of alternative splicing to venom protein diversity in our study is the serpin gene, P. puparum serpin 1 (PpSerpin1, OGS2.1 ID: Ppup140770). Our comprehensive sequencing and bioinformatics analysis revealed the presence of 31 isoforms from the PpSerpin1 gene locus, encoding 14 different proteins (Supplemental Fig. S15). Among these isoforms, 30 out of 31 were generated through alternative splicing of exons adjacent to the 3′ ends, whereas the remaining isoform (isoform PB.148.34) resulted from a distinct alternative RNA processing event. In the venom gland, 23 isoforms showed expression evidence supported by both Iso-Seq and RNA-seq (Fig. 3B), producing a total of 12 distinct proteins. One of these proteins lacked the serpin domain (Pfam ID: PF00079). Among the 11 serpin proteins, eight were confirmed by mass spectrometry, indicating their presence as venom components (Fig. 3B,C; Supplemental Figs. S15–S17). By aligning all serpin protein sequences encoded by PpSerpin1 and analyzing their predicted protein structures, we observed significant differences in the reactive center loop regions (Fig. 3D,E; Supplemental Fig. S18), which are known to govern the inhibitory function and specificity of serpins (Huntington et al. 2000; Huntington 2011). Hence, we hypothesized that these diverse venom serpin proteins, generated through alternative splicing of PpSerpin1, may have evolved different functions and potential targets during parasitism. In a previous study, we demonstrated that one splicing isoform of PpSerpin1 (PB.148.30) inhibited host prophenoloxidase activation by likely forming a complex with host hemolymph proteinases (Yan et al. 2017). However, further functional characterization is required to elucidate the potential roles of other venom isoforms in regulating host immune responses. Overall, the AS of the venom gene PpSerpin1 exemplifies how AS contributes to the diversification of protein functions.
Alternative isoforms may contribute to venom evolution in P. puparum
Although gene duplication followed by neofunctionalization, gene co-option, and lateral gene transfer are well-established mechanisms driving venom evolution in parasitoid wasps (Martinson et al. 2016, 2017; Huang et al. 2021; Yang et al. 2021; Ye et al. 2022), our understanding of the role of alternative isoforms in this process is limited, with only one documented case in a Ganaspis wasp (Mortimer et al. 2013). Theoretically, the venom recruitment by alternative isoforms would involve distinct isoform usage patterns between venom-related organs and other tissues. For instance, GeneA may predominantly use isoform1 in various nonvenom-related tissues, serving its “normal” function, whereas in the venom gland, GeneA primarily uses isoform2, to fulfill its venomous role. Building upon this concept, we compared the isoform fraction values of each gene in the venom gland and carcass samples to assess their differential usage (see Methods). We identified a total of 718 isoforms (from 446 genes) that exhibited significantly different usage patterns (difference in isoform fraction > 0.4, FDR-adjusted P < 0.05; Supplemental Table S15). Among these, 357 isoforms (from 354 genes) showed significantly increased usage in the venom gland, and only seven isoforms (corresponding to seven genes) were identified as venom genes in our study (Figs. 4A, 5A; Supplemental Figs. S19–S23; Supplemental Table S16). This observation suggests that alternative isoforms may have played a role in the venom evolution of these genes in P. puparum.
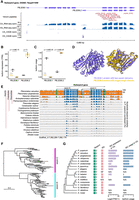
Isoform switching in the serpin3 gene contributes to venom recruitment in P. puparum. (A) Two distinct isoforms (PB.2036.1 and PB.2036.2) of PpSerpin3 are expressed in the venom gland (VG) and carcass (CA). From top to bottom: isoforms, venom peptides supported by mass spectrometry-based proteomics, RNA-seq, and CAGE-Seq data. (rpm) reads per million. For supporting evidence of the venom PB.2036.2 protein, please refer to Supplemental Figure S24. (B) Expression levels of the two PpSerpin3 isoforms in VG and CA, displaying adjusted P-values from the differential expression analysis conducted with DESeq2. (C) Isoform usage of the two PpSerpin3 isoforms in VG and CA, showing the FDR (Benjamini–Hochberg) adjusted P-values from the differential isoform usage analysis. Isoform usage is calculated as expression of a specific isoform divided by the expression of the parent gene. (D) AlphaFold2-generated protein structures of the two PpSerpin3 isoforms. The linker between the two individual domains is not depicted because of challenges in deducing its structure. (E) VISTA sequence conservation plot of the serpin3 gene in Hymenoptera, utilizing P. puparum as the reference. The genomic sequences displaying over 50% sequence identity compared to the reference are shown. Noncoding regions are represented in orange, exons in blue, and UTRs in green. Conserved regions with at least 70% identity over a 100 bp window are indicated in the corresponding color. The gene region, amino-terminal serpin domain (serpin-N), and carboxy-terminal serpin domain (serpin-C) are marked. (F) Maximum-likelihood protein phylogeny of the serpin domain sequences from 51 representative hymenopteran species. (G) Schematic representation of serpin3 genes and their isoforms containing only serpin-C (ISC), accompanied by a species tree of selected representative hymenopteran species. Filled boxes in the “serpin3” column represent genes with intact ORFs, and the numbers beside the boxes indicate the number of serpin domains in the genes. Filled boxes in the “ISC” column indicate the presence of ISC. (NF) not found, (N/A) not applicable. The expression (TPM) and isoform usage of ISCs in VG are visualized in the corresponding bar plots on the right. The N/A for O. abietinus and A. rosae indicates the absence of venom gland RNA-seq data for these two wasps.
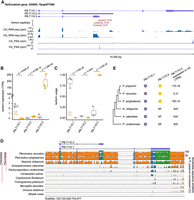
Evolutionary dynamics of the orcokinin gene and its venom isoform. (A) PpOrcokinin exhibits three isoforms (PB.7110.1, PB.7110.2, and PB.7110.3) expressed in the venom gland (VG) and carcass (CA). From top to bottom: isoforms, peptides supported by venom proteome, RNA-seq, and PAS-Seq. (rpm) reads per million. For supporting evidence of the venom PB.7110.3 protein, please refer to Supplemental Figure S26. (B) Expression levels of the three PpOrcokinin isoforms in VG and CA, displaying the adjusted P-values from the differential expression analysis conducted with DESeq2. (C) Isofrom usage of the three PpOrcokinin isoforms in VG and CA, showing the FDR (Benjamini–Hochberg) adjusted P-values from the differential isoform usage analysis. Isoform usage is calculated as expression of a specific isoform divided by the expression of the parent gene. (D) VISTA sequence conservation plot of the orcokinin gene in representative hymenopterans species, using P. puparum as the reference. Genomic sequences exhibiting over 50% sequence identity compared to the reference are depicted. Noncoding regions are indicated in orange, exons in blue, and UTRs in green. Conserved regions with at least 70% identity over a 100 bp window are shown in the corresponding color. The protein structures of two coding isoforms (PB.7110.1 and PB.7110.3) are displayed. A zoomed view of the exon3 alignment of PB.7110.3, including a specific DNA insertion in P. venustus, can be found in Supplemental Figure S27. (E) Schematic representation of orcokinin isoforms in six parasitoid wasps, presented to the right of the species tree. Filled boxes represent the presence of the corresponding isoform. (NF) not found. The isoform expression values (TPM) in VG are provided. (N/A) not applicable.
Shifts in isoform usage of serpin3 contributes to venom recruitment
A compelling example of differential isoform usage (isoform usage shift) between the venom gland and other tissues is observed in P. puparum serpin3 (PpSerpin3, OGS2.1 ID: Ppup011040). Our annotation revealed that PpSerpin3 expresses two isoforms with varying lengths because of alternative transcription initiation (Fig. 4A). In the carcass, the long isoform (PB.2036.1) predominated, accounting for 78.0% of isoform usage. This isoform encodes a protein with two intact serpin domains arranged in tandem (amino-terminal serpin: serpin-N, and carboxy-terminal serpin: serpin-C). However, in the venom gland, the utilization of the short isoform (PB.2036.2), which possesses only one intact serpin domain (serpin-C), significantly increased to 99.8% (P = 3.83 × 10−25, Mann–Whitney U test with FDR-adjust), making it the primary protein product of PpSerpin3 in the venom gland (Fig. 4C,D). Indeed, our mass spectrometry data confirmed the presence of peptides corresponding to serpin-C, providing further evidence that the product of PB.2036.2 is a venom protein, rather than the product of PB.2036.1 (Fig. 4A; Supplemental Fig. S24). Moreover, isoform expression analysis revealed significant differential expression of these two isoforms between the venom gland and carcass (p-adjusted < 0.001; Fig. 4B). In sum, PpSerpin3 serves as a representative case illustrating venom evolution associated with alternative isoforms, thus providing valuable insights into the evolutionary processes at play.
Evolution of serpin3 and its isoforms in Hymenoptera
To unravel the evolutionary history of this venom gene and its isoforms, we initially conducted a manual search for orthologs of PpSerpin3 in 50 Hymenoptera species with high-quality genome assemblies. Our findings revealed that this gene encodes proteins with two tandemly arrayed serpin domains, a feature conserved as a single copy gene across Hymenoptera. This conservation further supports the notion that the long isoform, containing two serpin domains, is generally maintained throughout Hymenoptera. Our analysis of whole-genome alignments also indicated that most exons exhibit conservation across Hymenoptera, whereas significant divergence was observed in the intron and UTR regions (Fig. 4E). Additionally, to explore the evolution of the two tandemly arrayed serpin domains within serpin3, we profiled these domains in 51 hymenopteran species using the Pfam domain annotation tool. Subsequently, a phylogenetic tree was constructed using the protein sequences and the maximum-likelihood method. Our results revealed two distinct and well-supported clades representing serpin-N and serpin-C, respectively, suggesting their ancient divergence (Fig. 4F).
Next, our investigation focused on the short isoform of serpin3 (the isoform containing serpin-C only, ISC) to determine its conservation across Hymenoptera and understand when it became predominantly expressed in the venom gland. We examined eight parasitoid wasps from Chalcidoidea, three parasitoid wasps from Ichneumonoidea, and four species from Aculeata, utilizing their high-quality genome assemblies and transcriptomes from both the venom gland and carcass/female adult. Additionally, we included Orussus abietinus (Orussidae) and Athalia rosae (Tenthredinidae) as outgroups, representing early branches of Hymenoptera. Consistent with our previous findings, serpin3 was maintained as a single copy gene in these 17 representative hymenopterans. With the exception of the honeybee Apis mellifera, we observed that serpin3 in all other species could generate proteins containing two tandemly arrayed serpin domains. Notably, the analysis of genome-guided transcriptome assemblies revealed the presence of a single ISC for each serpin3 in most species, with the exception of the braconid wasp Cotesia chilonis, where no ISC was detected. This observation suggests that, similar to the long isoform with two serpin domains, ISC may also be a conserved isoform of serpin3 in Hymenoptera. Next, we analyzed the isoform expressions and usages of serpin ISC and mapped this information to the phylogeny to explore the evolutionary trajectory of the venom ISC. Isoform quantification revealed that five chalcidoid species (P. puparum, P. venustus, P. qinghaiensis, Nasonia vitripennis, and Anastatus japonicus) exhibited high expression of ISC in the venom gland (TPM > 199), and the Asian giant hornet Vespa mandarinia showed moderate ISC expression in the venom gland with TPM = 96. On the other hand, other species displayed relatively low ISC expressions (TPM < 30) (Supplemental Table S17). This distinct expression pattern might suggest three independent events of expression elevation, occurring at the common ancestor of three Pteromalus and N. vitripennis, the terminal branch leading to A. japonicus, and the terminal branch of V. mandarinia, respectively (Fig. 4G). Furthermore, the usage of ISC in the venom gland showed a dynamic pattern consistent with the expression changes (Fig. 4G). To gain deeper insights, we focused on the evolution within the Pteromalidae family, which includes Pteromalus, N. vitripennis, Anisopteromalus calandrae, and Pachycrepoideus vindemmiae. This choice was motivated by two factors: (1) relatively recent species divergence, within 40 million years; and (2) the availability of high-quality RNA-seq data of the venom gland with three biological replicates for most wasps, except N. vitripennis. Among these six pteromalid wasps, A. calandrae and P. vindemmiae, which diverged early, exhibited low expression of ISC in the venom gland (median TPM = 17.1 and 6.6, respectively) with ISC usage of 57% and 59%, respectively. Conversely, N. vitripennis (TPM = 199.2) and three Pteromalus species (median TPM = 153.5–1552.7) showed significantly higher ISC expression, resulting in a substantial increase in venom ISC usage to over 97% in these species (Fig. 4G). Previous studies did not identify serpin3 as a venom gene in A. calandrae, P. vindemmiae, and N. vitripennis (Martinson et al. 2017; Yang et al. 2020; Chen et al. 2022). Additionally, we could not confirm serpin3 as a venom gene in P. venustus and P. qinghaiensis because of the lack of proteomic data, although high ISC expression was detected. Therefore, we were unable to accurately determine the evolutionary position for the recruitment of ISC as a venom component. Overall, our findings indicate that the single-copy gene serpin3, encoding two conserved isoforms, undergoes a shift in isoform usage in the venom gland by increasing the expression of a specific isoform, most likely in the common ancestor of Pteromalus and Nasonia, thereby contributing to the recruitment of this venom isoform in P. puparum. Moreover, we observed that after the recruitment of ISC to venom expression, the quantitative levels of ISC in the venom gland continued to evolve. Therefore, we suggest future research efforts to evaluate additional expression data from other parasitoid wasps to provide a more comprehensive understanding of ISC expression evolution.
Recruitment of a novel Pteromalus-specific isoform of the orcokinin gene into the venom repertoire
The orcokinin gene (PpOrcokinin, OGS2.1 ID: Ppup077490), encoding a venom neuropeptide, demonstrates distinct isoform usage between the venom gland and other tissues in P. puparum, providing an example of recruitment from the nervous system to venom through alternative isoforms. The PpOrcokinin locus generates three isoforms with different 3′ ends, one of which lacks predicted open reading frames (Fig. 5A). Multiple-sequence alignment reveals limited conservation between the protein products of the two coding isoforms (327aa for PB.7110.1 and 209aa for PB.7110.3), with only 51aa consistently aligned (Supplemental Fig. S25). Comparative analysis of isoform expression and usage demonstrates significant divergence between the venom gland and carcass for these two protein-coding isoforms (Fig. 5B,C; p-adjusted < 0.001). Notably, in the venom gland, PB.7110.3 is significantly up-regulated (average 465-fold, p-adjusted = 2.06 × 10−85), with usage exceeding 99%, confirming its protein product as a venom component, supported by mass spectrometry data (Supplemental Fig. S26). Conversely, PB.7110.1 is predominantly expressed in the carcass (84% usage, p-adjusted = 7.029 × 10−28), suggesting nonvenom functions. Analysis of the orcokinin gene's evolutionary history based on whole-genome alignment indicates rapid evolution among Hymenoptera, with no discernable genomic sequence conservation signals outside the superfamily Chalcidoidea, using P. puparum as a reference (Fig. 5D). Regarding the other coding isoform PB.7110.3, alignment and transcriptome assembly analyses reveal its presence only in three Pteromalus wasps, suggesting recent origin and species specificity (Fig. 5D,E). Further confirmation of this hypothesis comes from a BLASTP search against all identified sequences in the nr database. Expression analysis shows high expression of PB.7110.3 in the venom gland of P. puparum (median TPM = 170) and P. qinghaiensis (median TPM = 783), whereas it is barely expressed in the venom gland of P. venustus (median TPM = 0.21), implying expression turnover during Pteromalus evolution (Fig. 5E). In summary, these analyses unveil the recruitment of a novel isoform specifically in Pteromalus species, derived from a rapidly evolving neuropeptide gene, which is abundantly expressed and predominantly used in the venom gland, thus becoming part of the venom arsenal in P. puparum.
Discussion
Alternative RNA processing events play a crucial role in generating multiple transcripts from a single gene, thereby enhancing protein and phenotypic diversity and facilitating adaptation (Wright et al. 2022). Although alternative isoforms have been reported to contribute to venom protein diversity in a snake and a spider (Haney et al. 2019; Ogawa et al. 2019), their existence in parasitoid wasps, a megadiverse group with venom systems, has not been demonstrated. Thus, we hypothesized that the diversity of venom gene isoforms would contribute to the complexity of venom proteins in P. puparum. Our findings supported this hypothesis, as we discovered that 95 venom genes were capable of producing multiple proteins in the venom gland. Furthermore, mass spectrometry data provided additional evidence that eight venom genes produced at least two distinct venom proteins. Notably, the serpin1 gene served as an exemplary case with 23 venom-expressed isoforms, resulting in eight venom serpin proteins with highly variable reactive center loop regions that likely perform different functions during host-parasitoid interactions (Fig. 3B–E). It is worth noting that whereas one of these eight venom serpins had been previously identified using an assay-guided venom fractionation method paired with de novo, genome-independent, transcriptome assemblies from short-reads data (Yan et al. 2017), the remaining seven venom serpins were overlooked. This highlights the limitations of previous methods in discovering venom isoforms from a single gene, emphasizing the critical importance of our comprehensive approach involving full-length isoform sequencing with multiple RNA-seq techniques for uncovering the venom isoform landscape. In this study, our mass spectrometry data confirmed the existence of multiple venom proteins for a subset (8) of 95 venom genes that potentially produced such isoforms. This may be attributed to relatively low expression levels of some isoforms or the limitations of the proteomic approach in terms of depth and breadth of detection. Alternatively, it is possible that most isoforms produced by the venom genes do not result in venom proteins.
Alternative isoforms have been recognized as important contributors to key innovations in gene function or protein neofunctionalization over extensive evolutionary timescales in various organisms (Wright et al. 2022). Additionally, alternative isoforms have been suggested to facilitate rapid evolutionary innovations (Tovar-Corona et al. 2015; Howes et al. 2017; Fujikura et al. 2018; Wright et al. 2022). Although the rapid evolution of venom compositions in parasitoid wasps has been observed, potentially driven by variable host ranges or parasitoid strategies (Martinson et al. 2017; Mason et al. 2022; Ye et al. 2022), few studies have investigated the role of alternative isoforms in the origin of venom genes. Multiple models of venom evolution have been proposed over the years (Mrinalini and Werren 2016), including duplication followed by neofunctionalization (Chen et al. 2021; Yang et al. 2021; Ye et al. 2022), co-option (Martinson et al. 2017; Ye et al. 2022), and lateral gene transfer (Martinson et al. 2016), with three of these models well-characterized in parasitoid wasp venom systems. Only one example, based on short-read RNA-seq, has suggested a potential role of alternative isoforms in venom recruitment in a Ganaspis wasp (Mortimer et al. 2013). These observations prompted us to investigate whether alternative isoforms also contribute to the acquisition of venom genes in other parasitoid wasp species, potentially representing a relatively conserved mechanism, and to explore the evolutionary process underlying this phenomenon. In our study, focusing on P. puparum, we identified seven genes that exhibited significantly different isoform usage between the venom gland and carcass. These genes produced at least one isoform with venom-related function in the venom gland and at least one isoform associated with “normal function” in the carcass. We further examined the evolutionary trajectories of two examples, serpin3 and orcokinin, involving alternative isoforms. For the highly conserved serpin3, a shift in isoform usage was observed through the utilization of an internal TSS, resulting in an isoform that exhibited high expression and predominant usage in the venom gland of P. puparum. In the case of orcokinin, a rapidly evolving gene, a novel isoform emerged in Pteromalus species, which was likely co-opted for abundant expression and extensive usage in the venom gland, ultimately becoming a venom isoform. Another possibility for the origin of the venom isoform of orcokinin (PB.7110.3) is that it originated de novo in the venom gland of a Pteromalus ancestor. However, further RNA-seq samples of different tissues, demonstrating the venom gland-specific expression of PB.7110.3 are required to support this hypothesis. Overall, our findings provide clear examples of genes being actively recruited for venom functions through alternative isoforms in P. puparum, suggesting that this process might represent a relatively conserved mechanism in venom evolution among parasitoid wasps. However, considering that we only observed a small number of cases involving alternative isoforms in the venom evolution of P. puparum, we speculate that alternative isoforms may play a secondary role, compared to the widely reported models of duplication followed by neofunctionalization and gene co-option in shaping venom evolution in parasitoid wasps (Martinson et al. 2017; Chen et al. 2021; Yang et al. 2021; Ye et al. 2022).
We propose that the two case studies of venom evolution, serpin3 and orcokinin, through alternative isoforms can be considered as co-option of the pre-existing isoforms, representing co-option at the isoform level. This complementary model provides a resolution that the current gene co-option model lacks. Notably, these cases demonstrate isoform co-option occurring in isoforms of different ages, with the ancient serpin3 isoform and the newly evolved orcokinin isoform. Co-option enables a faster evolution of new gene functions compared to the classical duplication-neofunctionalization model. Furthermore, in contrast to the gene co-option model, the co-option of specific isoforms (alternative isoforms) likely facilitates functional diversification of a single gene by using different isoforms in different tissues. For example, an organism may recruit a specific isoform of a gene for venom function while retaining isoforms with other functions for basic processes. However, in cis-regulated gene co-option evolution, the recruited venom gene may be primarily limited to the venom gland, potentially losing its nonvenom function (Mrinalini and Werren 2016; Martinson et al. 2017). The emergence of multiple-function genes, which perform both “normal” and venomous functions, raises an intriguing question in evolutionary biology: How do they overcome pleiotropic constraints? We speculate that the highly tissue-specific isoform usage may alleviate significant pleiotropic constraints (Margres et al. 2017; Barua and Mikheyev 2020). Overall, our developed pipeline for full-length isoform analysis provides a high-resolution approach for studying venom evolution at the isoform level. Future investigations should explore the isoform evolution landscape of venom genes, particularly for genes with multiple isoforms.
The mechanisms underlying alternative isoform events during venom recruitment remain unclear. We hypothesize that various factors, such as changes in transcriptional regulatory elements and networks, posttranscriptional regulatory processes, and the origin of novel exons, may be involved in this process. For instance, the apparent shift of the short isoform of serpin3 to the venom gland in the comment ancestor of Pteromalus and Nasonia likely relates to changes in the potential cis-regulatory elements of the isoform, as evidenced by the high conservation of promoter regions only in Pteromalus and Nasonia based on whole genome alignment analysis (Fig. 4E). Overall, further comparative and functional studies are necessary to elucidate the mechanisms of alternative isoforms in venom recruitment. These investigations will serve as important complements to the theory of venom evolution and provide valuable insights into the molecular mechanisms underlying the origin of novel gene functions.
Methods
RNA sequencing
We used the laboratory strain of P. puparum (Ppup-ZJU) for sampling purposes. We used several RNA sequencing technologies, namely PacBio HiFi long-read sequencing, strand-specific Illumina RNA-seq, CAGE-Seq, and PAS-Seq. For full-length RNA sequencing using PacBio HiFi, we collected venom gland (100 tissues) and carcass samples of P. puparum, extracted the total RNA, and prepared the SMRTbell libraries, which were subsequently sequenced on the PacBio Sequel II platform. For Illumina RNA sequencing, we collected and extracted total RNA from six samples from venom gland (100 tissues per sample) and carcass. Each sample was prepared as three biological replicates. The sequencing libraries were constructed as strand-specific, paired-end libraries with an insert size of 300 bp, which were sequenced on the Illumina HiSeq X Ten platform. In addition, the total RNA for CAGE-Seq and PAS-Seq was also extracted from the venom gland (100 tissues) and carcass, and was subsequently sequenced on the NextSeq 500 system with a read length of 151 nt and the NovaSeq 6000 system with a read length of 151 nt, respectively. Detailed methods are provided in the Supplemental Methods.
Data processing and annotation of full-length transcripts
For quality control of the raw data from CAGE-Seq and PAS-Seq, we used FastQC v0.11.9 (https://github.com/s-andrews/FastQC). In the case of CAGE-Seq, read pairs that were concordantly mapped to reference sequences of rRNAs using Bowtie 2 v2.5 (Langmead and Salzberg 2012) were excluded. The remaining clean reads were then mapped to the genome using STAR v2.7.7a (Dobin et al. 2013). Regarding PAS-Seq data, once the library quality was confirmed, we used cutadapt v4.0 (Martin 2011) to trim the poly(A) reads. Subsequently, genome mapping was performed using STAR v2.7.7a. Paraclu (Frith et al. 2008) was used for CAGE/PAS peak clustering with the following parameters: minValue 5, -l 200, -d 2. For PacBio Iso-Seq data, we processed it using the SMRT Link v9.0, followed by the IsoSeq v3 pipeline (https://github.com/PacificBiosciences/IsoSeq). This included steps such as ccs generation, primer removal, and clustering. The resulting full-length nonchimeric (FLNC) reads underwent further refining using the ToFU pipeline from cDNA_Cupcake v28.0.0 (https://github.com/Magdoll/cDNA_Cupcake). The 5′ and 3′ ends of these isoforms were compared to the CAGE and PAS clusters to obtain the final isoforms (Sun et al. 2021). To generate updated annotations, we used SQANTI3 (Tardaguila et al. 2018), which involved comparing newly obtained isoform data with the former genome annotation (OGS2.0) in the Insectbase2.0 (Mei et al. 2022). To assess the coding potential of the isoforms defined in our study, we used TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder). This analysis integrated the results of the BLASTP (against the Uniport database released on June 2021) and Pfam (hmmer against the Pfam database 34.0 released in March 2021) searches to identify coding regions.
Quantification of genes and isoforms
We performed expression quantification for all genes and their isoforms using the six Illumina RNA-seq data sets from the venom gland and carcass samples. The TPM value was calculated using Salmon v 1.9 (Patro et al. 2017). With three biological replicates for each tissue, differentially expressed genes were identified using DESeq2 v1.30.1 (Love et al. 2014).
Alternative splicing analysis
We used SUPPA2 v2.3 (Trincado et al. 2018) to identify a total of seven types of alternative splicing events, including skipping exon (SE), alternative 5'/3′ splice sites (A5/A3), mutually exclusive exons (MX), retained intron (RI) and alternative first/last exons (AF/AL). We also used this tool to identify alternative splicing events that exhibited differential splicing between the venom gland and carcass samples. This analysis was conducted using three biological replicates for each tissue, and significance was determined using a threshold of P < 0.05.
Venom proteome
The venom proteome analysis was conducted following the previously described protocol (Ye et al. 2022). Approximately 100 venom reservoirs, which are the organs for venom storage, were isolated from P. puparum. The reservoirs were pierced and washed for three times in sterile PBS. After centrifugation at 12,000 g for 10 minutes, the supernatant was collected and digested into peptides using trypsin. LC-MS/MS analysis was performed on a timsTOF Pro mass spectrometer (Bruker) coupled to Nanoelute (Bruker Daltonics). The raw data were processed using MaxQuant v2.0.3.1 (Tyanova et al. 2016) and the annotated protein sequences of P. puparum were used as the search database. Detailed methods are provided in the Supplemental Methods.
Identification of venom genes
To identify venom genes, we used a strategy that combines transcriptomic and proteomic data, following our previously described methodology (Ye et al. 2022), which assumes that the highly expressed genes in the venom gland are more likely to be the venom genes. Genes with TPM values higher than the N90 value of the venom gland were considered as highly reliable venom gland expressed genes. Among these genes, a highly reliable venom gland expressed gene was defined as a venom gene if it had at least two (in the case of the top 50 expressed genes) or three proteomic peptides that matched completely. This criterion ensured the qualification of a gene as a venom gene.
Isoform analysis of venom genes
We used the GTF file to identify multiple isoforms of venom genes. To assess the isoform usage within each gene, we used IsoformSwitchAnalyzeR v1.21.0 (Vitting-Seerup and Sandelin 2019) to calculate the isoform fraction value. This value represents the fraction of the parent gene expression originating from a specific isoform and is computed as the ratio of isoform expression to gene expression. The expression data of isoforms and genes, obtained using Salmon, served as input for IsoformSwitchAnalyzeR. To identify venom genes that give rise to distinct proteins, we manually examined the venom peptides mapped and supported by mass spectrometry. A venom protein was defined as a protein that had at least one peptide completely matching the experimental data. The mapping results of the peptides were converted into a BED file, allowing visualization using the UCSC Genome Browser.
Differential isoform usage of venom genes and venom evolution
We focused on identifying venom genes that exhibit significant differential isoform usage between the venom gland and carcass, as these genes play a crucial role in understanding the impact of alternative isoforms on venom evolution. To accomplish this, we used IsoformSwitchAnalyzeR v1.21.0 to identify the differential isoform usage of venom genes between the venom gland and carcass. Genes with a difference in isoform fraction greater than 0.4 and an FDR-adjusted P-value below 0.05 were considered significant.
Additionally, we investigated the evolutionary history of two specific venom genes, PpSerpin3 and PpOrcokinin, along with their isoforms. Orthologs of these genes in other species were manually annotated. The P. puparum proteins were used as queries to perform TBLASTN searches against a set of 50 high-quality hymenopteran genomes (Supplemental Table S18) (e-value 1 × 10−5). Overlapping alignments were filtered and extended by 2,000 bp upstream and downstream. Fgenesh+ (Solovyev et al. 2006) was then used for predicting gene models within the alignment regions. Each candidate gene was manually inspected and subsequently used for further analyses. To study the evolution of the serpin domain in the serpin3 gene, the serpin domain sequences from 51 representative hymenopteran species were aligned using MAFFT v7.487 (Katoh and Standley 2013) with the L-INS- I model. After filtering with trimAl v1.2 (Capella-Gutiérrez et al. 2009), the resulting sequences were used to construct a maximum likelihood phylogenetic tree using IQ-TREE v2.0 (Minh et al. 2020) with 1,000 replicates for ultrafast bootstrap analysis. To investigate the evolution of the genomic sequences in the 20 hymenopterans (Supplemental Table S19), whole genome alignment analyses were conducted. Pairwise alignments were initially performed using LASTZ v1.04.03 (Harris 2007) with default parameters, using P. puparum as the reference genome. Subsequently, MULTIZ v012109 (Blanchette et al. 2004) was used to construct whole genome alignments. The resulting alignments were manually inspected and visualized using VISTA v1.4.26 (Frazer et al. 2004). For further evolutionary analyses on the diversity, expression, and usage of the isoforms of the two venom genes, we selected 15 hymenopteran species with high-quality genome assemblies and available transcriptomes of venom gland and carcass/female adults. Additionally, O. abietinus (Orussidae) and A. rosae (Tenthredinidae) were included as outgroups. RNA-seq data from venom gland and carcass/female adult tissue for each species were used to assemble the transcripts of the orthologs of the two venom genes, using HISAT2 v2.2.1 (Kim et al. 2015; Pertea et al. 2016) and StringTie v2.1.0 (Pertea et al. 2015). The resulting transcripts or isoforms were manually inspected, and the expression and usage of each transcript (or isoform) in each tissue sample were estimated using the methods described above. Finally, we mapped this information to the phylogeny obtained from (Peters et al. 2017; Ye et al. 2022) for comprehensive evolutionary analyses.
Prediction of venom protein structures by AlphaFold2
The prediction of serpin 3D structures was conducted using AlphaFold2 (Jumper et al. 2021). ColabFold (Mirdita et al. 2022), an implementation of AlphaFold2, was used. Multiple sequence alignments (MSAs) were generated using MMseqs2 (Steinegger and Söding 2017). The dual-serpin (PpSerpin3) was included as a single sequence input along with both paired and unpaired MSAs. For protein structure visualization, ChimeraX was used (Pettersen et al. 2021).
Enrichment analysis
GO enrichment analyses were performed using the Python library GOATOOLS v1.0.6 (Klopfenstein et al. 2018).
Software availability
Scripts used in the analysis are available as Supplemental Code.
Data access
All raw and processed sequencing data generated in this study have been submitted to the National Genomics Data Center (https://ngdc.cncb.ac.cn/) under accession number PRJCA013129. The annotation file of Pteromalus puparum OGS 2.1 is available in the Supplemental Dataset S1.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the Joint Funds of the National Natural Science Foundation of China (NSFC) (Grant No. U21A20225 to G.Y.Y.), the Key Program of NSFC (Grant No. 31830074 to G.Y.Y.), the Program of NSFC (Grant No. 32072480 to Q.F., Grant No. 32202376 to X.H.Y.), the Young Elite Scientists Sponsorship Program by China Association for Science and Technology (Grant No. 2022QNRC001 to X.H.Y.), the Program for Chinese Innovation Team in Key Areas of Science and Technology of Ministry of Science and Technology of the People's Republic of China (Grant No. 2016RA4008 to G.Y.Y.), the Fundamental Research Funds for Central Universities (Grant No. 2021FZZX001-31 to G.Y.Y. and Q.F.), and the China Postdoctoral Science Foundation (Grant No. 2021M700125 to X.H.Y.). We thank Dr. Mei Feng and Dr. Zhihua Wang at Shanghai Institute for Advanced Study (Zhejiang University) for comments on the manuscript. We thank Nextomics Biosciences Institute and ABLife (Wuhan, China) for their technical support on RNA sequencing. We thank Professor Haobo Jiang at Oklahoma State University for a valuable discussion on serpins.
Author contributions: G.Y.Y., F.W., Q.F., and X.H.Y. designed and managed the project. Y.Y. collected the samples for RNA sequencing; and X.H.Y., C.H., Y.Y., and Y.H.S. performed the sequencing, data processing, and full-length transcript annotation. X.H.Y., C.H., and Y.Y. identified venom genes and analyzed the venom evolution. X.H.Y. and K.C.C. performed venom protein structure predictions. X.X.Z., S.J.X., H.W.L., S.X., Y.M., Y.X.S., and Y.F.Y. participated in data collection and discussion. X.H.Y., C.H., Y.Y., Y.H.S., G.Y.Y., F.W., and Q.F. wrote and revised the manuscript. All authors read and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277707.123.
- Received January 16, 2023.
- Accepted August 8, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








