
A transcriptome-based single-cell biological age model and resource for tissue-specific aging measures
- Shulin Mao1,2,7,
- Jiayu Su2,3,7,
- Longteng Wang2,4,
- Xiaochen Bo5,
- Cheng Li2,6 and
- Hebing Chen5
- 1Yuanpei College, Peking University, Beijing 100871, China;
- 2Center for Bioinformatics, School of Life Sciences, Peking University, Beijing 100871, China;
- 3Department of Systems Biology, Columbia University, New York, New York 10032, USA;
- 4School of Life Sciences, Joint Graduate Program of Peking-Tsinghua-NIBS, Peking University, Beijing 100871, China;
- 5Institute of Health Service and Transfusion Medicine, Beijing 100850, China;
- 6Center for Statistical Science, Peking University, Beijing 100871, China
-
↵7 These authors contributed equally to this work.
Abstract
Accurately measuring biological age is crucial for improving healthcare for the elderly population. However, the complexity of aging biology poses challenges in how to robustly estimate aging and interpret the biological significance of the traits used for estimation. Here we present SCALE, a statistical pipeline that quantifies biological aging in different tissues using explainable features learned from literature and single-cell transcriptomic data. Applying SCALE to the “Mouse Aging Cell Atlas” (Tabula Muris Senis) data, we identified tissue-level transcriptomic aging programs for more than 20 murine tissues and created a multitissue resource of mouse quantitative aging-associated genes. We observe that SCALE correlates well with other age indicators, such as the accumulation of somatic mutations, and can distinguish subtle differences in aging even in cells of the same chronological age. We further compared SCALE with other transcriptomic and methylation “clocks” in data from aging muscle stem cells, Alzheimer's disease, and heterochronic parabiosis. Our results confirm that SCALE is more generalizable and reliable in assessing biological aging in aging-related diseases and rejuvenating interventions. Overall, SCALE represents a valuable advancement in our ability to measure aging accurately, robustly, and interpretably in single cells.
Aging is the gradual decline of biological and physiological functions that occurs in most living organisms over time. It is a complex process involving cumulative changes and damage at all levels, rendering it the primary risk factor of various human diseases, including cardiovascular disease, neurodegeneration, and cancer (López-Otín et al. 2013). By 2050, elderly individuals >60 yr old will make up >20% of the global population (Harper 2014). As the burden of aging-related diseases significantly increases in an aging world, there is a growing need to better understand the underlying biological process, quantify the impacts of aging, and provide rejuvenating interventions (Partridge et al. 2018; Scott et al. 2021).
People of the same chronological age have different aging states, which can be monitored using various biomarkers (Belsky et al. 2015). These markers are usually measurable indicators of a particular outcome or source of aging, such as phenotypical measures like frailty and molecular measures like DNA methylation dynamics (Schumacher et al. 2021; López-Otín et al. 2023). Although informative, they are not always quantitatively predictive of an individual's true biological age, nor are they easy to obtain. The advancement of high-throughput screening platforms and extensive longitudinal studies has greatly facilitated the search for new noninvasive and quantitative biomarkers of aging. For instance, high-throughput sequencing allows unbiased multiomics profiling of DNA, RNA, and epigenetic changes during aging, providing a comprehensive view of senescence at tissue and single-cell levels (Solovev et al. 2020; Aging Atlas Consortium 2021). These omics data sets contain vast and noisy measurements of potential candidate markers and, consequently, require carefully designed computational models to identify and extract predictive signals from the data. However, construction of such models is often highly degenerate, yielding little overlap of identified biomarkers between studies and thus making results difficult to interpret (Thompson et al. 2018; Galkin et al. 2020).
Among the many computational algorithms, linear regression and its variants have been widely used to select aging-related biomarkers and build “aging clocks,” namely, predictors of chronological age and biological age, in various omics data sets and aging systems (Hannum et al. 2013; Horvath 2013; Peters et al. 2015; Levine et al. 2018; Lu et al. 2019; Belsky et al. 2020; Moaddel et al. 2021; Buckley et al. 2023). To reduce overfitting and increase interpretability, regularization techniques are often used to shrink coefficients toward zero and highlight a sparse set of relevant features. In particular, the success of the first cross-tissue DNA methylation clock has popularized Elastic Net, a model that minimizes L1 and L2 penalties combined (Friedman et al. 2010; Horvath 2013).
Nonetheless, aging is a complicated phenomenon that impacts every aspect of an organism, and its roots and consequences spread across thousands of targets in a context-specific manner. There is likely no one-size-fits-all predictor of aging. Even if such a solution exists, the presence of multicollinearity makes it challenging to distinguish the true biologically relevant signatures driving the relationship with age. Because omics data often contain hundreds of thousands of redundant features but only one target variable to predict (chronological age), it is, therefore, conceptually and technically challenging to construct a consensus set of aging biomarkers de novo across data sets, let alone across modalities (Rutledge et al. 2022). Moreover, varying data quality and batch effects between sequencing platforms and cohorts may bias the feature selection and thus limit the generalizability of trained aging clocks.
Most of the existing omics-based aging clocks have been constructed using data from bulk tissues, which neglect the variations in cell compositions and cell-to-cell aging heterogeneity. To gain a more detailed and nuanced view of cell type–specific molecular changes during aging, several studies have applied machine-learning models to single-cell transcriptomics and DNA methylation data (Trapp et al. 2021; Buckley et al. 2023). Despite their success in predicting chronological age within specific training contexts, these clocks are constrained by their applicability to a limited number of cell types and tissues. Their generalizability to other cell types and disease data, particularly in cases with ambiguous cell type identities, remains uncertain. Additionally, problems like data sparsity and batch effects are more pronounced in single-cell omics data, further complicating the identification of consensus aging markers and the interpretation of model results. Furthermore, as chronological age is often the only available measure of biological age, it becomes critical to determine whether the features learned from single-cell omics data can capture other dimensions of biological aging.
Here, we developed single-cell aging-level estimator (SCALE), a tissue-specific measure that quantifies aging in individual cells using single-cell RNA-sequencing (scRNA-seq) data (Fig. 1). Unlike other clocks, SCALE adopts a knowledge-based forward feature selection strategy to prioritize genes that are well recognized to be associated with aging, making the results more explainable and less affected by technical noise. We applied SCALE to the Tabula Muris Senis (TMS) data set (Tabula Muris Consortium 2020) to identify tissue-level transcriptomic aging programs for more than 20 murine tissues and organs (Supplemental Fig. S1). We further evaluated the effectiveness of SCALE in accurately reflecting biological age under various conditions, including aging-related diseases and antiaging interventions.
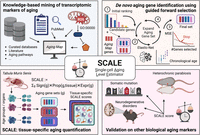
Schematic illustration of SCALE and the study design. We designed SCALE to select interpretable aging-related genes and quantify tissue-specific transcriptomic aging in a robust and easily generalizable manner. SCALE starts with a core set of manually curated aging-associated genes (Aging Map; top left), iteratively expands the list to include additional features that improve age prediction (top right), and eventually uses the gene set to quantify aging for each cell through weighted average of gene expression (bottom left). By explicitly introducing Aging Map genes as seeds to guide feature selection, SCALE encourages the inclusion of both well-documented genes and genes that contain complementary information on aging. SCALE also benefits from its indirect modeling of chronological age, in which the latter is only used to select genes rather than as the final prediction target. This makes SCALE less susceptible to technical noise and batch effects prevalent in single-cell RNA-seq data, allowing it to better capture biological age and the nuanced aging and antiaging effects of disease and interventions (bottom right).
Results
Knowledge-based mining of transcriptomic markers of aging in humans and mice
Extracting an informative subset of aging-related features out of high-dimensional transcriptomic data is challenging, owing to the intricate nature of aging itself as well as the interdependencies between genes. Existing regularization-based regression models (i.e., Elastic Net) for building aging clocks struggle with collinear factors, which results in unstable feature selection and fluctuating coefficients. These complications are amplified when the model is trained on or applied to different data sets (Supplemental Fig. S2A–C), undermining model interpretability and performance in biological applications (Supplemental Fig. S2D). To address this, we developed an alternative approach that aims to incorporate more well-documented aging genes and other genes with complementary information.
We began by creating “Aging Map” (Fig. 2A; Supplemental Fig. S3; http://sysomics.com/AgingMap/), a manually curated online database of human and mouse aging-related genes. Aging Map first integrated six existing databases focused on different aspects of aging, including “Gene Ontology (GO)” (The Gene Ontology Consortium et al. 2000), “CSGene” (Zhao et al. 2016), “Aging Atlas” (Aging Atlas Consortium 2021), “HAGR/GenAge” (Tacutu et al. 2018), “AgeFactDB” (Hühne et al. 2014), and “Longevity Map” (Fig. 2A,B; Budovsky et al. 2013). We also extracted and manually curated additional aging-related genes by mining PubMed abstracts to integrate more recent research efforts (Methods). This identified 636 literature-confirmed aging genes in humans and 460 genes in mice, including 162 and 49 novel hits that mostly encode histone proteins. Using GO analysis, we further verified that human and mouse literature-based genes were significantly enriched in well-established aging processes, such as cellular senescence, cell-cycle regulation, and telomere organization (Fig. 2C,D; Supplemental Fig. S3D,E). Together, Aging Map contains 1119 and 1459 curated human and mouse aging genes, respectively, covering almost all scales of aging, ranging from molecular damage to genetic predisposition. Cross-species comparison revealed a modest overlap between known human and mouse aging genes, suggesting both conservation of core senescence pathways and fundamental differences in aging between mice and humans (Fig. 2E).
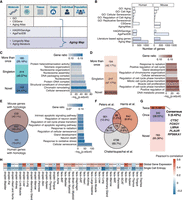
Aging Map: a manually curated database of aging-related transcriptomic features. (A) Summary of data sources used to build Aging Map, grouped by the scales of aging information covered by each collection. (B) Number of aging-related genes collected from each data source. (C) Characteristics of the 636 literature-confirmed aging-related genes in human. (Left) Overlaps with other databases listed in A. (Novel) Not covered in any of the existing database, (singleton) covered by one database, (≥2) covered by at least two databases. (Right) GO Enrichment analysis showing the top 10 GO terms associated with the literature-based aging genes, colored by the proportion of GO gene sets present in the list. (D) Similar to C but showing the results for 460 literature-confirmed aging-related genes in mouse. (E) Comparisons of human and mouse Aging Map genes. (Left) Venn diagram of homologous human and mouse genes. (Right) GO enrichment analysis showing the top 10 GO terms associated with aging genes shared between human and mouse, colored by the proportion of GO gene sets present in the list. (F) Venn diagram comparing human age-associated genes identified in three large cohort studies. For a full description of each data set, see Methods. (G) Overlaps between human Aging Map genes and the three data-driven gene lists in F. The five genes shared by all sets: CTSC, FOXO1, LMNA, PLAUR, and RPS6KA1. (H) Heatmap of correlation between chronological age and two high-level transcriptomic features (global gene expression and single-cell entropy) in each data set. (***) Adjusted P-value < 0.001, (**) adjusted P-value < 0.01, and (*) adjusted P-value < 0.05 using linear regression for global gene expression and Kruskal–Wallis test for single-cell entropy. All P-values were corrected by the Benjamini–Hochberg procedure.
Aging-associated genes can alternatively be identified in a data-driven manner (Finotello and Di Camillo 2015). However, most data-derived gene discoveries have not been tested experimentally or tested on independent cohorts, making it difficult to discern whether a gene is truly aging related or merely a reflection of technical noise (Rutledge et al. 2022). By integrating knowledge from the literature, Aging Map enables the prioritization of functionally important aging-associated genes from a vast pool of candidates. Here, we collected genes differentially expressed with age (age-DEGs) from three large longitudinal cohorts, including a pan-tissue study featuring 26 tissues from GTEx (Peters et al. 2015; Harris et al. 2017; Chatsirisupachai et al. 2019). These studies showed little agreement, in part owing to differences in study scope, sample source, and transcriptomic profiling technology, underscoring the need for more robust knowledge-based approaches to select aging markers (Fig. 2F). We then cross-referenced Aging Map human genes with the three age-DEG sets. Although the majority of data-driven age-DEGs did not have direct experimental support from existing aging literature, Aging Map helped identify five consensus genes (CTSC, FOXO1, LMNA, PLAUR, RPS6KA1) that have long been recognized for their roles in longevity (Fig. 2G).
Moving beyond individual aging genes, we explored the potential of using holistic transcriptomic features as quantitative aging biomarkers. Previous studies have reported an increased overall RNA abundance in aged murine hematopoietic stem cells (HSCs) (Flohr Svendsen et al. 2021). To test whether the finding generalizes to other systems, we used the TMS scRNA-seq data of more than 20 tissues and organs (Tabula Muris Consortium 2020). Here, we defined global expression of a cell as the log-transformed mean counts per gene and compared it across age groups sequenced in the same batch within each sample using a linear model (Methods). We observed a consistent significant positive correlation between global expression and chronological age in 27 out of the 37 analyzed samples. Two samples (droplet-based murine fat and limb muscle) showed the opposite trend (Fig. 2H; Supplemental Fig. S4), which was likely caused by measurement variability.
Another established phenomenon in aging is the increase in stochasticity and expression noise as cells gradually lose control over gene regulation (Martinez-Jimenez et al. 2017). To quantify the degree of expression uncertainty in each cell, we used entropy, a thermodynamic measure widely applied to describe cell differentiation potentials (Methods; MacArthur and Lemischka 2013; Chen and Teschendorff 2019). Cells with low entropy are generally more deterministic and express a narrower spectrum of genes. We found that entropy was significantly different across age groups in all samples (Fig. 2H; Supplemental Fig. S5). Nonetheless, we did not observe a monotonic increase of entropy over chronological age. As the regulation of gene expression is highly dynamic and comprehensive, further investigation is necessary to fully delineate the age-associated quantitative changes in transcriptional fidelity from noise generated by technical limitations and other biological processes. It is also possible to extend the analysis to other high-level systems-based transcriptomic signatures such as activities of gene regulatory networks and pathways using similar approaches.
Collectively, Aging Map provides a pan-tissue set of high-confidence genes associated with various aspects of aging, which can help prioritize targets in data-driven aging research. However, the substantial proportion of unannotated age-DEGs implies that there likely remain many more aging genes yet to be confirmed by experiments (Fig. 2F,G). Many of them, indeed, could represent tissue-specific aging programs driven by stress factors and cell–cell interactions in the local environment (Zhang et al. 2021). Furthermore, not all age associations are quantitative predictors of aging. To address these limitations, we proposed a knowledge-based machine-learning approach to identify novel quantitative aging markers from transcriptomic data sets.
Identification of tissue-specific genes as quantitative aging predictors using guided forward selection
To increase the statistical power of our study, we used scRNA-seq, which provides a larger number of data points and higher granularity compared with bulk RNA-seq. We analyzed more than 20 murine tissues and organs from the TMS data set to extract tissue-specific aging programs (Fig. 3A; Supplemental Fig. S1). Some tissues were sequenced by two different platforms (droplet-based 3′ biased and FACS-based full-length sequencing) at different time points and were thus analyzed separately, resulting in a total of 37 samples from two to six age groups for investigation. We assumed a generalized linear relationship between aging gene expression and chronological age and used Elastic Net to select a sparse subset of predictive genes. In particular, we used a guided forward selection strategy that iteratively incorporates new genes to improve regression performance while considering the expression of known aging-associated genes to enhance the robustness and the interpretability of feature selection (Methods; Supplemental Figs. S6, S7A,B). Based on this method, we generated tissue-specific aging gene sets for downstream analysis (Supplemental Table S1).
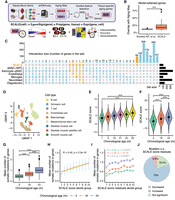
Tissue-specific quantification of aging in single-cell transcriptomic data using knowledge-guided feature selection. (A) Illustration of the SCALE pipeline applied to the Tabula Muris Senis data. (B) Boxplot showing overlaps between genes in Aging Map and aging genes selected by two methods, namely, aging clocks of Buckley et al. (2023) and SCALE. Each data point represents the gene set selected for a particular cell type (Buckley et al. 2023) or tissue (SCALE). (***) P-value < 0.001 using a two-sided unpaired Student's t-test. (C) UpSet plot illustrating the intersection of gene sets used by the six cell type–specific chronological aging clocks of Buckley et al. (2023) and the SCALE brain model. Genes selected by SCALE are highlighted in orange. (D) Uniform manifold approximation and projection (UMAP) plot of all cells in the kidney. Different colors show different cell types. (E) SCALE scores of kidney cells in each age group. Data are presented as the mean ± SD. (***) P-value < 0.001 using a two-sided unpaired Student's t-test. (F) SCALE scores of limb muscle cells in each age group. Data are presented as the mean ± SD. (***) P-value < 0.001 using a two-sided unpaired Student's t-test. (G) Boxplot showing the mutation burden (inferred from full-length scRNA-seq data) of limb muscle cells in each age group. Data are presented as the mean ± SD. (***) P-value < 0.001 using a two-sided unpaired Student's t-test. (H) Relationship between the mean number of somatic mutations in genes and the SCALE score of limb muscle cells. Cells were divided into 10 groups based on their SCALE scores (from low to high) for better visualization. The yellow line shows the linear regression model results fitted to the data with the corresponding R and P-value. Data are presented as the mean ± SD. (I) Relationship between the mean number of somatic mutations in genes and the SCALE score residual (after regressing out chronological age) of limb muscle cells. Cells were divided into 10 groups based on their SCALE score residuals (from low to high) for better visualization. Data are presented as the mean ± SE. R and P-value were calculated by fitting linear regression model for each age group individually. (J) Pie chart showing multitissue, multi–age group correlation analysis results between the mean number of somatic mutations in genes and the SCALE score residual. “Decreased” represents that mutation number significantly decreases with the SCALE score residuals, “Increased” represents that mutation number significantly increases with the SCALE score residuals, and “Not significant” represents that mutation number is not significantly linearly correlated with the SCALE score residuals.
Compared with genes directly selected by regression-based methods (Buckley et al. 2023), our aging gene sets showed a significantly higher overlap with well-documented aging genes (genes in Aging Map) (Fig. 3B), suggesting an increased likelihood of identifying genes involved in the mechanistic basis of aging. In addition, only seven out of 100 of our brain-aging genes were previously reported as cell type–specific aging genes in brain cells (Buckley et al. 2023). This shows that our approach uncovers novel aging programs shared across cell types, rather than simply combining cell type–level aging programs (Fig. 3C). Furthermore, most aging genes selected in different tissues were tissue specific (Supplemental Fig. S7C), aligning with our expectation of aging heterogeneity across tissues (Rutledge et al. 2022). Although there was little overlap in the aging genes across tissues, we found that these genes often contributed to the same biological processes (Supplemental Fig. S8A,B). In particular, these tissue-specific genes were frequently involved in “aging,” “ATP metabolic process,” and the regulations of protein translation and various other catabolic and metabolic processes, suggesting the importance and relative conservation of some aging programs across murine tissues and organs.
Among the pan-tissue aging genes (intersections of tissue-specific gene sets) identified in this study, some, such as Igf1 and Bcl2, have been confirmed in previous experiments (Junnila et al. 2013; Fernández et al. 2018; Rutledge et al. 2022). One of the novel discoveries, Lars2, was found to be broadly down-regulated across mouse tissues during aging (Supplemental Fig. S9). Clinical reports show that loss-of-function mutations in the human homolog LARS2 gene are present in one-third of patients with Perrault syndrome, which can cause hearing loss, motor and sensory neuron damage, decreased learning ability, and ovarian dysfunction (Kosaki et al. 2018). We therefore speculate that decreased expression of Lars2 may induce neurological and reproductive system damage, resulting in the aging phenotype. Another example is Rpl13a. We found it to be up-regulated in most tissues during aging (Supplemental Fig. S10). Studies in yeast have shown that individuals carrying the yeast homolog RPL13A loss-of-function mutations have longer lifespans than wild-type (WT) yeasts (Steffen et al. 2008). Also, a high expression level of RPL13A could negatively affect translation regulation and protein biogenesis, leading to discordance between transcriptome and proteome and thereby inducing cellular aging (Janssens et al. 2015).
Aging quantification at single-cell resolution using SCALE
With tissue-specific aging genes selected in the previous section, we constructed a statistical measure named SCALE to quantify the relative aging status of each cell residing in the same tissue (Methods). SCALE weights each aging gene according to its expression level, the direction in which its expression changes with chronological age, and the proportion of cells expressing the gene.
Applying the model to the TMS data set, we observed a positive correlation between SCALE scores and chronological age across all tissues (Fig. 3D–F; Supplemental Fig. S11). Pairwise comparisons between chronological age groups in each tissue confirmed that 130 out of the total 134 pairs showed significant differences in their SCALE scores (t-test, P-value < 0.05). The remaining four pairs may have insufficient statistical power owing to limitations in the data set or individual cell variability. To further validate our findings, we constructed empirical null distributions of the correlation by randomly selecting genes and replacing the aging genes in our model (Methods). SCALE outperformed the null in 35 of the 37 samples (adjusted P-value < 0.05) (Supplemental Fig. S12A), suggesting that the aging genes selected by SCALE are highly informative and still predictive even in the absence of trained regression models.
To show SCALE's robustness against technical artifacts, we conducted additional sensitivity tests. We downsampled the scRNA-seq data to simulate dropout events and reduce available aging signals. Even after zeroing-out expression of 50% of the genes, which effectively reduced the sequencing depth by half, the SCALE score still presented impressive resilience, with a Pearson's correlation of >0.8 (Supplemental Fig. S12B–D). We also experimented with the number of aging genes used to build SCALE and found the score to be highly correlated (median Pearson's correlation > 0.5) (Supplemental Fig. S13).
The accumulation of somatic mutations in cells owing to various internal and external stress during aging is a renowned hallmark of biological aging (Lodato et al. 2018; Zhang et al. 2019; Brazhnik et al. 2020). To evaluate SCALE's predictive performance regarding biological age, we conducted mutation analysis on samples in which full-length RNA coverage was available for exonic somatic mutation detection (Fig. 3G; Supplemental Fig. S14; Tabula Muris Consortium 2020). In all 24 tissues, SCALE scores showed a significant positive linear relationship with the mean number of somatic mutations in genes (P-value < 0.05) (Fig. 3H; Supplemental Fig. S15). Because both the SCALE score and the mutation number also positively correlate with chronological age, we regressed out the effect of chronological age and examined whether SCALE can reflect complementary aspects of biological aging not captured by chronological age. Indeed, we found that the residual SCALE scores (ΔSCALE) were still significantly positively associated with the mean number of somatic mutations in 54 out of the total 72 age groups (75%) in 24 tissues (P-value < 0.05) (Fig. 3I,J; Supplemental Fig. S16), indicating that SCALE can distinguish nuanced differences in biological aging within the same chronological age group. Samples in which ΔSCALE had no significant positive association with mutations were mostly from 3-mo-old mice (11 out of the total 18 groups), implying that the number of mutations in young mice may not accurately reflect aging status.
SCALE outperforms other single-cell aging clocks in assessing impacts of disease and rejuvenating interventions on aging
Aging is often accompanied by skeletal muscle atrophy and subsequent decline in mobility, which can be attributed to the decreased regenerative capacity of muscle stem cells (Blau et al. 2015; Shcherbina et al. 2020). To test whether our tissue-level model records the aging of stem cells, we applied the SCALE model trained on the TMS limb muscle tissue to an external multiomics data set of mouse muscle satellite cells (Fig. 4A; Hernando-Herraez et al. 2019). Although the two data sets differ substantially in sequencing technology and data quality, SCALE generalized well in the external stem cell data and recovered the aging difference between cells from 2-mo-old and 24-mo-old mice (P-value = 0.016) (Fig. 4B). Leveraging the simultaneously measured single-cell DNA methylation data, we compared SCALE with scAge, an epigenetic aging clock that exploits methylation-age associations learned from bulk data to estimate age at the single-cell level (Trapp et al. 2021). We found that scAge predicted more outliers with extreme age values and had difficulty differentiating cells from different age groups (P-value = 0.053; Methods) (Fig. 4C), probably because of sparsity and CpG coverage variation in single-cell DNA methylation data. This suggests that SCALE may be more stable and reliable in capturing biological age when applied to noisy single-cell omics data.
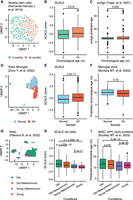
SCALE outperforms other single-cell aging clocks in assessing impacts of disease and rejuvenating interventions on aging. (A) UMAP plot of muscle stem cells. Different colors show different chronological ages. (B) SCALE scores of muscle stem cells (using single-cell transcriptomic profiles from single-cell M&T-seq). Data are presented as the mean ± SD. (C) ScAge predicted age of muscle stem cells (using single-cell DNA methylation data from single-cell M&T-seq). Data are presented as the mean ± SD. (D) UMAP plot of brain microglia cells from wild-type mice (normal; blue) and AD mouse models (AD; red). (E) SCALE scores of brain microglia cells from wild-type mice (normal; blue) and AD mouse models (AD; red). (F) Predicted chronological age of brain microglia cells by the method reported by Buckley et al. (2023). (G) UMAP plot of brain cells from old mice (green), young mice (magenta), old heterochronic mice (orange), and young heterochronic mice (purple). (H) SCALE scores of brain cells from parabiosis mice. P-values using a two-sided unpaired Student's t-test are shown. (I) Predicted chronological age of brain cell types (ependymal cell, macrophage, neuron, oligo pre cell, pericyte, and T cell) lacking clocks trained by Buckley et al. (2023). This box plot shows the results calculated by the aNSC_NPC clock, which incorrectly predicted cells from young heterochronic mice with lower ages than these of young mice and was not able to distinguish cells from old heterochronic mice and old mice, developed by Buckley et al. P-values using a two-sided unpaired Student's t-test are shown.
Alzheimer's disease (AD) is an aging-related degenerative neurological disorder that affects vision, hearing, speech, and memory (Ballard et al. 2011). The risk of AD increases with chronological age, with most cases occurring in people over 65 (Trevisan et al. 2019). To test if SCALE could reflect disease-associated aging changes, we applied the model trained on the TMS brain tissue to a single-nucleus RNA-seq data set of brains from the AD mouse model (Fig. 4D; Supplemental Fig. S17A; Zhou et al. 2020). SCALE scores were significantly higher in both brain microglia cells and nonmicroglia cells of AD mice than those in WT mice (Fig. 4E; Supplemental Fig. S17B), which is consistent with the previous finding that AD-associated genes are up-regulated during aging and early-onset AD (Zhou et al. 2020). To further benchmark the robustness of SCALE over other measures, we evaluated the performance of a collection of cell type–specific transcriptomic clocks built on scRNA-seq data of the mouse brain neurogenic region (Buckley et al. 2023). Out of the three clocks in which the corresponding cell type was presented in the AD data (microglia, endothelial, and oligodendrocytes), two failed to distinguish cells from AD mice versus from the control group (Fig. 4F; Supplemental Fig. S17C,D). Additionally, the clocks of Buckley et al. (2023) were highly cell type specific and could not generalize across cell types. When applied to three other abundant brain cell types (neurons, astrocytes, and OPCs) in the AD data set, five out of the six clocks failed to faithfully distinguish AD and WT cells (Supplemental Fig. S17F). On the contrary, the tissue-level SCALE model correctly reflected the significant aging differences between the AD and WT groups (Supplemental Fig. S17E). Together, our results indicate better generalizability of SCALE and highlight the importance of model robustness in protecting aging quantifiers from batch effects.
Heterochronic parabiosis (joining the circulatory systems of two individuals of different ages) has been recognized as an effective way to counteract aging in older animals and improve various physiological functions, including tissue regeneration, bone repair, and cognition improvement (Castellano et al. 2015). To investigate whether SCALE could capture the effect of rejuvenating interventions, we applied it to a multitissue scRNA-seq data set of mice receiving the heterochronic parabiosis surgery (Pálovics et al. 2022). We observed a significant decrease of SCALE scores in old mice and a significant increase in young mice following 5 wk of the parabiosis procedure in 19 of the total 20 tissues (P-value < 0.01) (Fig. 4G,H; Supplemental Fig. S18), confirming that SCALE can recapitulate the aging order of cells in old and young, control, and heterochronic mice. In brain samples, we again compared our model to Buckley's clocks. Although the two approaches each identified distinct aging programs at the tissue and cell type levels, respectively (Fig. 3C), both achieved good performance in quantifying rejuvenation on matching data (Fig. 4H; Supplemental Figs. S18, S19). However, similar to the AD results, we also observed that these cell type–specific clocks had low predictive accuracy when applied to previously unseen cell types (Fig. 4I; Supplemental Fig. S20A). Moreover, the models of Buckley et al. (2023) require explicit cell type annotations and are only trained for a few brain cell types, which constrains their applicability in aging-related disease study and other scenarios with ambiguous cell type identity. In contrast, although SCALE, too, was not trained on the intervention data set, it clearly distinguished intervention effects in all cell types (Fig. 4H; Supplemental Fig. S18), including cell types in which Buckley's models underperformed (Supplemental Fig. S20B).
Generalizing mouse SCALE models to human and rat data
Although our SCALE models were trained on mouse tissues, we speculated that the conservation of the core aging programs (Fig. 2E), as well as the generalizability of SCALE, might enable us to accurately measure aging across species. To test this hypothesis, we replaced selected mouse aging genes with corresponding homologs in other species and similarly calculated the SCALE scores in scRNA-seq data from humans and rats.
Our first experiment involved a human brain data set with seven chronological age groups (Hodge et al. 2019). We found that SCALE was able to distinguish different human age groups (linear regression, P-value < 0.001) (Supplemental Fig. S21). We then applied SCALE to a multitissue scRNA-seq atlas of rats undergoing aging and calorie restriction (CR), which is known to be one of the most effective antiaging interventions (Ma et al. 2020). We observed that in the brown adipose tissue and muscle, cells from young rats had significantly lower SCALE scores than those from older individuals. Furthermore, CR significantly reduced the SCALE scores of cells in older individuals compared with those in the control (Supplemental Fig. S22). However, we did not observe similar trends in other rat tissues, suggesting that cellular responses to aging and CR are tissue and species specific. In general, we recognize that training SCALE on one species and applying it to another has many limitations, as there may be some discrepancies in aging genes and pathways between species (Fig. 2E). Therefore, if possible, we recommend training individual SCALE models directly in the species of interest.
SCALE reveals cell type–specific aging patterns across tissues
SCALE's ability to learn and quantify single-cell aging programs within a tissue enables us to compare aging patterns of different cell types residing within the same tissue. Stem cells have the potential to proliferate and differentiate, which plays a significant role in rejuvenation and regeneration. We observed that SCALE scores of stem cells were usually lower than those of other cells. For instance, the SCALE scores of adipose mesenchymal stem cells in all age groups were significantly lower than that of other cells in the adipose tissue (Fig. 5A). Additionally, we observed that leukocytes in the pancreas were significantly older than others in the same tissue (Fig. 5B). We suspected that this finding was related to the inflammatory response and therefore decided to systematically examine immune cell populations across tissues.
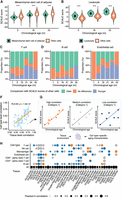
SCALE reveals cell type–specific aging patterns across tissues. (A) SCALE scores of mesenchymal stem cells from adipose cells in subcutaneous adipose tissue (SCAT). Data are presented as the mean ± SD. (***) P-value < 0.001 using a two-sided unpaired Student's t-test. (B) SCALE scores of leukocytes in pancreas. Data are presented as the mean ± SD. (***) P-value < 0.001 using a two-sided unpaired Student's t-test. (C) Differences between the SCALE scores of T cells and the tissues in which they are located. (Older) The SCALE scores of T cells are significantly larger (P-value < 0.05) than all cells in the tissue, (Younger) the SCALE scores of T cells are significantly smaller (P-value < 0.05) than all cells in the tissue. P-values were calculated using two-sided unpaired Student's t-tests. (D) Differences between the SCALE scores of B cells and the tissues in which they are located. (Older) The SCALE scores of B cells are significantly larger (P-value < 0.05) than all cells in the tissue, (Younger) the SCALE scores of B cells are significantly smaller (P-value < 0.05) than all cells in the tissue. P-values were calculated using two-sided unpaired Student's t-tests. (E) Differences between the SCALE scores of endothelial cells and the tissues in which they are located. (Older) The SCALE scores of endothelial cells are significantly larger (P-value < 0.05) than all cells in the tissue, (Younger) the SCALE scores of endothelial cells are significantly smaller (P-value < 0.05) than all cells in the tissue. P-values were calculated using two-sided unpaired Student's t-tests. (F) Schematic overview of the relationship between SCALE scores and chronological age. For each cell type or all cells in one tissue, we calculated the correlation coefficient to define the relationship between the SCALE score and chronological age. We observed cell type–specific aging characteristics and tissue environment may influence cell type aging patterns. (G) Correlation coefficient between SCALE scores and chronological age of each cell type. The size of the circle shows the Pearson correlation coefficient. P-values were calculated using a permutation-based test. Orange indicates that the coefficient is significantly larger, and blue indicates that the coefficient is significantly smaller (P-value < 0.05). (H) Relationship between the correlation coefficient of each cell type and the correlation coefficient of their counterpart tissues. The R value and P-value are shown.
The immune system is vital for organisms to resist infections and maintain homeostasis and is associated with aging. In particular, lymphocytes play a major role in chronic inflammation, and defects in their functional responses have been linked with known aging phenotypes (Nikolich-Žugich 2018). We found that, in most tissues of young mice (1-mo-old and 3-mo-old), the SCALE scores of B cells and T cells were significantly higher than the scores of the whole tissues in which they were located. Although in old mice (21-mo-old, 24-mo-old, and 30-mo-old), there became more tissues where the SCALE scores of B cells and T cells were lower than the tissue background (Fig. 5C,D), possibly indicating a slower aging rate in these cells. To determine if it was a B cell–specific and T cell–specific phenomenon, we investigated the SCALE scores of the endothelial cells, which also exist in multiple tissues. However, we did not observe a similar pattern in endothelial cells (Fig. 5E). One possible explanation for the different aging patterns between B and T cells and other cells in the same tissue is that immune cells are highly mobile and are constantly renewed by HSCs rather than being produced locally.
To further explore how different cell types change during aging in various tissue environments, we calculated the SCALE scores for all 145 cell types in the mouse samples. Generally, we found that the correlation between SCALE scores and chronological age varies across different cell types, even within the same tissue. This prompted us to use the correlation coefficient as an indicator of aging programs. For example, a low correlation between the SCALE score and chronological age within a cell type could imply stronger unique cell type aging characteristics. However, different tissues provide distinct environments with various stressors and thus have different aging programs (Supplemental Fig. S7B; Supplemental Table S1).
To compare one cell type across tissues, we first need to assess the aging baseline in each tissue. We focused on 13 cell types that were detected in at least five samples and investigated if their aging characteristics (i.e., the strength of the correlation between SCALE scores and chronological age) were related to their tissue location. Our results indicated that cells located in tissues with higher correlation coefficients (i.e., stronger aging programs) generally show higher coefficients at the cell type level, suggesting a tissue influence (Fig. 5F).
Still, deviations in cell type correlations from the tissue background suggest distinct aging characteristics in several cell types. To infer the significance of deviations (i.e., the influence of cell type–specific aging characteristics) from the tissue baseline, we developed a permutation-based approach and classified cell types in a given tissue into three categories (Methods): (1) cell types with a significantly higher correlation than all other cells in the same tissue, (2) cell types with a comparable correlation coefficient to the tissue as a whole, and (3) cell types with a significantly lower correlation coefficient (Fig. 5G). These categories have straightforward interpretations. For example, aging in cells of category 3 may have unique aging characteristics that cause the deviation from the tissue baseline (category 2).
Using this metric, we investigated whether the aging pattern is consistent across different tissues for each cell type. We observed that, for a given cell type, it tended to be classified in the same category of the aforementioned three types across diverse tissue environments. For example, immune cells (B cells, T cells, CD4+ T cells, CD8+ T cells, macrophages, and NK cells) had significantly lower correlation coefficients than the tissue as a whole (category 3) in most nonimmune organs where they were detected in our data (Fig. 5H). Furthermore, we found that other cell types that were present in multiple tissues, such as endothelial cells and epithelial cells, also tended to belong to the same category across tissues (Supplemental Fig. S23). These findings indicate that inherent characteristics of cell types play an important role in shaping cell aging patterns, even when situated in different tissue environments.
Discussion
Here we show that tissue-specific aging programs can be learned from scRNA-seq data and applied to describe aging heterogeneity within single-cell populations. From the Aging Map, a curated database of aging-associated genes, we extended our search for transcriptomic features that could quantitatively predict chronological age, generating a multitissue resource of mouse genes as aging biomarkers. We then developed a statistical measure of aging called SCALE, which we applied to scRNA-seq data sets from various murine tissues and organs to evaluate differences in biological aging. As an internal validation, we compared SCALE to the number of somatic mutations and confirmed that SCALE can distinguish aging statuses beyond just predicting chronological age. When quantifying the aging and antiaging effects of diseases and rejuvenating interventions in external data, SCALE shows high robustness to usual challenges in single-cell omics data such as batch effects and sparsity. We further considered the SCALE scores of each cell type and observed conserved cell type–specific aging patterns across tissues. In conclusion, our study provides a useful tool for revealing tissue-level aging processes and evaluating the benefits of potential interventions.
Our results suggest that SCALE has great potential in various applications (Fig. 6): (1) predicting relative biological age and helping to avoid bias when using chronological age as the target in aging studies; (2) prioritizing tissue-specific aging genes and advancing our understanding of aging biology; (3) assessing the efficacy of rejuvenation interventions; (4) predicting AD and other aging-related diseases; (5) quantifying aging at the single-cell level, identifying aging cell populations, and aiding in the development of stem cell therapies; and (6) Being integrated as a functional health assessment tool for the elderly.
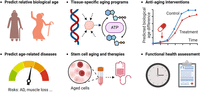
Potential applications of SCALE.
SCALE is an aging quantifier that explicitly makes interpretability a priority and addresses the multilinearity problem using knowledge from literature. By encouraging the inclusion of well-recognized and experimentally confirmed aging genes (Fig. 3B), SCALE is more explainable and robust to spurious correlation and technical variability, as shown in the external validation in aging-related diseases and rejuvenating interventions data (Fig. 4). Indeed, we showed that SCALE became less interpretable (reduced overlap with well-documented aging-associated genes) (Supplemental Fig. S24A) and less accurate (subpar performance on rejuvenation data) (Supplemental Fig. S24B,C) when the Aging Map initialization was replaced by randomly selected genes in the iterative feature selection step. This confirms the importance of including validated aging genes during training. Still, caution should be exercised when interpreting the SCALE scores. Although SCALE and other cellular measures of aging use known aging-associated pathways, such as the accumulation of somatic mutations or DNA methylation, they are most likely only capturing a small aspect of biological aging. It remains an open question whether the predictive power of SCALE and other quantifiers implies causation and whether a change in the SCALE score reflects the shifts most relevant for therapeutic purposes. In the future, it is therefore necessary to test out aging clocks on more data in larger cohorts and expand our understanding of antiaging interventions in a quantitative manner.
From a method development perspective, SCALE can be further improved in many ways. First, all single-cell-level aging clocks, including SCALE, face a common problem: There are often more cells in the training data set than individuals, meaning the model treats the chronological age as discrete rather than a variable continuously sampled from the population. Although the problem may be mitigated by using carefully designed cohorts with larger sample sizes, it is also possible to increase the granularity of age by computationally inferring unobserved sample points. As a proof-of-concept experiment, we showed that optimal transport, a mathematical framework that measures the distance between distributions (also known as the Wasserstein distance), can be applied to smoothly interpolate between discrete chronological age groups and simulate the expected distribution of the feature of interest (e.g., the SCALE score) at a new time point (Methods; Supplemental Fig. S25). Second, SCALE improves robustness by down-weighting genes that are more affected by sampling noise in scRNA-seq data. It is, however, possible to directly model dropouts as missing data and incorporate uncertainty when building the predictor (Ibrahim et al. 2005). Alternatively, technical noise and outliers can be removed through data imputation before training and applying the model (Hou et al. 2020). Third, an increase or decrease in expression during aging does not mean that the gene is responsible for aging. Although our guided forward selection approach encourages selection of well-documented aging genes, it does not directly model causality. Improving the quality of casual aging gene databases and applying casual inference methodology may help to build better aging models. Finally, a better measure of biological aging is likely to be an ensemble of clocks operating at different scales (Belsky et al. 2017; Li et al. 2020). This requires integrating aging quantifiers trained on various data type and aspects of aging, including multiomics clocks, frailty index, iAGE (Sayed et al. 2021), 3D facial-image (Xia et al. 2020), etc. Designing such an integration approach in an interpretable way is critical for understanding fundamental aging biology and for guiding rejuvenation strategies.
Methods
Collection of single-cell aging omics data sets
In this study, we collected and analyzed publicly available single-cell aging RNA-seq data sets across multiple tissues and organs in mice, rats, and humans, covering aging, rejuvenations, and age-related diseases. These data sets include the TMS data set of mice (Tabula Muris Consortium 2020), the heterochronic parabiosis data sets of mice (Pálovics et al. 2022), the single-nucleus data set of mouse AD model (Zhou et al. 2020), and the caloric restriction (CR) data set of rats (Ma et al. 2020). To compare transcriptomic aging clocks with epigenetic clocks, we additionally collected and analyzed the scM&T-seq data of mouse muscle stem cells that contain simultaneously measured single-cell DNA methylation and transcriptome profiles (Hernando-Herraez et al. 2019).
Specifically, for the TMS and heterochronic parabiosis data sets, we downloaded the preprocessed and annotated H5AD files from the corresponding data portals (https://figshare.com/articles/Processed_files_to_use_with_scanpy_/8273102 and https://figshare.com/projects/Molecular_hallmarks_of_heterochronic_parabiosis_at_single_cell_resolution/127628, respectively). For the other two single-cell mouse brain aging data sets (Dulken et al. 2019; Buckley et al. 2023), we downloaded the preprocessed and annotated files from Zenodo (https://doi.org/10.5281/zenodo.7145399).
For the AD data set and the CR data set, we downloaded the count and barcode matrices from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) using the accession numbers GSE137869 and GSE140511, respectively. For the scM&T-seq data set, we downloaded the raw expression and methylation profiles from GEO using the accession numbers GSE121364 and GSE121436, respectively.
Single-cell RNA-seq data preprocessing
We used R version 3.6.0 (R Core Team 2019) and R package Seurat version 3.1.5 (Stuart et al. 2019) to analyze all scRNA-seq data. Because different aging data sets were generated by different platforms for different biological systems, we generally followed the preprocessing steps reported in the original publications for each data set and compared (e.g., disease vs. healthy control) within data sets only. For data sets in which preprocessed data were available, such as TMS, we proceeded with the provided preprocessed and annotated data in downstream analyses. For data sets with raw data and annotations only (AD, CR, scM&T-seq), according to the methods and codes from their original articles, we used Seurat to conduct quality control, normalization (using method “LogNormalize,” set “scale.factor” = 10000), and dimension reduction (PCA, t-SNE, and UMAP) for visualization. The heterochronic parabiosis data sets provide filtered raw counts data, which were then processed following the same normalization and visualization steps.
Searching aging-related mouse and human genes in existing databases and the literature
We first downloaded and combined five existing aging gene databases, namely “CSGene” (Zhao et al. 2016), “Aging Atlas” (Aging Atlas Consortium 2021), “HAGR” (Tacutu et al. 2018), “AgeFactDB” (Hühne et al. 2014), and “Longevity Map” (Budovsky et al. 2013), for mouse and human genes individually. Next, we searched for genes associated with aging-related GO terms, including “aging,” “cell age,” “cellular senescence,” “regulation of cell aging,” and “replicative senescence” (The Gene Ontology Consortium et al. 2000; The Gene Ontology Consortium 2021). To incorporate more recent research efforts, we screened PubMed abstracts for additional aging-associated genes that have been verified experimentally. Specifically, we first searched for potential aging-related genes through the NCBI Gene database (https://www.ncbi.nlm.nih.gov/gene/) using the keywords “aging” and “senescence.” For each of the potential genes, we searched through PubMed using the gene name and “aging” or “cell senescence” as keywords. We then downloaded the abstracts of the top 20 related articles and curated them manually to generate the final list of literature-based, experimentally verified aging-related genes. Together, we created Aging Map (http://sysomics.com/AgingMap/), a manually curated online database of human and mouse aging-related genes.
Global gene expression
To measure the global transcriptional activity of a cell, we defined the global gene expression as the log-transformed mean
counts per gene in a cell:
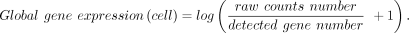 Note that the global gene expression measure depends both on cellular translational activities and the sequencing depth. In
the absence of spike-in controls, we only compared the global expression value across samples generated by the same sequencing
platform (e.g., droplet-based scRNA-seq) within the same data set (e.g., TMS tissues) to mitigate potential variability in
sequencing depth. We evaluated the relationship between global gene expression and chronological age (month) in each condition
using linear regression and assessed the statistical significance of the coefficient using two-sided Student's t-test.
Note that the global gene expression measure depends both on cellular translational activities and the sequencing depth. In
the absence of spike-in controls, we only compared the global expression value across samples generated by the same sequencing
platform (e.g., droplet-based scRNA-seq) within the same data set (e.g., TMS tissues) to mitigate potential variability in
sequencing depth. We evaluated the relationship between global gene expression and chronological age (month) in each condition
using linear regression and assessed the statistical significance of the coefficient using two-sided Student's t-test.
Single-cell entropy
We quantified single-cell transcriptional variability using Shannon entropy. Specifically, in each sample, we first removed
genes that were expressed in <5% of cells. Then, in each cell, we divided the total n expressed genes into m groups with equal group intervals based on their expression value. The number of gene groups m was determined by the Rice rule and was the same for all cells. Further, we considered each gene as a categorical random
variable with m potential outcomes and, in each cell, calculated the event probability as the proportion of genes in each of the m expression group. The single-cell Shannon entropy can then be defined as
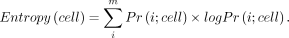 Here Pr(i;cell) is the frequency of the ith gene expression group in the given cell. We again calculated the single-cell entropy for each cell in each sample and evaluated
the association between entropy and chronological age using the Kruskal–Wallis test.
Here Pr(i;cell) is the frequency of the ith gene expression group in the given cell. We again calculated the single-cell entropy for each cell in each sample and evaluated
the association between entropy and chronological age using the Kruskal–Wallis test.
Selecting tissue-specific aging genes
The first step of the SCALE pipeline is to select aging-associated genes for each tissue using guided forward feature selection. At each iteration, we fitted an Elastic Net model using the R package glmnet version 3.0.2 (Friedman et al. 2010) to evaluate the predictive performance of the current aging gene set. To avoid overfitting and underfitting, we divided all samples into training and test sets, tuned model hyperparameters using 10-fold cross-validation on the training set, and internally evaluated the prediction accuracy on the test set. Our procedure can be summarized into the following steps: (1) remove genes that were not expressed in a sample (i.e., detected in <5% of cells); (2) perform an initial Elastic Net regression to rank gene by their predictive performance on chronological age; (3) starting with Aging Map genes as the initial gene set (seed), at each iteration, add more genes to the set according to their rankings and fit a new Elastic Net model against chronological age using raw expression counts of genes in the new set; (4) repeat step 3 until the mean square error (MSE) decreases to a turning point (i.e., defined using the Elbow method or by loss convergence); and (5) select the final aging genes in the last regression model. In our study, we selected the top 100 genes (by coefficient) of each tissue for downstream analysis (Supplemental Table S1).
Calculating the SCALE score
After aging gene selection, for each tissue we now have a set of aging genes with known expression direction of age association
(up-regulated or down-regulated during aging). For each single cell in a given tissue, we calculated the SCALE score using
the weighted average of normalized expression of genes in the tissue-specific aging gene set:
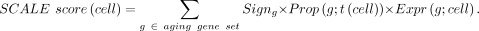 Here the Signg represents the age-association direction (+1 for up-regulated genes and −1 for down-regulated genes) learned from the training
data in the gene selection step; Prop(g;t(cell)) is the proportion of cells in the target data set that expressed gene g; and Expr(g;cell) is the Z-score (zero-centered, variance-standardized) of the normalized gene expression level after preprocessing.
Here the Signg represents the age-association direction (+1 for up-regulated genes and −1 for down-regulated genes) learned from the training
data in the gene selection step; Prop(g;t(cell)) is the proportion of cells in the target data set that expressed gene g; and Expr(g;cell) is the Z-score (zero-centered, variance-standardized) of the normalized gene expression level after preprocessing.
Different from the final Elastic Net prediction and other chronological-age-based aging quantifiers, SCALE generates a relative aging score for each cell based on tissue-specific aging programs. Alternatively, we can view SCALE as a prediction model that replaces regression coefficients learned during feature selection with gene-specific weights to mitigate dropout and other batch effects. Our benchmark showed that the final SCALE score was less sensitive to the training set and other technical variability and therefore more generalizable across data sets (Supplemental Fig. S2). We also hypothesized that the slight decoupling of SCALE and the chronological age help it better capture the biological aging effects in real-world applications.
Evaluating the statistical significance of the SCALE score
We constructed a permutation test to estimate the amount of aging-related information captured by the selected set of aging genes and the statistical significance of SCALE scores in each sample. We first replaced the aging gene set with the same number (100 by default) of expressed genes, which were randomly selected through bootstrap sampling without replacement. Next, we applied the same procedure used to calculate the SCALE score to compute a score based on these genes. By repeating this process 1000 times, we obtained a permutation-based null distribution of the SCALE score and used it to calculate the empirical P-value of any observed score.
Testing the robustness of the SCALE score
Initially, we set raw count values of given proportions (ranging from 10% to 90%) of genes to zero. Next, we renormalized the expression matrix to adjust for varying sequencing depths (using method “LogNormalize” in Seurat, set “scale.factor” = 10000) and then recomputed SCALE scores of cells. Finally, we determined the correlation between the original scores and the newly computed ones.
Evaluating the statistical significance of the correlation between the SCALE score and chronological age
We again used permutation as an empirical statistical approach to compute the null distribution of the correlation coefficient between the SCALE score and chronological age in each set of cells. For each set of cells to inspect (e.g., T cells in the limb muscle), we randomly sampled the same number of cells from the same sample (e.g., limb muscle). Then, we calculated the correlation coefficient between SCALE scores and chronological ages of randomly selected cells. By repeating this process 1000 times, we obtained a permutation-based null distribution of the correlation coefficient and used it to calculate the empirical P-value of the observed correlation in the set of interest. We further categorized the set of interest into three groups based on whether its correlation is significantly higher or lower than the tissue background (P-value < 0.05) (Fig. 5F).
Single-cell methylation data analysis and epigenetic age calculation
We followed the pipeline and scripts provided by the original publication (Trapp et al. 2021; https://github.com/alex-trapp/scAge) to reconstruct the scAge model and analyzed the single-cell methylation data of muscle stem cells (obtained from GEO accession number GSE121436) (Hernando-Herraez et al. 2019) to predict the single-cell epigenetic ages. To compare the robustness against extreme outliers, we did not filter out cells with lower CpG coverage, as opposed to the quality control steps described in the original paper.
Interpolation of SCALE scores by optimal transport
Optimal transport is a mathematical framework that can be applied to describe the distance between probabilistic distributions. The goal of interpolation is to infer the distribution of a variable of interest at an unobserved data point given the distributions observed at the start and end points. Starting with the observed distributions of SCALE scores at various chronological ages, here we aimed to interpolate the SCALE score distribution at the middle point of a given time interval for which no real observation was available. We assumed that the distribution of the SCALE score at the middle point of an interval was the optimal transportation barycenter of distributions at the start and end points. In the one-dimensional case, we solved the barycenter problem using an iterative Bregman algorithm with entropic regularization (Benamou et al. 2015).
Software availability
The SCALE pipeline and associated codes and scripts for analyses described in this study are available on GitHub (https://github.com/ChengLiLab/SCALE) and as Supplemental Codes.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by National Key Research and Development Program of China (2021YFA1100300 to C.L.), National Natural Science Foundation of China (32288102 to C.L., 32025006 to C.L., 31871266 to C.L., 62173338 to H.C.), and Beijing Nova Program of Science and Technology (20220484198 to H.C.). Part of the data analysis was performed on the High Performance Computing Platform of the Center for Life Sciences, Peking University. J.S. was supported by the Edward P. Evans Center for Myelodysplastic Syndromes at Columbia University. We thank Yao Yao for critical comments on the paper. We also thank the aging research community for generating and making data sets available online.
Author contributions: C.L. and H.C. conceptualized and supervised the study. S.M. collected resources and created the Aging Map database. S.M., J.S., and L.W. performed data analysis and interpreted the results. S.M. and J.S. prepared and revised the manuscript. All authors read and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277491.122.
-
Freely available online through the Genome Research Open Access option.
- Received November 19, 2022.
- Accepted July 12, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








