
A Chinese indicine pangenome reveals a wealth of novel structural variants introgressed from other Bos species
- Xuelei Dai1,8,
- Peipei Bian1,8,
- Dexiang Hu1,8,
- Funong Luo1,8,
- Yongzhen Huang1,8,
- Shaohua Jiao1,
- Xihong Wang1,
- Mian Gong1,
- Ran Li1,
- Yudong Cai1,
- Jiayue Wen1,
- Qimeng Yang1,
- Weidong Deng2,
- Hojjat Asadollahpour Nanaei1,3,
- Yu Wang1,
- Fei Wang1,
- Zijing Zhang4,
- Benjamin D. Rosen5,
- Rasmus Heller6 and
- Yu Jiang1,7
- 1Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology, Northwest A&F University, Yangling, Shaanxi 712100, China;
- 2Faculty of Animal Science and Technology, Yunnan Agricultural University, Kunming 650201, China;
- 3Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran 1983969412, Iran;
- 4Institute of Animal Husbandry and Veterinary Science, Henan Academy of Agricultural Sciences, Zhengzhou 450002, China;
- 5Animal Genomics and Improvement Laboratory, USDA-ARS, Beltsville, Maryland 20705, USA;
- 6Department of Biology, University of Copenhagen, 2200 Copenhagen, Denmark;
- 7Key Laboratory of Livestock Biology, Northwest A&F University, Yangling, Shaanxi 712100, China
-
↵8 These authors contributed equally to this work.
Abstract
Chinese indicine cattle harbor a much higher genetic diversity compared with other domestic cattle, but their genome architecture remains uninvestigated. Using PacBio HiFi sequencing data from 10 Chinese indicine cattle across southern China, we assembled 20 high-quality partially phased genomes and integrated them into a multiassembly graph containing 148.5 Mb (5.6%) of novel sequence. We identified 156,009 high-confidence nonredundant structural variants (SVs) and 206 SV hotspots spanning ∼195 Mb of gene-rich sequence. We detected 34,249 archaic introgressed fragments in Chinese indicine cattle covering 1.93 Gb (73.3%) of the genome. We inferred an average of 3.8%, 3.2%, 1.4%, and 0.5% of introgressed sequence originating, respectively, from banteng-like, kouprey-like, gayal-like, and gaur-like Bos species, as well as 0.6% of unknown origin. Introgression from multiple donors might have contributed to the genetic diversity of Chinese indicine cattle. Altogether, this study highlights the contribution of interspecies introgression to the genomic architecture of an important livestock population and shows how exotic genomic elements can contribute to the genetic variation available for selection.
Cattle are one of the most important livestock species owing to their production and role in human culture (Felius et al. 2014). Domestic cattle are mainly divided into indicine cattle (Bos indicus) and taurine cattle (Bos taurus), which originated from independent domestication events in the Indus Valley and the Near East, respectively. These have spread around the world to form six commonly accepted cattle groups (Park et al. 2015; Verdugo et al. 2019), including European taurine, African taurine, Asian taurine, Indian indicine, African indicine, and Chinese indicine. Among these, Chinese indicine cattle is thought to have spread into China between 3500 and 2500 years before present (YBP) (Naik 1978; Payne and Hodges 1997; Chen et al. 2010). In general, range expansions tend to lead to the loss of genetic diversity through the effect of serial bottlenecks or founder events (Ramachandran et al. 2005; Liu et al. 2006). However, Chinese indicine cattle show at least twofold higher genetic diversity than other known cattle groups (Chen et al. 2018a; Kim et al. 2020). A previous study found that introgression from other Bos species could account for this increased genetic diversity, and estimated that the average Chinese indicine cattle genome contained ∼2.93% of genetic ancestry from the banteng (Chen et al. 2018a). More recently, another study found that ∼10% of the Chinese indicine cattle genome derives more broadly from “exotic” ancestry admixture (Sinding et al. 2021). Therefore, the amount of introgressed genetic material in Chinese indicine cattle, as well as the impact of this introgression in shaping the genetic diversity in this cattle population, remains unresolved, and in general, the contribution of introgression to genetic diversity and functional variation in admixed livestock populations remains understudied to date.
Any single linear reference genome is unable to capture the genetic diversity contained in a population and therefore leads to the oversight of millions of relevant sequence and structural variants (SVs), leading to various types of reference biases (Crysnanto et al. 2021). Conceptually, a pangenome that represents structural and sequence variation segregating in the population can alleviate many of these biases (Tettelin et al. 2005). Pangenome approaches have already been used to improve the completeness and representation of variants across various cattle breeds. Three different approaches have been used to construct cattle pangenomes: read mapping to the reference (iterative assembly) (Zhou et al. 2022), alignment of de novo assembled genomes (Gong et al. 2022), and pangenome graphs (Crysnanto and Pausch 2020). Advances in long-read sequencing technology such as Nanopore sequencing and PacBio single-molecule real-time (SMRT) sequencing have been used to resolve novel sequences and SVs in the cattle genome (Crysnanto et al. 2021; Leonard et al. 2022). For example, a bovine multiassembly graph was integrated with multiple taurine genomes and two close relatives, revealing an extra 70 Mb of novel sequence absent from the B. taurus reference genome (Crysnanto et al. 2021). Similarly, 294 diverse cattle genome sequences were integrated into a global cattle graph genome that recovered 116.1 Mb nonreference sequence (Talenti et al. 2022). These initial bovine breed-specific augmented and pangenome graphs improved sequence read mapping and removed biases in variant discovery (Crysnanto and Pausch 2020). Despite these improvements, the unique genetic diversity and SVs of Chinese indicine cattle are not reflected in any high-quality genomic resources currently available, despite the fact that Chinese indicine cattle are among the most genetically diverse of all domesticated cattle breeds (Kim et al. 2017; Chen et al. 2018a; Zhang et al. 2022b). This is important, because SVs are a rich source of genetic variation accessible for selection, and they can, in some cases, be subject to stronger selection pressures than single-nucleotide polymorphisms (SNPs) (Hsieh et al. 2019; Ho et al. 2020). Yet, the role of SV introgression in highly reticulated evolutionary history of the Bos genus (Wu et al. 2018) has not been systematically investigated by using population-based detection methods.
In the present study, we generated high-quality partially phased genomes for 10 representative Chinese indicine cattle breeds across southern China, with the aim of identifying the genomic landscape of non-Hereford sequence. By taking advantage of these partially phased assemblies, we comprehensively assessed the introgression landscape of Chinese indicine cattle to shed light on the unique genetic composition and diversity contained in this cattle population. Additionally, we generated a comprehensive novel nonredundant SV set and explored the evolutionary fate of introgressed SVs. This research provides fundamental new insights into the adaptive potential of SV introgression in general and the contribution of archaic introgression to the genetic diversity in Chinese indicine cattle in particular.
Results
High-quality de novo assemblies of 10 representative Chinese indicine breeds
We generated 18- to 24-fold coverage accurate circular consensus sequencing (PacBio HiFi) for 10 geographically distant female Chinese indicine cattle (Fig. 1A; Supplemental Table S1). We produced 20 de novo assembly genomes using phased assembly graphs with hifiasm (Cheng et al. 2021), which can render a primary assembly contig and two haplotype contigs (haplotype 1 and haplotype 2) for each sample. Reference-guided scaffolding with RagTag (Alonge et al. 2022) produced the chromosome-scale genome assembly. The size of the primary genome was 2679–2714 Mb, consisting of 110–367 contigs with an N50 of 18–91 Mb at ∼95.5% benchmarking universal single-copy ortholog (BUSCO; cetartiodactyla_odb10) (Waterhouse et al. 2018) completeness. The haplotype genomes ranged in size from 2585–2698 Mb, consisting of 1057–4286 contigs with an N50 of 1.24–13.45 Mb at 89.3%–93.8% BUSCO completeness (Supplemental Fig. S1; Table 1; Supplemental Table S2). The assembly quality values (QVs) were further estimated using HiFi reads and 18-fold-coverage Illumina data per individual by merqury (Rhie et al. 2020) and showed an average QV score of 61.31 and 39.13, respectively, and a base accuracy >99.99% (Supplemental Table S3).
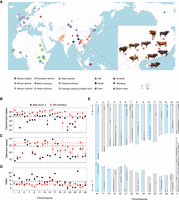
Geographic distribution of the cattle breeds used in this study and the centromeric and telomeric completeness of 10 Chinese indicine assemblies. (A) Each circle represents a cattle breed collected from public databases, with circle size relative to sample size. The squares indicate the PacBio HiFi sequencing data used in our study. (B) Statistical distribution of centromeric bases on each chromosome of 10 Chinese indicine assemblies (red dots) and reference genome ARS-UCD1.2 (black dots), where error bars indicate the 95% confidence interval. Dashed lines represent the mean value for the corresponding color of dots. (C) Similar to B, but the telomeric bases within 10 kb of chromosome ends. (D) The number of scaffold gaps on each chromosome. (E) The chromosome ideogram of Jinjiang cattle primary assembly (left) and ARS-UCD1.2 (right). Scaffolded contigs alternated by white/gray regions across gapped regions, which are colored red. Black arrows show the centromere to telomere of the chromosome, and those generally considered to be end-to-end are blue frames.
Summary statistics of the 10 assembled Chinese indicine genomes
Bovine chromosomes are acrocentric except for sex chromosomes (Blazak and Eldridge 1977), and so the goal for complete assemblies should be “centromere-to-telomere” completeness. The primary assemblies of the 10 individuals contained an average of 962.18 kb of centromeric sequences per autosome, compared with 88.52 kb in the current ARS-UCD1.2 (Rosen et al. 2020) reference (Fig. 1B; Supplemental Table S4). Moreover, we also evaluated telomeric sequences of the autosomes using vertebrate telomeric repeats (TTAGGG) within 10 kb of the chromosome end, compared with 0.82 kb for the ARS-UCD1.2 reference and the average 2.26-kb telomere length of all our assemblies (Fig. 1C; Supplemental Table S5). Consistent with previous assessments on end-to-end of chromosomes (Leonard et al. 2022), there were five for the ARS-UCD1.2 reference and a mean of 13.2 autosomes for our primary assemblies (Supplemental Table S6). The gap content of our primary assemblies was slightly lower than the ARS-UCD1.2 reference at around five gaps per autosome (Fig. 1D; Supplemental Table S7). The example of our assembly from a Jinjiang breed individual shows the distribution of assembly gaps (Fig. 1E), as well as “centromere-to-telomere” markers, which indicates that the majority of assembly scaffolds approach chromosome-level and shows that our new reference genomes are high quality.
Constructing a Chinese indicine multiassembly graph
To obtain nonreference sequences contained in Chinese indicine cattle, we used the Hereford-based linear reference genome (ARS-UCD1.2) (Rosen et al. 2020) as the backbone and integrated the 20 partially phased assemblies of the Chinese indicine into a multiassembly graph using minigraph (Li et al. 2020). The regions of synteny were ignored, whereas sufficiently diverged subsequences (>50 bp) were used to augment the graph with new nodes (“bubbles”). The resulting multiassembly graph contained 160,000 nonreference nodes spanning 148.5 Mb with ∼22.21% of the resulting pangenome being flexible (i.e., not shared by all assemblies) (Fig. 2A), which is more than previous studies (see Methods) (Crysnanto et al. 2021; Leonard et al. 2022). A total of 74,907 nonreference alleles (>100 bp) were extracted from our multiassembly graph. When compared with the whole-genome resequencing of Bos species (see Methods), 53.96% of these novel sequences cannot be detected in other indicine/taurine cattle populations, whereas 26.21% were identified in wild Bos species among these undetected sequences (Supplemental Table S8). Variants in our graph are enriched with repetitive elements and LINE/L1 is dominant, and variable number tandem repeat (VNTR) is the leading type of multiallelic variants (Fig. 2B). To validate the nonreference nodes and variants, we mapped the PacBio HiFi reads from 10 Chinese indicine cattle to the multiassembly graph using GraphAligner (Rautiainen and Marschall 2020) and calculated the coverage (number of reads aligned) at each node and edge in the graph; 98.80% nonreference nodes and 98.49% of the variation breakpoints had support (more than one read in any one sample) from HiFi reads. A pangenome constructed using the Brahman (UOA_Brahman_1) reference assembly (Low et al. 2020) as the backbone produced similar results compared with using the Hereford-based reference genome (ARS-UCD1.2) (Fig. 2C).
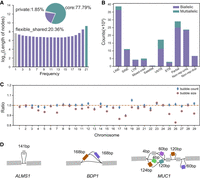
Chinese indicine multiassembly graph. (A) Composition of the multiassembly pangenome graph. (B) Variations are stratified by repeat class and by the number of alleles of each variation. The repeat annotation was obtained from the longest allele of each variation. (VNTR) Variable-number tandem repeat, a tandem repeat with the unit motif length ≥7 bp; (STR) short random repeat, a tandem repeat with the unit motif length ≤6 bp; (mixed-inter) a variation involving two or more types of interspersed repeats. (C) Comparing the number and mean size of bubbles present in Brahman-backed pangenome to ARS-UCD1.2-backed pangenome. The number of bubbles is highly consistent, but the size of bubbles is slightly larger on average in ARS-UCD1.2-backed pangenome compared with Brahman-backed pangenome. (D) The genes with alternative complex coding structures because of tandem repeat. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids. The repeat unit in the VNTR corresponds to a 168- and 60-nucleotide sequence in BDP1 gene and MUC1 gene, respectively.
The extended graph coordinate system further helps to recover genes with alternative complex structures compared with ARS-UCD1.2. We found 381 bubble breakpoints overlapping with Hereford genome coding sequences (Supplemental Table S9), which indicates substantial variability in the length of amino acid sequences. These genes often contain a highly polymorphic VNTR domain, such as ALMS1 (Liao et al. 2003), BDP1 (Liao et al. 2003), RTP4 (Boys et al. 2020), CYLC2 (Hess et al. 1995), PRDM9 (Zhou et al. 2018), QRICH2 (Hiltpold et al. 2022), and mucin genes (MUC1, MUCL1, MUC6, MUC16, MUC20) (Fig. 2D), which leads to intra- and inter-breed/species amino acid variation with the potential to affect phenotypes.
We used a combination of de novo and homology-based approaches to evaluate the presence of genes and genic structures with novel sequences (see Methods). The AUGUSTUS (Stanke et al. 2008) software de novo predicted 1153 complete gene models from the nonreference sequences. The nonreference alleles and predicted protein sequences were aligned with the protein reference database using DIAMOND's BLASTP and BLASTX, respectively. A total of 456 genes were found of which 271 are novel compared with previous studies (Crysnanto et al. 2021; Talenti et al. 2022). RNA-seq read alignments provided additional support for 260 of these gene models (Supplemental Table S10). These predicted protein-coding genes are mostly represented in multigene families and could play a role in the following processes: olfactory transduction (olfactory receptor 9K2, 10X1, 1134, and 5M9-like), immune response (BOLA class I histocompatibility antigen alpha chain BL3-7-like, interleukin-3 receptor subunit alpha, interferon omega-1, low affinity immunoglobulin gamma Fc region receptor II-like, lymphokine-activated killer T-cell-originated protein kinase), signaling (mitogen-activated protein kinase kinase kinase 7 [MAP3K7], ras-related protein Rap-1b, inhibitor of nuclear factor kappa-B kinase subunit alpha isoform X1, E3 ubiquitin-protein ligase RNF146), endogenous retrovirus-K proteins, and some ribosomal proteins.
Generating and characterizing a catalog of SVs in Chinese indicine cattle
Previous studies have shown that HiFi sequencing significantly increases SV discovery (Audano et al. 2019; Ebert et al. 2021). To obtain reliable SVs, we used four SV callers: pbsv (https://github.com/PacificBiosciences/pbsv), SVIM (Heller and Vingron 2019), Sniffles (Sedlazeck et al. 2018), and cuteSV (Jiang et al. 2020), all specifically designed for SV detection by long-read mapping-based approaches for each genome. Consistent with previous studies (Wu et al. 2021), we retained the SVs identified by at least two methods for each sample. Consequently, we merged the high-confidence SVs detected from all the samples and constructed a set of 156,000 nonredundant SVs (comprising 73,889 deletions and 82,120 insertions with a length ≥ 50 bp) (Fig. 3A). We annotated all SVs and found that most cover intergenic and intronic regions, whereas a smaller number intersect with functional elements and exons (Fig. 3B; Supplemental Fig. S2). We then classified the final nonredundant SV catalog of Chinese indicine cattle into four categories: shared (identified in all samples), major (identified in ≥50% of samples, but not all), polymorphic (identified in 1%–50% of samples), and singleton (identified in only one sample) (Fig. 3C). The accumulated size of the nonredundant catalog increased rapidly at low sample sizes but gradually reached a plateau with the accumulation of samples, which indicates that a considerable proportion of common SVs was detected by our approach. Meanwhile, the set of shared SVs decreases quickly as samples are added, and only 8958 SVs (5.74%) were observed in all samples.
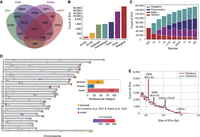
Discovery and distribution of structural variation in 10 Chinese indicine. (A) Venn diagrams show the total number of SVs in the Chinese indicine population identified by pbsv, Sniffles, cuteSV, and SVIM software and their overlap. (B) The numbers of SVs overlapping with different genomic features. (C) The number of variants in each discovery class is shown per sample. The SVs can be classified into four classes: shared, major, polymorphic, and singleton. (D) Genome-wide distribution of SV hotspots detected from previously published SV data sets by Crysnanto et al. (2021) and Talenti et al. (2022) (blue to red heatmap). SV hotspots identified in this study were divided into three groups: “terminal,” the last 5 Mb of chromosome end (yellow); “known,” overlapping with published data (blue); and “novel,” unique for this study (red). The bar plot (inset) shows the total of the number of hotspots in each group. (E) The length distribution of deletions and insertions. Expected two SINE BOV-A2, LTR, and LINE-1 mobile element insertion peaks are marked at ∼145 bp, 285 bp, 1.3 kb, and 8.5 kb, respectively.
In the final catalog, the SV size distribution shows that most are relatively short, and two peaks with lengths of ∼145 bp and 285 bp are mainly annotated to BOV-A2 (SINEs), a peak at 1295 bp corresponds to ERV2-LTR-BT, and a peak at 8500 bp corresponds to LINE/L1 (Fig. 3E). This observation supports the idea that transposable elements are an important source of SVs in cattle. The SVs are mainly enriched in LINE and LINE/L1, which differs from variations in the corresponding human graph constructed with minigraph enriched with Alus (SINE) and VNTRs (Li et al. 2020). This is consistent with reports of similar patterns in sheep (Li et al. 2023) and could therefore be a bovid-specific phenomenon. We also found that SVs are nonrandomly distributed across the genome (Audano et al. 2019; Ebert et al. 2021), and we identified 206 SV hotspots spanning ∼195 Mb of the genome (Fig. 3D; Supplemental Fig. S3; Supplemental Table S11). Of these hotspots, 61 are within the last 5 Mb of chromosome arms. Excepting the terminal regions of chromosomes, 28 hotspots overlap with hotspots identified in previously published long-read-based SV data sets from 294 diverse cattle graphs (VG5) (Talenti et al. 2022) and a bovine multiassembly graph (Crysnanto et al. 2021), whereas the 119 remaining hotspots are novel (Fig. 3D, inset). We then used the generic annotation of ARS-UCD1.2 to detect protein-coding genes overlapping with our SV hotspots. A permutation result showed a significant enrichment of SV hotspots (n = 206) in protein-coding genes (P-value = 0.001, Z-score = 4.203) (Supplemental Fig. S4A). In contrast, a permutation test using all SVs showed depletion in protein-coding genes (P-value = 0.001, Z-score = −10.174) (Supplemental Fig. S4B). Finally, a total of 2533 protein-coding genes overlapped at least one SV hotspot, and these genes are mainly associated with the immune system and olfactory transduction (Supplemental Fig. S5).
Genetic contribution of different Bos species to Chinese indicine cattle
To characterize the genomic landscape of archaic introgression in Chinese indicine, we applied a two-state hidden Markov model (Skov et al. 2018) to detect segments of individual genomes of archaic origin, without using an archaic reference genome, by identifying regions with a high density of derived alleles. We used the 20 partially phased assemblies as targets (see Methods) and 321 non-Chinese indicine domestic cattle as an outgroup (Supplemental Table S12). For all subsequent analyses, we retained 34,249 fragments with a posterior probability of >90% of being archaic, and we ruled out archaic fragments that were likely caused by incomplete lineage sorting (ILS) according to a probability calculation (Supplemental Table S13).
We next analyzed the distribution of tree topologies of each archaic fragment across several species belonging to the Bos genus. We encountered five tree topologies among our introgressed fragments, and in contrast to previous studies (Chen et al. 2018a), we found evidence that Chinese indicine genomes contain significant numbers of fragments from other external sources than banteng; in particular, ancient kouprey-like fragments were numerous (Fig 4A). The introgressed fragments that are banteng-like, kouprey-like, gayal-like, gaur-like, and unknown cover 45.7%, 37.9%, 20.5%, 7.9%, and 9.3% of the genome, respectively, and in total, these covered 1.928 Gb (73.3%) of the genome. The X Chromosome is noticeably depleted in introgressed elements compared with the autosomes (Fig. 4B), which may be owing to stronger natural selection or the susceptibility to incompatible foreign alleles (Presgraves 2018). We further explored the proportion and length of introgressed fragments. Three percent to 16% sequences of each Chinese indicine genome were identified as introgressed, and an average of 3.8%, 3.2%, 1.4%, 0.5%, and 0.6% of each individual's genome was assigned to banteng-like, kouprey-like, gayal-like, gaur-like, and unknown origin, respectively (Supplemental Fig. S6). The average length of introgressed elements was 195 kb, 135 kb, 157 kb, 117 kb, and 66 kb for each donor species, respectively (Fig. 4C). We applied MultiWaver 2.0 (Ni et al. 2019) to reconstruct the admixture history of each donor species into Chinese indicine cattle based on the length distribution of introgressed fragments. The admixture pulses with banteng-like, kouprey-like, gayal-like, gaur-like, and unknown origins were estimated to have occurred 532–560, 763–807, 656–713, 912–1039, and 1517–1721 generations ago, respectively (see Methods; Fig. 4F; Supplemental Fig. S7; Supplemental Table S14). The youngest of these inferred admixture pulses—that from the banteng-like source—occurred 3360–3192 years ago, assuming a generation time of 6 yr, whereas the other admixture pulses are all older than 3500 yr. This suggests that the introgression from various sources happened at different temporal and geographical stages of the cattle dispersal into East Asia, consistent with dispersing cattle having come into contact with different Bos species in different localities (Fig. 4F). It also suggests that most of the introgression predates the presumed arrival of indicine cattle into China between 3500 and 2500 YBP (Naik 1978; Felius et al. 2014), which indicates that almost all admixture waves predate the presumed introduction of indicine cattle into China. These results suggest a more complex evolutionary history of Chinese indicine cattle than previously suggested, in which introgression from multiple donor Bos species occurred during possibly nonoverlapping bursts, whereas limited introgression from one or more currently unknown sources may even have occurred before the domestication of indicine cattle.
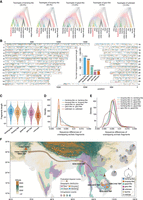
The genomic landscape of introgression in 20 Chinese indicine partially phased genomes. (A) DensiTree (Bouckaert 2010) visualization of five tree topologies: banteng-like, kouprey-like, gayal-like, gaur-like, and unknown origin. The value in brackets is the percentage of the same topologies. (B) The genome-wide distribution of archaic fragments in 20 Chinese indicine haplotypes, colored according to the topologies: banteng-like (blue), kouprey-like (golden), gayal-like (green), gaur-like (purple), and unknown (dark orange). The inset shows the total introgression-covered length of each origin. (C) Length distribution of banteng-like, kouprey-like, gayal-like, gaur-like, and unknown archaic fragments. The sequence differences between overlapping archaic fragments from the same origin are shown in D (KDE plot) and from different origins are shown in E. (F) The postulated dispersal routes of Chinese indicine cattle and admixture with wild Bos species. Introgression events with different wild Asian Bos species are marked with numbers in colored circles. The pie charts show the geographical distribution and genetic make-up of 15 Chinese indicine cattle breeds (45 samples), with each circle sized proportionally to the number of samples per breed.
We further explored how introgression has shaped the genetic diversity in Chinese indicine cattle. Divergent genomic regions introduced by admixture can result in an increased genetic diversity of the admixed population (Pan et al. 2022). Consistent with previous studies (Chen et al. 2018a), the nucleotide diversity (θπ) was significantly higher in Chinese indicine than other domestic cattle populations (Supplemental Fig. S8), and the number of variants in the introgressed regions was significantly higher than those in the nonintrogressed regions (Supplemental Fig. S9). Introgressed regions of the genome that had two haplotypes with shared inferred ancestry showed an average sequence difference of 1.92–2.23 variants per kilobase (Fig. 4D), whereas regions with haplotypes of different inferred ancestry had a higher average difference of 4.24–6.57 variants per kilobase (Fig. 4E), again indicating that multiple Bos donors were involved in the admixture. We added another 35 Chinese indicine cattle short-read sequencing samples to calculate the introgressed content of different donors using the same method as above and found that the global content of banteng-like and kouprey-like fragments was significantly negatively correlated with altitude (Fig. 4F; Supplemental Figs. S10, S11), suggesting either population structure or different post-admixture selection pressures in different cattle habitats. Taken together, these observations indicate that Chinese indicine cattle experienced extensive admixture with the surrounding species of the Bos genus, which was instrumental for the increased genetic diversity in this cattle population.
Adaptive introgression of Chinese indicine population-stratified SVs
To supplement the above SNP-based introgression results, we also assessed the role of SVs in local adaptive introgression into Chinese indicine cattle. We genotyped 402 domestic and wild Bos genomes with Illumina reads with a graph-based approach (see Supplemental Methods; Supplemental Fig. S12; Supplemental Tables S15, S16). To ensure a high-quality set of autosomal SVs for population genetic analysis, we excluded SVs that failed to be genotyped in >90% of the samples and those whose minimum frequency was less than 0.01, leaving 114,387 (77.3%) reliably genotyped autosomal SVs for downstream analysis (Supplemental Fig. S13). In the six commonly assumed domestic cattle groups, principal component analysis (PCA) revealed that indicine and taurine cattle were separated into two major clusters, and each lineage had a clear genetic structure consistent with the geographical region (Supplemental Fig. S14). The same population structure was recovered by a phylogenetic tree, in which the Chinese indicine cattle formed the most basal lineage (Supplemental Fig. S15). The six major genetic clusters were confirmed by the best-fitting admixture model (Supplemental Fig. S16).
We searched Chinese indicine cattle population–stratified SVs introgressed from other Bos species, using similar methods described in previous studies (see Methods; Almarri et al. 2020; Quan et al. 2021), identifying a total of 3136 (2.74%) introgressed SVs (Fig. 5A; Supplemental Table S17). We uncovered a 6.3-kb insertion (a full-length transposable element) downstream from ASIP (13:63675338) (Supplemental Fig. S17), which is a high-frequency introgressed SV that is shared with banteng and kouprey (Fig. 5B,C; Supplemental Fig. S18). Based on previous studies, the agouti signaling protein (ASIP) gene is a potential candidate gene that encodes proteins affected by coat color in mammals (Mohanty et al. 2008; Han et al. 2011; Bannasch et al. 2021; Trigo et al. 2021), and the ASIP gene region of Chinese indicine has introgressed only from banteng, which may partly explain the tan coat color patterns of Chinese indicine cattle (Chen et al. 2018a). This insertion was segregating at 55.6% frequency in Chinese indicine and at 100% frequency in banteng and kouprey but was absent from other cattle populations (Fig. 5D). Meanwhile, the flanking region of the insertion showed particularly high signals of the fixation index (FST; Chinese indicine cattle vs. Indian indicine cattle) (Fig. 5B; Supplemental Fig. S19), indicating that this region might undergo positive selection in Chinese indicine cattle. We extracted this region from Chinese indicine cattle and outgroup species and investigated the differences between haplotypes of Bos species. Through haplotype heatmap and network analysis (see Methods), we found that all haplotypes with the insertion were clustered together and that almost all of this region is introgressed from kouprey-like origin rather than banteng as previously reported (Fig. 5C,E; Chen et al. 2018a). The admixed Chinese indicine cattle individuals show a tan coat color, whereas other Chinese indicine cattle that do not harbor this haplotype have a predominantly yellow coat color (Fig. 5C), suggesting the coat color of the Chinese indicine cattle population may have been under artificial selection for purely aesthetic or other cultural reasons. These results provide insights into the adaptive introgression of coat color in Chinese indicine at the SV level.
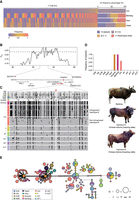
Candidate introgressed SV of Chinese indicine cattle and a case of adaptive introgressed haplotype associated with coat color. (A) The heatmap shows the frequency distribution of 3136 population-stratified SVs in Chinese indicine population and shared with the banteng, gaur, and gayal populations. Each vertical line represents an SV in the corresponding population, and the color ranging from dark blue to red represents frequency varying from zero to one. The bar plot at the right of the heatmap shows the percentage of the SV frequency in the corresponding population. (B) The schematic map indicates the selective signal (FST) and the genomic location on Chromosome 13 (62.37–64.83 Mb). The horizontal dashed line of SNPs (the 1% right tail of the empirical FST distribution) was identified as the selected regions for Chinese indicine cattle and Indian indicine cattle. (C) The haplotypes of the insertion and its flanking SNP in each cattle population. The missing genotypes of the ancient kouprey sample in the haplotype are indicated by blank blocks. (D) The frequency of the 6.3-kb insertion in each cattle group. (E) Haplotype network based on pairwise differences around the insertion region; the haplotypes with the insertion (introgressed haplotype-A) in Chinese indicine population are predominantly clustered with kouprey-like origin, whereas other haplotypes without the insertion (nonintrogressed haplotype-B) tend to cluster with domestic cattle population. (AFB) African buffalo; (EUB) European bison; (AMB) American bison; (CHI) Chinese indicine; (II) Indian indicine; (AFI) African indicine; (AFT) African taurine; (AST) Asian taurine; (EUT) European taurine.
SVs may impact the expression of nearby genes by changing the state of the cis-regulatory elements (Chiang et al. 2017; Alonge et al. 2020). To explore SV-associated gene expression changes, we generated blood transcriptome and whole-genome sequencing data from 19 PiNan cattle (hybrids of Piedmontese × Nanyang cattle). These data enabled us to identify the candidate expression-associated SVs by allelic-specific expression (ASE) mapping, and we found 2237 heterozygous SVs (SVs near ASE genes) that would putatively play cis-regulatory elements and cause ASE in associated genes (Supplemental Table S18). Among these, 52 SVs in the Chinese indicine population were introgressed from wild Asian Bos species whose corresponding ASE genes are functionally relevant to disease resistance and energy metabolism (e.g., MALT1, PRKAR2A, etc.) (Supplemental Table S18; Supplemental Fig. S20; London et al. 2020; Gu et al. 2022). We additionally performed SV-eQTL mapping and identified 250 SVs with genotypes that correlate with expression levels of a nearby gene (P value < 0.05, Benjamini–Hochberg adjust) (Supplemental Table S19), of which 15 introgressed SVs. Taken together, these results may partly explain the effect of introgressed SVs on gene expression and might contribute to the environmental adaptability for Chinese indicine cattle.
Discussion
In this study, we take advantage of HiFi reads to accurately represent the haplotype information of Chinese indicine cattle in a phased assembly graph, which provides better assemblies than previous tools and outperforms older methods (Cheng et al. 2021). Our assemblies of Chinese indicine cattle are of high quality, contain more centromeric and telomeric sequence content, and will likely be an indispensable resource for future bovine genomic studies, facilitating the comparison of haplotypes to identify heterozygous variants and determining genetic diversity at the level of single individuals. By identifying 5.6% novel genomic content that is not present in the Hereford reference genome, we not only provide physical coordinates for these nonreference sequences but also add substantial new genomic resources with important implications for cattle research. Among the 74,907 nonreference alleles identified here, we predict up to 1153 complete genes using in silico approaches, of which 271 are novel compared with previous studies (Crysnanto et al. 2021; Talenti et al. 2022). Overall, we conclude that Chinese indicine cattle contain a large number of genes not previously known or characterized.
Compared with previous reports based on various short-read and long-read sequencing technologies to detect SVs (Talenti et al. 2022; Zhou et al. 2022), we leveraged PacBio HiFi sequencing to greatly improve the precision and recall rates (Wenger et al. 2019). In our study, we took a stringent filtering strategy and only retained the SVs identified by at least two methods for each sample, ensuring that we obtain a reliable SV data set that can be used in subsequent studies. In doing so, we present a nonredundant SV data set and a new map of 119 novel SV hotspots from the Chinese indicine population compared with previous studies (Crysnanto et al. 2021; Talenti et al. 2022). The large number of novel SV hotspots identified in our study may be owing to (1) improvements in SVs detection methods using PacBio HiFi technology instead of Oxford Nanopore or short-read sequencing technology, (2) the Chinese indicine population having the highest genetic diversity of all domestic populations (Chen et al. 2018a), or (3) unique SVs private to Chinese indicine cattle being poorly represented among SV data set in previous studies.
The population divergence process and admixture history of the Chinese indicine population is complex according to previous studies (Chen et al. 2018a; Sinding et al. 2021). We took advantage of the high accuracy and continuity of the haplotype for partially phased assemblies, which significantly improves the accuracy of haplotype introgression inference compared with those based on short-read data, especially in complex regions of the genome (see Supplemental Methods; Supplemental Figs. S21, S22). Furthermore, a two-state hidden Markov model (Skov et al. 2018) allowed us to identify introgressed fragments that derive from highly divergent donor populations (i.e., other Bos species), and then determine the donor based on tree topologies of each archaic fragment across the Bos genus. This enabled us to investigate the introgression landscape of Chinese indicine cattle without a large reference panel and without making a priori assumptions about the specific donor species. Hence, we identified an average introgressed genome proportion of 9.5% among the sequenced Chinese indicine cattle, which is similar to the ∼10% “exotic” ancestry proportion in one previous study (Sinding et al. 2021). The significant difference in introgression ranging from 3%–16% among breeds of Chinese indicine cattle is mainly owing to two factors: (1) Chinese indicine continued to mix with Asian taurine as it spread to northern China, causing the introgressed genome proportion to become diluted in various proportions (Supplemental Fig. S10), and (2) some of the introgression regions might be also under further negative selection in altitude adapted breeds (Supplemental Fig. S11). In contrast to previous studies, we provide the first detailed results regarding the contribution of different Bos species to this exotic admixture (see Supplemental Methods; Supplemental Table S20). Furthermore, we show that the admixture history of each donor Bos species into Chinese indicine cattle could have occurred in nonoverlapping pulses, providing an important new insight into the history of Asian cattle. Although we have taken full advantage of partially phased assemblies in our analysis, the available wild Asian Bos genomes are a significant limitation, and we acknowledge that further insights may be challenged by the fact that the various introgression sources may be only partially represented by the available genome data from wild Bos. Future research with the inclusion of more wild Bos genomes is needed to further address these shortcomings.
Although SVs are known to shape population diversity and local adaptive introgression (Hsieh et al. 2019), few studies have examined this aspect of the complex admixture history of Chinese indicine cattle. In this study, we performed the first investigation of introgressed SVs across the genome and found an SV adjacent to the well-known coat color–associated genes ASIP that may have a functional role and may have been under artificial selection in the Chinese indicine cattle population. Coat color traits are known from previous studies to have been some of the traits under strong artificial selection in livestock owing to selective breeding for preferred color variants (Innan and Kim 2004; Li et al. 2010; Bannasch et al. 2021). Taken together, our results provide a valuable new resource for future bovine introgression and SV studies.
Methods
Sample collection
Fresh blood samples were collected from 10 representatives of Chinese indicine cattle across different regions in southern China, which were used for Illumina and PacBio HiFi sequencing (Supplemental Table S21). We also collected fresh blood of 19 PiNan cattle (hybrids of Piedmontese [male] × Nanyang cattle [female]) from Nanyang County, Henan Province, China, which was leveraged for whole-genome and transcriptome resequencing by the Illumina platform (Supplemental Table S21). In addition, we downloaded 394 representative whole-genome resequencing data from species of the Bos genus for population analysis (including two African buffalo, four American bison, five European bison, three yaks, 10 gayals, four gaurs, eight bantengs, two ancient koupreys, 110 European taurines, 43 African taurines, 47 Asian taurines, 78 African indicines, 43 India-Pakistan indicines, and 35 Chinese indicines). Summary information and overview of above samples, including breeds, country of origin, geographical location, sequencing depth, and contributors, are detailed in Supplemental Tables S15 and S16.
Short-read sequencing
In addition to the above, we extracted DNA from the same 10 Chinese indicine cattle and 19 PiNan cattle as above using a standard cetyltrimethylammonium bromide (CTAB) extraction protocol. This DNA was used to construct the sequence libraries according to the Illumina library preparation protocol and was sequenced on the Illumina HiSeq platform to generate 150-bp paired-end reads (Supplemental Tables S1, S21). The RNA of 19 PiNan cattle was extracted with a Qiagen RNeasy mini kit for transcriptome sequencing, and a total of 19 paired-end libraries were prepared and sequenced on an Illumina HiSeq platform with 150-bp paired-end reads (Supplemental Table S21).
PacBio HiFi sequencing
We extracted high-quality gDNA from the fresh blood of the above 10 Chinese indicine to construct libraries using the SMRTbell express template prep kit 2.0 (PacBio) and fractionated them on the SageELF (Sage Science) into narrow library fractions. Before sequencing, library fractions were bound to polymerase with the sequel II binding kit 2.0 (PacBio) and then sequenced with the PacBio sequel II sequencing kit 2.0 and 8 million SMRT cells on the sequel II (PacBio) with 30-h movie times for each sample. The HiFi reads were generated from raw data using the CCS algorithm (version 6.0.0; parameters: ‐‐minPasses 3 ‐‐minPredictedAccuracy 0.99 ‐‐maxLength 21000). Generating CCS reads does not include or require alignment against a reference sequence but does require at least two full-pass subreads from the insert (Supplemental Table S1).
De novo genome assembly and quality assessment
Firstly, we used a fast haplotype-resolved de novo assembler, hifiasm (version 0.13-r308; with default parameters) (Cheng et al. 2021) to assemble the PacBio HiFi reads. Next, we applied the reference-guided software RagTag (v2.0.1) (Alonge et al. 2022) to scaffold the contigs to chromosome level. The completeness of the assemblies was assessed with BUSCO (version 5.4.5), using the metaeuk backend (6.a5d39d9) and cetartiodactyla_odb10 database. The assembly base QVs were calculated with merqury (version 1.3) (Rhie et al. 2020), with k-mer databases constructed from short reads and HiFi reads using meryl (version 1.4; https://github.com/marbl/meryl). RepeatMasker (v4.1.1) (Chen 2004) was used to search known TEs by mapping sequences against the ruminant repeat library to annotate repeat sequences on each chromosome (see Supplemental Methods; Supplemental Fig. S23).
Detection of non-Hereford sequence
Minigraph (version 0.15) (Li et al. 2020) was used to integrate 21 genome assemblies into a multiassembly graph. The Hereford-based linear reference genome (ARS-UCD1.2) was used as the backbone of the Chinese indicine multiassembly graph, and 20 partially phased assemblies from the 10 Chinese indicine individuals were integrated into the multiassembly graph with minigraph. To determine the support of the nodes, we aligned (with minigraph parameters “‐‐cov -x asm”) individual assemblies back to the multiassembly graph. Nodes with nonzero coverage were then color-labeled according to which assembly path traversed them. We used the bubble popping algorithm of gfatools (version 0.5) (Crysnanto et al. 2021) to derive structural variations from the multiassembly graph, traversing all possible paths in the bubble and retaining only paths with color-consistent labels. We then classified a path as a reference path if all nodes and edges were part of the Hereford-based reference assembly, and classified as nonreference otherwise (Crysnanto et al. 2021). We collected all non-Hereford alleles originating from bubbles (excluding complete deletions, paths without non-Hereford bases, and paths with length <100 bp) to obtain a comprehensive set of non-Hereford bases from the multiassembly graph. The Brahman-backed pangenome was integrated using the same pipeline.
To exclude the nonreference nodes and variations caused by assembly errors and to validate the nonreference nodes and variations, we mapped the PacBio HiFi reads from 10 Chinese indicine cattle to the multiassembly graph using GraphAligner (version 1.0.13) (Rautiainen and Marschall 2020) with the command “GraphAligner -t 40 -x vg.” We then calculated coverage (number of reads aligned) at each node and edge in the graph based on the graphical alignment format output from GraphAligner; 98.80% nonreference nodes and 98.49% of the structural variation breakpoints had support (more than one read in any one sample) from HiFi reads.
Whole-genome resequencing data of 84 cattle (16 African taurines, 16 African indicines, 16 Asian taurines, eight European taurines, eight Indian indicines, eight gayals, four gaurs, eight bantengs) were downloaded for analyzing the presence/absence of nonreference sequences. We appended the non-Hereford sequences as additional contigs to the ARS-UCD1.2 reference, making an extended reference genome. The reads were aligned to the extended ARS-UCD1.2 reference genome sequence. The presence and absence of each novel sequence were then determined according to the sequence coverage and depth. Novel sequences with a depth greater than one-third of the whole-genome depth were identified as present.
Repeat analysis
Centromeric repeats were identified as Satellite/centr family by RepeatMasker (v4.1.1) (Chen 2004) using a modified Repbase database (release 20181026). Repeats with a Smith–Waterman score < 20 and substitution percentage > 40% were filtered out. Vertebrate telomeric repeats (TTAGGG) were identified within 10 kb of the chromosome end. According to a previous study (Leonard et al. 2022), the end-to-end was considered where the proximal end of a chromosome contains at least 50-kb centromeric repeat sequences and the distal end contains at least 500-bp telomeric repeat sequences. Repeats of nonreference sequences were analyzed with a method combining homology comparison and de novo structure analyses. We applied RepeatMasker (v4.1.1) (Chen 2004) using the database of repetitive DNA elements from Repbase (release 20181026) to classify interspersed repeats in the longest allele sequence of each variation. We identified tandem repeats composed of a motif occurring twice or more by Tandem Repeats Finder (TRF) (Benson 1999).
Detection of high-confidence SVs
To obtain high-confidence SVs, we performed a long-read mapping-based approach to call SVs with multiple tools and strict filtering steps. For each genome, we aligned PacBio HiFi reads to the cattle reference genome ARS-UCD1.2 (Rosen et al. 2020) by pbmm2 (version 1.4.0; with parameters: ‐‐sort ‐‐preset HiFi ‐‐rg “@RG\tID:SampleID”; https://github.com/PacificBiosciences/pbmm2). Subsequently, pbsv v2.4.0 (https://github.com/PacificBiosciences/pbsv), SVIM v1.4.2 (Heller and Vingron 2019), Sniffles v1.0.12 (Sedlazeck et al. 2018), and cuteSV (Jiang et al. 2020) v1.0.10 were used to detect SVs in the aligned BAM files for each sample. pbsv was used with parameters “discover, call ‐‐ccs -m 50,” and we set the following parameter for SVIM.
We merged the SV call sets of each individual derived from the above four SV callers for each sample. We applied the SURVIVOR (Jeffares et al. 2017) software to merge SVs for each SV type based on the variant position and length, and retained SVs detected by at least two callers for each individual. The maximum distance between breakpoints of SVs is equal to 10, and the minimum size of SVs to be taken into account is equal to 50; hence, the parameters were “merge $Sample.vcf_files_raw_calls.txt 10 2 1 0 0 50 $Sample.merged.vcf.” As suggested by benchmark analysis of LRS callers (Dierckxsens et al. 2021), we chose results that have a priority of pbsv > SVIM > Sniffles > cuteSV. Finally, we merged the above high-quality SV call set for each sample using SURVIVOR with the parameters “merge vcf_files_calls.txt 50 1 1 0 0 50 Total_sample.merged.vcf.”
SV annotation and hotspot identification
Annotation of the SV call set was performed using SnpEff (version 5.0e) (Cingolani et al. 2012) and ANNOVAR (Wang et al. 2010) software. According to a previous study (Ebert et al. 2021), we selected the middle position of each SV and used the “hotspotter” function of the primatR package for hotspot analysis with the parameters “bw=200000, pval=1e-08, num.trial=2000.” Previously published long-read-based SV data sets by Crysnanto et al. (2021) and Talenti et al. (2022) were also used for hotspot detection and comparison. We classified our inferred SV hotspot (n = 206) into three catalogs: “terminal,” residing in the last 5 Mb of the chromosome end (n = 61); “known,” overlapping with previous studies (Crysnanto et al. 2021; Talenti et al. 2022) (n = 26); and “novel,” unique for this study (n = 119). We further investigated whether our SV hotspots overlap with protein-coding genes. For this, we extracted 20,271 unique protein-coding genes from the generic feature file of ARS-UCD1.2 (obtained from the NCBI Assembly database [https://www.ncbi.nlm.nih.gov/assembly] under accession number GCF_002263795.1). To test for overrepresentation of SV hotspots in coding regions, we ran a permutation test using the regioneR package with 1000 iterations; the permTest function was used for calculation with the circularRandomizeRegions function. We obtained the repeat file (GCF_002263795.1_ARS-UCD1.2_rm.out.gz) and gap file (GCF_002263795.1_ARS-UCD1.2_genomic_gaps.txt.gz) from NCBI. Then, we extracted “Satellite/centr” entries from the repeat file and merged them with the gap regions to create a comprehensive mask file. As a comparison, we also performed the same permutation test with all SVs.
Read mapping and variant calling
The short-read data were filtered by the fastp program (version 0.20.0) (Chen et al. 2018b) with default parameters. All passed quality-filtered reads were aligned to the reference genome (ARS-UCD1.2) with BWA-MEM (version 0.7.17) (Li and Durbin 2009) using default parameters. After sorting the BAM files and removing PCR duplicates using Picard (version 2.1.1) tools (http://broadinstitute.github.io/picard/), SNPs were called using the Genome Analysis Toolkit (GATK) (version 2.1.1) (McKenna et al. 2010), and then, we filtered raw SNPs with the parameters “QUAL < 40.0, MQ < 25.0, MQ0> = 4 & (MQ0/(1.0 * DP)) > 0.1, -cluster 3 -window 10.” The final obtained variants were filtered using BCFtools (version 1.1) (Danecek et al. 2021) with the parameters “-i ‘MAF>0.01 & F_MISSING<=0.1’ -v”.
Graph genotyping of structural variation
We downloaded a large amount of short-read sequence data (about 13× coverage) from the ARS-UCD1.2 cattle reference using BWA-MEM (version 0.7.17-r1188) (Li and Durbin 2009) with default parameters. Using the above SVs set by the long-read mapping-based approach identified from HiFi sequencing data from 10 Chinese indicine cattle, we used Paragraph (v2.4a; with default parameters) (Chen et al. 2019) to genotype SVs in each individual. To ensure a high-quality SV set for population genetics analysis, we excluded SVs that failed to be genotyped in >90% of the samples, and an excess of heterozygotes deviated from Hardy–Weinberg equilibrium using VCFtools (Danecek et al. 2011); 41,622 SVs were removed after filtering, leaving 114,387 variants for downstream analysis.
Identifying archaic introgressed fragments
To more accurately identify the archaic fragments of Chinese indicine, we constructed a high-quality SNP collection. For partially phased assemblies, we aligned each partially phased assembly to the reference genome (ARS-UCD1.2) using minimap2 (version 2.17-r941; with parameters: ‐‐paf-no-hit -a -x asm5 ‐‐cs -r2k) (Li 2018) and SAMtools (version 1.12; with default parameters) (Danecek et al. 2021), and then, the SNPs were called by bcftools mpileup (version 1.12; with parameters -Q 20 -q 20 ‐‐annotate FORMAT/AD,FORMAT/DP) (Danecek et al. 2021) and bcftools call (with parameters -m -v -O z). For the whole-genome resequencing data SNP set, we used the method in the SNP calling part above. To reduce any bias from combining these two SNP sets, we also added the resequencing data of the corresponding samples of the partially phased assembly; then Beagle (version 5.1; with default parameters) (Browning et al. 2018) was used to phase the SNPs, and according to the evaluation results of phasing accuracy below in the Methods section, we removed the resequencing Chinese indicine SNPs from the phased set. The SNP set of both partially phased assemblies and resequencing data was integrated into the final SNPs collection with BCFtools (version 1.12) (Danecek et al. 2021) and in-house scripts, which were used for subsequent analysis (see Data access).
To infer archaic introgressed fragments in Chinese indicine, we used a two-state hidden Markov model (Skov et al. 2018) to remove the variation found in outgroup and then used the remaining variants to group the genome into regions of different variant density, in which the introgressed regions have higher variant density than nonintrogressed. According to the major update pipeline of this model (https://pypi.org/project/hmmix/), (1) we first found SNPs that are derived in 321 non-Chinese indicine domestic populations (Indian indicine, African indicine, African taurine, Asian taurine and European taurine as outgroup; with the command hmmix create_outgroup); (2) then we used the number of variants in the outgroup to estimate the substitution rate as a proxy for mutation rate for each independently partially phased assembly (with the parameter hmmix mutation_rate); (3) we kept variants that were not found to be derived in the outgroup for each individual in Chinese indicine (with the parameter hmmix create_ingroup); and (4) we trained the parameters for haploid data and then decoded (with the parameters hmmix train -haploid; hmmix decode -haploid). The introgressed fragments were retained with a posterior probability of >90% of being archaic.
Finally, to determine the possible source of introgression for each archaic fragment, we constructed a tree topology for each fragment across Bos genus samples. We extracted the corresponding SNPs of other species of the Bos genus (the populations were African buffalo, Indian indicine, Asian taurine, European taurine, yak, American bison, European bison, ancient kouprey, banteng, gayal, and gaur) on each archaic introgressed fragment, where heterozygous SNPs sites were represented using the standard International Union of Pure and Applied Chemistry chemical nomenclature (IUPAC) codes (Cornish-Bowden 1985). We inferred the tree topology by using the maximum-likelihood statistical method for each archaic fragment using the MEGA program (version 11.0.10; with the parameters Bootstrap 100 replicates; Model Tamura-Nei model) (Tamura et al. 2021). The Environment for Tree Exploration (ETE) package (V3) (Huerta-Cepas et al. 2016) was used to identify the closest branch for each archaic fragment as a possible donor.
Estimation of admixture time based on length distribution of introgressed fragments
We applied MultiWave 2.0 (Ni et al. 2019) to infer the optimal model without prior model assumptions or estimated parameters and admixture time but based on the length distribution of introgressed fragments in admixed genomes. The program first applies a likelihood ratio test and an exhaustion method to choose an optimal admixture model based on the length distribution of introgressed fragments and then uses an EM algorithm to estimate the corresponding admixture times and proportions under the above optimal model. The length distributions for introgressed fragments were taken from the results of hmmix above. The results of MultiWave 2.0 showed that the “HI model” fitted the Chinese indicine population well, with two discrete admixture events and 100% confidence intervals of the admixture dates for each donor population obtained from 1000 bootstrapping repeats (Supplemental Table S14). Additionally, to ensure the reliability of the results, we used MultiWaverX (Zhang et al. 2022a) to infer the admixture time for each individual of Chinese indicine cattle with each donor Bos species, and the results were similar to the above (see Supplemental Methods, Supplemental Fig. S24; Supplemental Table S14).
Identification of population-stratified and introgressed SVs
To determine Chinese indicine cattle candidate population-stratified SVs, we calculated the fixation index (FST) of SVs between Chinese indicine cattle and Indian indicine cattle using VCFtools (Danecek et al. 2011) We then performed permutation tests 1000 times to calculate the empirical P-values. We finally applied screening to retain 16,584 SVs in the Chinese indicine cattle population-stratified SVs that satisfy (1) FST > 0.1, (2) empirical P-value < 0.05, and (3) maximum missingness rate < 0.1 and minimum allele frequency > 0.01.
For the validation of introgressed SVs in Chinese indicine cattle population derived from other Bos species, we considered the following: (1) SVs uniquely shared between wild Asian Bos species and Chinese indicine cattle, but absent in other indicine and taurine cattle, and (2) SVs that are positioned on the archaic introgressed fragments (see above). A total of 14,716 introgressed SVs were retained. Finally, the intersection of population-stratified and introgressed SVs of Chinese indicine cattle identified a total of 3136 shared SVs (Supplemental Table S17).
Haplotype pattern and network analysis
To visualize the specific genotype pattern of the introgressed regions flanking the 6.3-kb insertion (13:63675338), we extracted the phased SNPs in the 20-kb region flanking the variant from 389 individuals and visualized their genotypic pattern in a heatmap (Fig. 5C). We also constructed haplotype networks for this region using R package PEGAS based on the pairwise differences (Fig. 5E; Paradis 2010). We retained at most four individuals with consistent haplotypes in the same population and excluded removed SNPs with minor allele frequency < 0.05. In this introgressed region, we retained 173 SNPs in 118 samples from 14 Bos populations. In addition, by combining the introgressed SV in the section Identifying Archaic Introgressed Fragments, we found haplotypes where insertions were clustered together and defined two main haplotypes in Chinese indicine population as introgressed haplotype-A and nonintrogressed haplotype-B.
Detecting expression-associated SVs
To reduce potential mapping bias caused by non-Hereford sequences, we constructed a custom genome by merging the non-Hereford sequences and the reference genome (ARS-UCD1.2). RNA sequencing reads of PiNan cattle were aligned to the custom genome by STAR (version 2.2.1) (Dobin et al. 2013). In an effort to minimize mapping bias, we also used the “mappability filtering” module of WASP software to filter our BAM files (van de Geijn et al. 2015; https://github.com/bmvdgeijn/WASP).
Then, the ASE genes were identified following the pipeline described in our previous study (Wang et al. 2022). Briefly, the ASE SNPs were identified from RNA-seq data using the following parameters: (1) at least 20 total reads, (2) allele ratio (allele reads count/total reads count) > 0.65 or <0.35, and (3) significant allelic imbalances with FDR < 0.05 (chi-squared test). Those genes with at least two ASE SNPs on gene exons and found in at least three samples were defined as candidate ASE genes. For each SV–gene pair, the following conditions need to be met: (1) The SV is located 100 kb upstream of and downstream from the gene; (2) the SV is heterozygous; and (3) the ASE gene corresponds to the heterozygous state of the associated SV in at least three RNA-seq samples. Finally, we considered these SVs as plausibly expression-associated. In addition, we have also identified the ASE of the novel genes (see Supplemental Methods; Supplemental Fig. S25).
Gene-level transcript abundance was estimated as transcripts per million (TPM) using the StringTie (version 1.2.2) (Kovaka et al. 2019). We considered a gene as detected/transcribed when it had an expression value >1 TPM in at least three samples. Local eQTLs were calculated based on linear regression models using FastQTL (v2.0) (Ongen et al. 2016). The mapping window to detect local eQTLs was defined as 1 Mbp upstream of and downstream from the gene. To account for confounding effects, we incorporated gender and population structure (the top three principal components derived from PCA analysis of the 19 individuals) as covariates in our analysis. FastQTL was run in normal mode to calculate nominal P-values of all local gene–variant associations. Statistical significance was considered with P < 0.05 (Benjamini–Hochberg adjusted).
Data access
The data sets supporting the conclusions of this article are included within the article and its Supplemental Files. The de novo assemblies, HiFi data, and WGS data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA786777. The details are provided in Supplemental Tables S21 and S22. The multiassembly graph and the VCF file of the SV catalog are available at Zenodo (https://doi.org/10.5281/zenodo.7607407). Workflow and analysis scripts are available at GitHub (https://github.com/Xuelei-Dai/Chinese_indicine_pan-genome_project_scripts_and_piplines) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Key R&D Program of China (2022YFF1000100) and the National Natural Science Foundation of China to Y.J. (U21A20247, 31822052). R.H. was supported by a European Research Council Starting grant (no. 853442). We thank the High-Performance Computing platform of Northwest A&F University for providing computing resources.
Author contributions: Y.J. conceived the project, designed the experiments, and managed components of the project; X.L.D., P.P.B., D.X.H., F.N.L., B.D.R., R.H., and Y.J. organized the manuscript; X.L.D. and S.H.J. performed de novo assembly, SV calling, and population analyses; P.P.B. constructed the Chinese indicine cattle multiassembly graph; X.L.D., M.G., and D.X.H. performed archaic fragment analysis; X.L.D. and D.X.H. performed the adaptive introgression of Chinese indicine population-stratified SVs; F.N.L. performed ASE and eQTL analyses; X.L.D. and P.P.B. analyzed the RNA-seq; Y.Z.H., Z.J.Z., and W.D.D. collected the samples; Y.J., B.D.R., Y.W., F.W., H.A.N, R.L., X.H.W., and R.H. revised the manuscript. X.L.D., S.H.J., J.Y.W., Q.M.Y., and Y.D.C. prepared the WGS data; and all the authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277481.122.
-
Freely available online through the Genome Research Open Access option.
- Received November 8, 2022.
- Accepted June 30, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








