
Loss of epigenetic suppression of retrotransposons with oncogenic potential in aging mammary luminal epithelial cells
- Parijat Senapati1,6,
- Masaru Miyano2,
- Rosalyn W. Sayaman2,3,
- Mudaser Basam1,2,
- Amy Leung1,
- Mark A. LaBarge2,4,5 and
- Dustin E. Schones1,4
- 1Department of Diabetes Complications and Metabolism, Beckman Research Institute, City of Hope, Duarte, California 91010, USA;
- 2Department of Population Sciences, Beckman Research Institute, City of Hope, Duarte, California 91010, USA;
- 3Department of Laboratory Medicine, Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, California 94143-0981, USA;
- 4Irell & Manella Graduate School of Biological Sciences, City of Hope, Duarte, California 91010, USA;
- 5Center for Cancer Biomarker Research, University of Bergen, 5021 Bergen, Norway
Abstract
A primary function of DNA methylation in mammalian genomes is to repress transposable elements (TEs). The widespread methylation loss that is commonly observed in cancer cells results in the loss of epigenetic repression of TEs. The aging process is similarly characterized by changes to the methylome. However, the impact of these epigenomic alterations on TE silencing and the functional consequences of this have remained unclear. To assess the epigenetic regulation of TEs in aging, we profiled DNA methylation in human mammary luminal epithelial cells (LEps)—a key cell lineage implicated in age-related breast cancers—from younger and older women. We report here that several TE subfamilies function as regulatory elements in normal LEps, and a subset of these display consistent methylation changes with age. Methylation changes at these TEs occurred at lineage-specific transcription factor binding sites, consistent with loss of lineage specificity. Whereas TEs mainly showed methylation loss, CpG islands (CGIs) that are targets of the Polycomb repressive complex 2 (PRC2) show a gain of methylation in aging cells. Many TEs with methylation loss in aging LEps have evidence of regulatory activity in breast cancer samples. We furthermore show that methylation changes at TEs impact the regulation of genes associated with luminal breast cancers. These results indicate that aging leads to DNA methylation changes at TEs that undermine the maintenance of lineage specificity, potentially increasing susceptibility to breast cancer.
Aging leads to a progressive decline in tissue and organ function and is associated with many diseases, including cancer. At the genomic level, alterations to the epigenome contribute to the development of age-associated diseases (López-Otín et al. 2013; Booth and Brunet 2016). DNA methylation is a relatively stable component of the epigenome that is established in early development and maintained through successive cycles of cell division to preserve cell identity (Bird 2002). DNA methylation patterns, however, are known to change with age (Maegawa et al. 2010; Rakyan et al. 2010; Heyn et al. 2012; Horvath 2013). DNA methylation levels of specific CpG sites show a significant correlation with chronological age; this “epigenetic clock” can predict the biological age of diverse human tissues (Horvath 2013). It remains unclear, however, how these changes contribute to increased disease risk. Moreover, pan-tissue clocks do not capture tissue-specific drivers of aging, which are essential to understand disease risk (Bell et al. 2019).
Age is the most significant risk factor for breast cancer, as >75% of breast cancers are diagnosed in women over the age of 50 (Jemal et al. 2007). This is especially true for luminal subtypes of breast cancer, which account for ∼80% of all age-related breast cancers (Jenkins et al. 2014). The mammary epithelium is composed of two principal lineages, myoepithelial cells (MEps) and luminal epithelial cells (LEps), with the latter being the chief suspect cell of origin for luminal breast cancers (Prat and Perou 2009; Fu et al. 2020). Aging in breast tissue accompanies significant physiological changes that lead to changes in molecular properties as well as the proportions of cell types (Benz 2008; Garbe et al. 2012; Pelissier et al. 2014; Miyano et al. 2017). Human mammary epithelial cells (HMECs) from older postmenopausal women, when transformed in vitro, show gene expression patterns characteristic of luminal breast cancer subtypes (Lee et al. 2015). In contrast, transformed HMECs from younger premenopausal women show more properties of basal breast cancers (Lee et al. 2015). Age-dependent changes in abundance and phenotype occur in LEps, which lose lineage specificity by gaining expression of characteristic MEp genes (Garbe et al. 2012; Pelissier Vatter et al. 2018; Sayaman et al. 2021) and signaling molecules consistent with those of immortalized LEps (Pelissier Vatter et al. 2018), suggesting that age-associated epigenetic changes in LEps are directly related to elevated cancer risk.
Although numerous studies have characterized the DNA methylomes of individual epithelial cell populations in human and mouse breast tissue (Bloushtain-Qimron et al. 2008; Dos Santos et al. 2015), there are not many studies that have interrogated the effect of age on DNA methylation profiles of mammary epithelial cell types. Earlier examinations of DNA methylation changes with age in breast tissue (Hofstatter et al. 2018; Castle et al. 2020) either were performed in bulk mammary tissue with heterogeneous cell populations or interrogated only a subset of CpGs in the genome (Johnson et al. 2017; Song et al. 2017). A complete understanding of age-associated alterations to DNA methylation in human mammary LEps and of how this relates to age-related breast cancer has been lacking.
One of the major functions of DNA methylation in mammalian genomes is to suppress transposable elements (TEs). The repetitive content of human genomes, mainly consisting of retrotransposons such as SINEs, LINEs, and ERVs, accounts for approximately half the genome (Cordaux and Batzer 2009). Although most of these retrotransposons have lost their ability to propagate, they still carry latent regulatory potential and can serve as regulatory elements binding transcription factors (Sundaram et al. 2014). The global loss of DNA methylation observed in cancer cells has been shown to result in the loss of epigenetic repression of TEs in many cancer types (Burns 2017; Ishak and Carvalho 2020) and can drive the expression of oncogenes (Jang et al. 2019).
In aging, a genome-wide loss of methylation with age has been observed in human CD4+ T cells and skin keratinocytes (Heyn et al. 2012; Vandiver et al. 2015; Jenkinson et al. 2017). Several early studies analyzing the methylation of repetitive elements in bulk suggested a decline in methylation of Alu and HERV-K elements with age in human blood cells (Bollati et al. 2009; Jintaridth and Mutirangura 2010). Increased expression of retrotransposons and active retrotransposition has furthermore been reported in aging mouse tissues (De Cecco et al. 2013; Benayoun et al. 2019). Outside of these early studies, a thorough examination of epigenetic changes at TEs in aging has not been previously performed. Given that TEs contain latent regulatory potential, loss of epigenetic repression with age has the potential to lead to altered gene regulation and potentially impact disease risk. We describe here our investigation into the impact of aging on the epigenetic repression of TEs in normal mammary LEps, the primary suspected cell of origin for luminal breast cancers.
Results
Evolutionarily recent LTR elements function as regulatory elements in mammary LEps
We performed whole-genome bisulfite sequencing (WGBS-seq) on human mammary LEps from younger (<30 yr or premenopausal) and older (>55 yr or postmenopausal) women (Supplemental Fig. S1A; Supplemental Table S1). These age groups were chosen to represent samples from women who were clearly premenopausal or postmenopausal to avoid confounding issues of peri-menopause. We previously showed that LEps from these primary cultures maintain gene/protein expression and DNA methylation profiles that mirror those in vivo (Miyano et al. 2017; Pelissier Vatter et al. 2018; Sayaman et al. 2021). Moreover, these are nonsenescent cells and can therefore be used to interrogate age-dependent changes in the breast tissue that are distinct from those arising from cellular senescence.
WGBS libraries were sequenced to ∼30× genome coverage, generating approximately 1 billion 150-bp paired-end reads (∼86 Gb uniquely mapped sequence per sample) (Supplemental Table S2) to obtain a representation of ∼95% of individual CpG sites in our samples (see Methods; Supplemental Tables S2, S3). We compared our DNA methylation profiles with previously published WGBS studies on distinct HMEC types from reduction mammoplasty (RM) tissue of a young (19-yr-old) donor (Pellacani et al. 2016). The cultured LEps resemble the ER-negative (ER−) LEp population (Supplemental Fig. S1B). Moreover, young and old LEps clustered separately, indicating differences in methylomes (Supplemental Fig. S1B) that were not apparent when considering total methylation levels (Supplemental Fig. S1C,D). We did not observe any changes in the global methylation levels across the genome with age, as has been reported in other cell types (Supplemental Fig. S1C,D; Cole et al. 2017).
We first focused our analysis on the methylation levels of TEs in young LEps by examining the average methylation of each TE subfamily. Because evolutionarily recent TEs (inserted <100 million yr ago) (Burns and Boeke 2012) are more likely to be silenced by DNA methylation (Edwards et al. 2017; Greenberg and Bourc'his 2019), we plotted average DNA methylation with the evolutionary age of each subfamily (Supplemental Fig. S1E) in young LEps alone. As expected, most TE subfamilies showed average methylation of >70% in young LEps regardless of evolutionary age, similar to the global methylation levels of the LEp genome (Supplemental Fig. S1E, S1C). TEs that have been recently active, such as AluYa5, AluYb8, and AluYb9 elements, showed the highest average methylation. Many evolutionarily older LINE and ERV subfamilies (>100 million yr) had lower overall methylation (<70%), consistent with the loss of CpG density over evolutionary time (Supplemental Fig. S1E,F; Gonzalgo and Jones 1997). Given the regulatory potential of TEs, the observation that LTRs of some evolutionarily recent ERV subfamilies had low average methylation (<70%) (Supplemental Fig. S1E) suggested that these LTRs could be functioning as regulatory elements in luminal cells.
To assess the potential for TEs to function as regulatory elements in LEp genomes, we identified overrepresented TE subfamilies among ATAC-seq peaks in young LEps. We overlapped ATAC-seq peaks with the RepeatMasker annotation (Smit et al. 2023) and compared the overlap frequency at each TE subfamily with random control regions (see Methods). We found many ERV/LTR subfamilies to be enriched at sites of open chromatin, consistent with these elements being bound by transcription factors and functioning as regulatory elements (Fig. 1A; Supplemental Table S4; Tsompana and Buck 2014). Many of these are LTR elements of evolutionarily recent ERVs, including MER11D (28.46% overlap with ATAC-seq peaks), LTR13 (45.28%), and LTR10C (24.08%) (Fig. 1A; Supplemental Table S4). We further confirmed that individual elements from these subfamilies were hypomethylated (<30% methylation) (Fig. 1B) and had enrichment of ATAC-seq signal (Fig. 1C; Supplemental Fig. S1G). Within each subfamily, hypomethylated LTRs (<30% methylation) consisted of unmethylated CpGs spanning the entire LTR (Supplemental Fig. S1H, unmethylated panels), whereas the partially methylated LTRs (30% > methylation > 70%) overlapping ATAC-seq peaks had only a subset of CpGs in the LTR unmethylated (Supplemental Fig. S1H, partially methylated panels). Moreover, subfamilies LTR13 and LTR10F had relatively fewer fully unmethylated copies compared with MER11D (Supplemental Fig. S1H, cf. ii, iv [unmethylated panels] and i [unmethylated panel]). For each subfamily, specifically the hypomethylated LTRs had open chromatin signals (Supplemental Fig. S1G, i–iv [unmethylated panels]). For example, a completely unmethylated MER11D element near the SERHL gene locus harbors an open chromatin peak distinct from the SERHL TSS peak in young as well as old LEps (Fig. 1D). The low methylation levels of these elements combined with presence of an open chromatin site is consistent with them functioning as potential regulatory elements. We then asked if these hypomethylated LTRs have specific TF binding sites (see Methods). Binding sites for SOX4 and ELF5, two ER− luminal lineage-specific TFs (Sayaman et al. 2021) were enriched among the hypomethylated LTRs (Fig. 1E,F). Binding sites for other luminal expressed TFs, such as NFIA and EGR1, were also detected among LTR18A-C and MER11D. GATA3 sites were most enriched among MER11D elements in addition to STAT5B and SOX4 (Fig. 1E,F). Taken together, these results suggest that specific subfamilies of evolutionarily recent LTRs have the potential to function as regulatory elements in normal (noncancer) LEps.
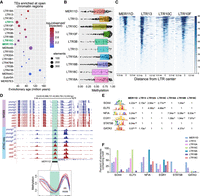
Evolutionarily recent LTR elements have signatures of regulatory elements in mammary luminal epithelial cells (LEps). (A) Top 20 TE subfamilies (mostly LTRs) enriched in open chromatin peaks in young LEps. Evolutionary age is indicated for each subfamily. The size of the dot indicates the number of elements overlapping ATAC-seq peaks, and the color indicates log2 enrichment (observed/expected). (B) Boxplot of DNA methylation levels for each copy of the 10 most evolutionarily recent LTR subfamilies that were enriched among open chromatin regions. (C) ATAC-seq profile across all elements of MER11D, LTR13, LTR10C, and LTR10F subfamilies in young LEps. (D) Genome Browser tracks show a representative example of a hypomethylated (<30% methylation) MER11D element in LEps. The bottom panel shows smoothed line plots of methylation levels at the same element highlighted in green. (E) Transcription factor binding motifs enriched among hypomethylated (<30% methylation) LTR sequences of evolutionarily recent subfamilies. The P-values for enrichment over scrambled sequences are listed. (F) Bar plots show the percentage of hypomethylated TE sequences (<30% methylation) that contain indicated TF binding motifs.
Age-dependent methylation changes occur at TE subfamilies that contain lineage-specific transcription factor binding sites
Having established the methylation status of TEs in young LEps, we next assessed how TE methylation changes with age. We first identified differentially methylated regions (DMRs) by comparing the methylomes of old versus young LEps (see Methods; Supplemental Fig. S2A,B; Supplemental Table S5). We overlapped DMRs with ATAC-seq peaks and found that DMRs with methylation loss showed an overall gain in chromatin accessibility in old LEps. Conversely, DMRs with methylation gain showed decreased chromatin accessibility (Supplemental Fig. S2C).
We next examined the presence of specific TE subfamilies at DMRs. Although many TEs in the genome overlapped DMRs, those of the ERV class were particularly enriched over background (Fig. 2A,B). At DMRs with age-dependent methylation loss, HERVH, MER52A, MER52D, and LTR2752 elements were overrepresented (Fig. 2A; Supplemental Table S6A). LTR2752 elements are annotated as MER52A elements in Repbase (Bao et al. 2015) so we did not include them as a separate group for further analysis. DMRs with methylation gain were enriched for LTR10C, HERVE, LTR16C, and MER11D elements (Fig. 2B; Supplemental Table S6B). We next examined TF binding motifs enriched in these elements and found that among TEs that lose methylation, HERVH-int elements showed enrichment for TEAD4 and TEAD1 binding sites (Fig. 2C,D), and MER52A and MER52D elements were enriched with EGR1 binding sites (Fig. 2C,D). The location of the DMR within HERVH-int elements was also consistent with the presence of a TEAD binding site (Supplemental Fig. S3B). Similarly, DMRs within MER52A and MER52D elements were located around EGR1 binding sites (Supplemental Fig. S3C,D). Enrichment of TEAD (family) and EGR1 binding sites is known to occur at active regulatory elements in basal cells/MEps (Pellacani et al. 2016). DMRs with methylation loss at non-TE regions also showed enrichment of TEAD and EGR1 binding sites (Supplemental Fig. S3A; Supplemental Table S7A). These results indicate that methylation changes at TEs may impact gene regulatory networks. Given that open chromatin at transcription factor binding sites can be used to infer TF binding (Tsompana and Buck 2014), we next assessed the ATAC-seq signal at these binding sites. We found increased ATAC-seq signal in old LEps at TEAD binding sites within HERVH-int elements (Fig. 2E,F). These data are in line with previous studies suggesting that aging LEps acquire a basal-like gene expression signature (Pelissier Vatter et al. 2018; Sayaman et al. 2021) and that LEps specifically dysregulate YAP, a transcriptional coactivator that binds DNA through TEADs (Garbe et al. 2012; Pelissier et al. 2014).
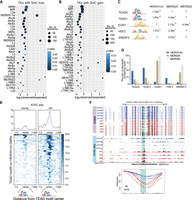
Age-dependent methylation changes occur at TE subfamilies that contain lineage-specific transcription factor binding sites. (A,B) TE subfamilies enriched at DMRs with methylation loss (A) and methylation gain (B). The size of the dot represents the number of elements with methylation loss or gain. All TEs with more than 10 copies overlapping DMRs are shown for completeness. (C) Transcription factor binding motifs enriched in HERVH-int, MER52A, and MER52D elements with methylation loss. The P-values for enrichment over scrambled sequences are shown. (D) Percentage of elements containing indicated transcription factor motifs across TEs with methylation loss. (E) ATAC-seq profile at TEAD motifs within HERVH-int elements showing methylation loss with age. (F) Genome Browser tracks show a representative example of a HERVH-int element losing methylation in old samples. The bottom tracks are from ATAC-seq data showing a gain of accessibility at the DMR. The bottom panel shows kernel-smoothed line plots of the highlighted region.
Among TEs that gain methylation, LTR10C elements showed enrichment of NF-Y (NFYA and NFYC), SMAD3, and SOX4 binding sites (Supplemental Fig. S3E). HERVE elements were enriched for YY1, TFAP2C, and SMAD3, whereas MER11D showed enrichment of SOX4 and SMAD3 binding sites (Supplemental Fig. S3E). SOX4 and GATA3 are luminal lineage-specific TFs (Asselin-Labat et al. 2007; Sayaman et al. 2021), and their binding sites are enriched within active regulatory elements in the luminal lineage (Pellacani et al. 2016). As shown above, LTR10C and MER11D elements have low methylation levels and signatures of regulatory function in young LEps (Fig. 1; Supplemental Table S4). Moreover, SOX4 expression decreases with age in LEps (Sayaman et al. 2021), suggesting that loss of TF binding at these luminal lineage-specific sites leads to epigenetic suppression by DNA methylation. Furthermore, we observed additional lineage-specific TF binding sites to be enriched at other TE subfamilies, such as MIRb and L2(a-c), that overlap DMRs (Supplemental Fig. S3G). Specifically, binding sites of ELF5, a luminal lineage-specific TF, are abundant among MIRb, MIR, and L2(a-c) elements (Supplemental Fig. S3G), as well as non-TE DMRs that gain methylation with age (Supplemental Fig. S3F; Supplemental Table S7B). Altogether, these results suggest that consistent methylation changes at TEs reflect changes in lineage-specific regulation in LEps with age.
Stochastic methylation loss at TEs and methylation gain at CpG islands with age
Aging is associated with increased cell-to-cell variation in gene expression (Martinez-Jimenez et al. 2017) and DNA methylation (Hernando-Herraez et al. 2019). Increased cell-to-cell variation in DNA methylation can lead to changes in methylation patterns that may not be detected as significant methylation changes in populations of cells (Jenkinson et al. 2018). Because we used a FACS-enriched LEp population, we quantified this cell-to-cell variation in methylation patterns using methylation entropy, a measure of methylation discordance between adjacent CpGs (Jenkinson et al. 2018). We calculated the methylation entropy levels for 150-bp windows across the genome in young and old LEps to examine whether there was increased methylation heterogeneity in old LEps. Methylation entropy increases either when unmethylated regions have a stochastic gain in methylation or when methylated regions undergo stochastic loss of methylation (Fig. 3A,B). Considering both these scenarios, we observed an overall increase in methylation entropy in old LEps (Supplemental Fig. S4A). We identified regions with greater methylation entropy in older individuals and found a net decrease in methylation levels from young to old LEps at these regions (Supplemental Fig. S4A–C), consistent with the fact that most of the genome is methylated in normal cells, with the exception of CpG islands (CGIs) (Edwards et al. 2017). Of these, regions that are unmethylated in young LEps and display increased methylation entropy with methylation gain in aging LEps were mainly at CGIs (69.2%; 1017 of 1469) (Fig. 3C; Supplemental Fig. S4D). Conversely, regions that are methylated in young LEps and display increased entropy with methylation loss were found primarily at TEs (70.3%, 76,575 of 108,877; compared with random regions [simulated 1000×] 57.03%, 62,094.7 of 108,877) (Fig. 3D; Supplemental Fig. S4E). These results indicate that increased DNA methylation heterogeneity with age leads to stochastic methylation gain at CGIs and methylation loss at TEs.
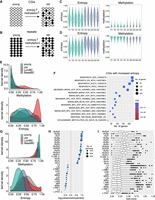
Increased methylation entropy with age in normal LEps predicts CpG island (CGI) methylation gain and Alu methylation loss in breast cancer. (A) Schematic depicting changes in entropy associated with stochastic methylation gain at CGIs. (B) Schematic depicting changes in entropy associated with stochastic methylation loss at fully methylated regions such as repeat elements. (C) Methylation and entropy levels at unmethylated CGIs with an increase in entropy. (D) Methylation and entropy levels at fully methylated regions with an increase in entropy. (E) Kernel density plots of methylation and entropy levels at unmethylated CGIs with an increase in entropy with age. Luminal breast cancer (LumBC) samples (red) show an increase in DNA methylation and entropy at these CGIs, whereas basal breast cancer (BasalBC) samples (brown) do not. (F) MSigDB gene sets enriched in CGI promoters that gain methylation entropy with age. BENPORATH gene sets represent PRC2 target genes identified in embryonic stem cells. (G) Kernel density plots of methylation and entropy levels at fully methylated regions with an increase in entropy with age. Both luminal breast cancer (red) and basal breast cancer samples (brown) show a decrease in DNA methylation with an increase in entropy at these regions. (H) TE subfamilies enriched at regions that show increased entropy with methylation loss. The size of the dot represents the number of elements. (I) Boxplot shows the CpG score of TEs that show stochastic methylation loss. TEs are grouped by subfamily.
To examine the potential consequences of age-associated stochastic methylation changes in breast cancer, we used a published WGBS data set of 30 primary tumors containing ER+ (luminal breast cancer) and ER− (basal) breast cancer samples (Brinkman et al. 2019). We checked the entropy and methylation levels of the set of CGIs that showed increased entropy with age in the breast tumor samples. Luminal breast tumors showed higher methylation and entropy than old LEps (Fig. 3E; Supplemental Fig. S4F), whereas the basal tumors did not show such an increase in methylation and entropy at the same set of CGIs (Fig. 3E). This result suggests that the increased methylation and entropy observed at CGIs is specific to the luminal subtypes of breast cancer. These CGIs were furthermore enriched for Polycomb repressive complex 2 (PRC2) target genes, which are known to be hypermethylated in luminal breast cancer (Fig. 3F; Holm et al. 2010). CGIs showing methylation gain in luminal breast tumors were more likely to be marked by H3K27me3 signifying PRC2 occupancy in ER− luminal cells compared with those that remained unmethylated (Supplemental Fig. S4G). In contrast, the CGIs that remain unmethylated in luminal breast cancer are predominantly marked by H3K4me3 in normal luminal cells (Supplemental Fig. S4G; ER− luminal H3K27me3 and H3K4me3 data were obtained from Pellacani et al. 2016). These results indicate that increased DNA methylation entropy with age leads to stochastic methylation gain especially at PRC2 target CGIs, which is further exacerbated in luminal breast cancers.
We also examined the methylation levels from the breast cancer samples for the set of TEs that have increased methylation entropy with age. These TEs displayed a general decrease in methylation in both basal and luminal breast tumors compared with old LEps, unlike CGIs, which gained methylation only in luminal tumors (Fig. 3G). Methylation loss was also more subtle than methylation gain, possibly owing to other protection mechanisms, such as KZFPs, that might be expressed during the process of tumorigenesis. We then examined whether any TE subfamilies are particularly affected by stochastic methylation loss and found that Alu elements such as AluS, AluY, and AluJ were most enriched among these regions (Fig. 3H; Supplemental Table S8). We mapped the regions of increased entropy at AluSz6 elements and found that these regions are randomly located throughout the length of the Alu elements (Supplemental Fig. S4H) in contrast to the specific locations of hypomethylated regions and DMRs in LTRs. These Alu elements also showed a higher CpG score than other TEs (Fig. 3I), indicating that CpG-dense Alu elements lose methylation stochastically with age and in breast cancer.
Evolutionarily recent TEs show variable methylation loss with age
Although methylation entropy measures the discordance between the methylation status of neighboring CpGs on DNA strands of the same individual (Xie et al. 2011), inter-individual variability measures differences in methylation levels at the same CpG/region across multiple individuals (Gunasekara et al. 2019). Given our observation of increased methylation entropy with age, we next examined whether aging would lead to increased inter-individual variability. Comparing single CpG methylation for all CpGs with at least 3 × coverage, we observed a higher methylation variability in older individuals (Supplemental Fig. S5A). However, differences at single CpG sites are prone to high false-positive rates (Bock 2012), and some amount of DNA methylation variability is expected in human populations owing to genetic variation. We therefore used the systemic interindividual variation (SIV) approach (see Supplemental Methods; Gunasekara et al. 2019) to identify systemic interindividual epigenetic variants, and found these regions to be enriched at TEs. Among the variably methylated TEs, the evolutionarily recent elements tended toward DNA methylation loss, whereas the older TEs (>100 million yr) tended to gain methylation in old LEps (Fig. 4A–D). Although evolutionarily recent Alu elements, such as AluY and AluS, are highly abundant in the genome, several AluS subfamilies, such as AluSc, AluSp, and AluSq, were enriched over background for variable methylation loss (Fig. 4E). In addition, primate-specific LINE elements (L1PA2-6) and HERVH elements were also enriched among sites of variable methylation loss (Fig. 4E).
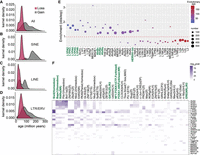
Evolutionarily recent TEs show variable methylation loss with age. (A–D) Evolutionary age of all TE (A), SINE (B), LINE (C), and LTR/ERV (D) elements that are variably methylated with methylation loss or gain. Evolutionary ages of only the significantly enriched TE subfamilies are plotted. (E) TE subfamilies enriched in regions with variable methylation loss. Dots show the observed/expected ratio for each TE subfamily. The expected number of overlaps in shuffled controls was used to calculate the ratio. The color of the dots represents evolutionary age, whereas size represents the number of elements. (F) TF binding motifs enriched within each TE subfamily with variable methylation loss. The color scale is –log10(P-value) of TF motif enrichment.
We next examined the TF motifs enriched at the TE subfamilies with variable methylation loss in old LEps. We found that Alu subfamilies contain motifs for Homeobox TFs such as NKX3-2 (previously known as BAPX1) and nuclear receptor TFs such as RARA (Fig. 4F), which are up-regulated in breast cancers as observed from analyzing expression levels across The Cancer Genome Atlas (TCGA) breast cancer cohort (TCGA-BRCA) RNA-seq data (Supplemental Fig. S5G,H). The younger L1PA subfamilies L1PA(2-4) have motifs for many TFs, including binding sites for SIX4, GATA, FOXA1, and estrogen response elements (EREs), which are bound by estrogen receptor 1 (ESR1) (Fig. 4F; Supplemental Fig. S5I–L). GATA3, FOXA1, and ESR1 are highly up-regulated in luminal breast cancers (Perou et al. 2000), as confirmed in the TCGA-BRCA RNA-seq data (Supplemental Fig. S5I–L). Therefore, the methylation loss at TEs harboring GATA, FOXA, and ESR1 binding sites could lead to binding by the respective TFs and promote aberrant TE activity in breast cancer.
TEs that lose methylation with age are activated in breast cancer
Given our observations that evolutionarily recent transposons lose methylation with age and contain motifs for TFs overexpressed in luminal breast cancer, we examined whether breast cancers show loss of epigenetic suppression at these same TEs. Loss of TE suppression can lead to aberrant TE-derived transcripts and alternative isoforms owing to splicing with neighboring genes in cancer cells (Jang et al. 2019). To investigate the potential of this happening in breast cancer, we examined published RNA-seq data from high-grade ductal carcinoma in situ (DCIS), a form of precancer in breast (Abba et al. 2015). We quantified gene and TE expression changes in DCIS compared with normal tissues; 554 age-hypomethylated TEs showed higher expression in DCIS. Of these, 468 (84%) were TEs that show variable hypomethylation with age, and the additional 86 TEs were within DMRs with methylation loss. As with the loci that show variable methylation loss in older samples, HERVH-int elements and L1PA2 elements were most common among the TE subfamilies that showed aberrant expression in DCIS. A representative example of a HERVH-int element that loses methylation in aging LEps and breast cancers is shown (Fig. 5A). Aberrant transcription from this HERVH-int element was observed in DCIS samples and not in normal breast tissue (Fig. 5A). We also found examples of exonization of TEs, such as a MIRb element within an exon of a noncoding RNA, DRAIC, that is expressed in DCIS (Supplemental Fig. S6A). Although aberrant transcription from hypomethylated TEs in DCIS was clear, we did not observe breast cancer subtype specificity in the expression of these TEs.
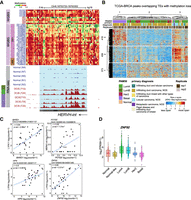
Transposable elements (TEs) with age-dependent methylation loss are activated in breast cancer. (A) A representative HERVH element with variable loss of methylation in old samples and breast cancer (BC) samples. The estrogen receptor (ER) expression status of the tumors is shown for the BC WGBS tracks. The bottom tracks are RNA-seq tracks from DCIS samples showing cryptic transcription of the HERVH element. All RNA-seq tracks are scaled to the same value, and the PAM50 subtype is indicated on the left. (B) Heatmap showing signals across 74 TCGA-BRCA samples for ATAC-seq peaks that overlap TEs hypomethylated with age. ATAC-seq peaks (rows) and TCGA BRCA samples (columns) are split into three clusters, each using k-means clustering. (C) Dot plot of RNA-seq counts at indicated TE and gene pairs that are correlated across DCIS samples. Each dot represents an individual sample. Spearman's correlation coefficients and P-values are indicated. (D) Expression of ZNF92 in normal tissues and five PAM50 subtypes of breast tumors from the TCGA BRCA collection (P-value luminal A [LumA] vs. basal and luminal B [LumB] vs. basal; P-value < 2.2 × 10−16, using a two-tailed Wilcoxon signed-rank test).
We next examined whether TEs showing methylation loss with age (DMRs), function as regulatory elements in breast cancers. To this end, we used ATAC-seq data from TCGA-BRCA. We identified 1197 TCGA-BRCA ATAC-seq peaks that overlap TEs with methylation loss in aging LEps (Fig. 5B). Although some TEs were accessible in basal subtype tumors (418 of 1197), a majority of them were accessible across luminal tumors (779 of 1197 peaks) (Fig. 5B). Breast tumors that were diagnosed as lobular carcinomas had particularly high chromatin accessibility across all TEs (Fig. 5B, cyan color in primary diagnosis), including those that lose methylation in aging LEps, such as HERVH-int elements. These results indicate that TEs hypomethylated with age may be primed to function as aberrant regulatory elements in breast cancers.
To determine whether methylation loss at TEs can lead to changes in the expression of genes that are thought to be important in breast cancers, we mapped the age-hypomethylated TEs to genes within 500 kb. This window was chosen based on data indicating that >75% of all 3D promoter interactions occur within 500 kb (Javierre et al. 2016). We calculated the correlation of TE expression with each gene mapped within this window in DCIS samples. After filtering out TEs that were intragenic and had reads on the same strand as the host gene, we mapped 256 TEs to 2176 genes. We identified 462 genes that were significantly correlated (adjusted P-value < 0.05) with the expression of a nearby TE (Supplemental Table S9). A randomly selected set of 256 TEs with no methylation change with age yielded 331 genes significantly correlated with the TE expression. Four representative examples of significantly correlated TE–gene pairs are shown (Fig. 5C). Genome Browser tracks show the RNA-seq signal at the L1PA4 element and the nearby ZNF92 gene (Spearman's correlation: 0.65, P-value: 2.21 × 10−5) (Supplemental Fig. S7A). ZNF92 is also highly expressed in luminal subtype breast tumors compared with basal tumors (Fig. 5D). These results suggest that TEs in deleterious locations of the genome are reawakened through variable loss of methylation, potentially contributing to the regulation of genes associated with breast cancers. Overall, our data suggest that the reawakening of TEs is directly related to increased breast cancer susceptibility with age.
Discussion
TEs, which are ubiquitous in the human genome, are potentially harmful to the host genome when not silenced. The primary function of DNA methylation in eukaryotic genomes is in the silencing of these elements. Reactivation of TEs has been shown in cancer cells, coincident with a global loss of DNA methylation (Burns 2017; Ishak and Carvalho 2020). Previous studies using PCR-based quantification strategies reported an overall decline in methylation of Alu and HERV-K elements with age in human blood cells (Bollati et al. 2009; Jintaridth and Mutirangura 2010). Increased expression of retrotransposons and active retrotransposition has been reported in aging mouse tissues (De Cecco et al. 2013; Benayoun et al. 2019), and increased LINE-1 expression has been reported in senescent human fibroblasts in vitro (De Cecco et al. 2019). However, these studies used bulk quantitative strategies such as RT-qPCR, which are not ideal owing to high sequence similarity between multiple active and inactive copies of repetitive elements. A WGBS study comparing DNA methylation levels of CD4+ T cells between a neonate and a centenarian reported that 12% of DMRs that lost methylation with age overlapped Alu elements (Heyn et al. 2012). However, the methylation changes at TEs and any downstream consequences were not systematically investigated in these individuals. Our approach allowed us to investigate the impact of aging on DNA methylation at specific TEs using nonsenescent mammary epithelial cells. Our results show that evolutionarily recent (e.g., primate and hominid-specific) transposons are more likely to be silenced by DNA methylation in young cells. These same elements show stochastic methylation loss with age. In agreement with previous reports, we showed that several recent Alu insertions (primate-specific) lose methylation stochastically with age. This stochastic loss of TE silencing is potentially an important component of understanding differential susceptibility to cancer. Our data also underscore the need for the use of human cells or organoids for future studies.
We further show that the methylation loss observed at TEs with age becomes exacerbated in frank luminal breast cancers. The derepressed TEs lead to aberrant transcription and potentially function as regulatory elements in breast cancer cells. We show that hypomethylated TEs regulate genes associated with luminal subtype breast cancers. TEs also contribute lineage-specific TF binding sites consistent with loss of lineage fidelity with age. Retrotransposon activation has been widely observed in nearly every human cancer, including breast cancer (Rodić et al. 2014; Rooney et al. 2015). TE activation has been observed in premalignant lesions as well (Ishak and Carvalho 2020), suggesting that early events in tumorigenesis may be driven by retrotransposon activity. We show here, for the first time, that loss of transposon silencing is evident in aging LEps that could predispose toward breast cancer. Future studies are needed to characterize the role of these TEs in the early events of mammary cancer initiation.
Previous studies analyzing aging-associated DNA methylation changes in human mammary LEps either have used array-based technologies (Miyano et al. 2017; Hofstatter et al. 2018; Castle et al. 2020; Sayaman et al. 2021) or were performed using composite tissues that represented a mixture of cells (Johnson et al. 2017; Song et al. 2017; Hofstatter et al. 2018; Castle et al. 2020). Array probes are mainly enriched at genic regions, and intergenic elements, including repeats and distal regulatory elements, are underrepresented (Bell et al. 2019; Gunasekara et al. 2023). Furthermore, different cell lineages have unique responses to aging (Miyano et al. 2017; Kimmel et al. 2019), making pure cell populations critical for interpretation. Our use of WGBS on sorted normal HMECs addresses both shortcomings.
Previous DNA methylome studies in human cells suggested a global loss of methylation with age (Heyn et al. 2012; Vandiver et al. 2015; Jenkinson et al. 2017). Those studies were performed using CD4+ T cells from a neonate and a centenarian (Heyn et al. 2012), sorted CD4+ T cells (Jenkinson et al. 2017), and skin keratinocytes (Vandiver et al. 2015) from young (<25 yr) and old (>70 yr) individuals. The global methylation loss with age is accompanied by a global increase in entropy or methylation discordance (Jenkinson et al. 2017). We did not observe a global methylation loss or a global increase in entropy in mammary LEps with age. This could be because of the higher fidelity of DNA methylation maintenance in luminal cells or because of a smaller age gap between young (<30 yr) and old (>55 yr) individuals in our study. DNA methylome studies in aging mouse livers also showed no global methylation loss, but specific changes at regulatory elements were detected (Cole et al. 2017). Whereas global methylation loss was not detected, we did detect methylation changes at regulatory elements and detect variable methylation changes at evolutionarily recent transposons.
Our results provide a comprehensive analysis of the methylation changes at TEs in mammary LEps with age. As LEps are the putative cell of origin for luminal breast cancers, these findings are relevant in understanding the role of TEs in increasing breast cancer risk. However, because of the limited availability and culturing potential of normal LEps, we were unable to perform mechanistic experiments to show the regulatory role of TEs in aging cells and tumorigenesis. Future studies are needed to fully understand the DNA methylation of candidate TEs and their regulatory activity during the process of tumorigenesis. Furthermore, because of a higher cost associated with WGBS experiments, we were able to profile a limited number of individual samples. In the future, a more targeted approach will be needed that will use candidate regions identified in our study to detect DNA methylation changes in a larger number of samples in a more cost-effective manner. Additionally, it has been previously shown that pregnancy alters the methylome of mammary epithelial cells (Choudhury et al. 2013; Dos Santos et al. 2015; Huh et al. 2015). Pregnancy-related DNA methylation changes have been observed in all epithelial cell types including LEps in mouse breast tissue (Dos Santos et al. 2015). That study found that pregnancy induced persistent methylation loss at genes associated with mammary gland development, lactation, and involution. Moreover, those hypomethylated sites were found to be enriched for STAT5A/B binding sites. We did not find any significant enrichment of STAT binding sites within DMRs with methylation loss (Supplemental Fig. S2C; Supplemental Table S7), nor did we find any DMRs at lactation or mammary gland development genes (Supplemental Table S5). For these reasons, we believe our observations regarding DNA methylation changes with aging are unlikely to be confounded by parity-related changes. However, because of the lack of parity information for our samples, we are not able to conclusively delineate pregnancy-related changes from age-related changes.
Global loss of methylation and focal increase in methylation, termed “epigenetic drift,” has been reported to occur with age and in cancer (Issa 2014). Focal increases in methylation occur mainly at CGIs. We observed increased methylation entropy and methylation gain at CGIs with age, which is further enhanced during the transition to luminal breast cancers. These CGIs were predominantly PRC2 targets known to gain methylation in other cancer types (Easwaran et al. 2012). Loss/repositioning of PRC2 has been proposed to lead to increased DNA methylation at PRC2 targets. Presumably, CGIs with increased methylation entropy are in the process of becoming hypermethylated in luminal breast cancers. Hypermethylation of PRC2 targets seems specific to the luminal subtype of breast cancer (Holm et al. 2010), although the reasons for this are unclear. Altogether, our results provide further evidence in support of the observation that epigenetic changes with age could determine susceptibility toward breast cancer.
Methods
Isolation of mammary LEps
LEps were isolated from fourth passage (p4) cultures of prestasis finite-lifespan HMECs from normal reduction mammoplasty (RM) tissues (Garbe et al. 2012). Primary nonimmortalized HMECs were generated and maintained, as described previously (LaBarge et al. 2013). Cells were grown in M87A medium with cholera toxin and oxytocin at 0.5 ng/mL and 0.1 nM, respectively. These primary cultures retain lineage-specific and age-dependent gene expression profiles that are consistent with uncultured organoid tissues indicating minimal culture-induced artifacts (Miyano et al. 2021; Sayaman et al. 2021). Moreover, p4 HMEC cultures contain ER− luminal cells and MEps; ER+ luminal cells are lost in primary cultures as they are postmitotic. As ER− LEps represent the bulk of the mammary epithelial cell population (Petersen et al. 1987) and as our previous studies showed significant age-dependent changes in ER− LEps compared with MEps (Sayaman et al. 2021), we used ER− LEps for this study.
p4 HMECs were stained with anti-human CD133-PE (Biolegend, clone 7) and anti-human CD271 (Biolegend, clone ME20.4) by following standard flow cytometry protocol. The LEp population was enriched using an Aria III (Becton Dickinson) flow sorter by gating on forward and side scatter to eliminate abnormal or senescent cells and by selecting the CD133+/CD271− fraction. Because LEps show loss of lineage fidelity with age, we use CD133+ and CD271− as LEp markers. We have previously shown that these markers are not impacted by age or senescence (Garbe et al. 2012; Pelissier Vatter et al. 2018). The different HMEC strains used for WGBS and ATAC-seq are described in Supplemental Table S1. The type of analysis was chosen based on the strain that had enough cells after flow sorting on the day of the experiment to avoid potential batch effects.
WGBS
We examined the DNA methylome of primary LEps from younger (<30 yr) premenopausal and older (>55 yr) postmenopausal women. Genomic DNA from FACS-enriched LEps was extracted using the quick-DNA microprep kit (Zymo Research). WGBS-seq was performed at Admera Health Biopharma Services. Genomic DNA was bisulfite-converted using the Zymo EZ DNA methylation kit (Zymo Research) per the manufacturer's protocol.
Library preparation was then performed using the Accel-NGS methyl-seq DNA library kit (Swift Biosciences) per the manufacturer's recommendations. Equimolar pooling of libraries was performed based on QC values. Samples were sequenced on an Illumina NovaSeq S4 (Illumina) with a read length configuration of 150-bp paired-ends. We obtained about 38× coverage per genome to an average of 414 million paired-end reads (Supplemental Table S2).
Sequencing reads were hard-trimmed 15 bp from both ends using Trim Galore! (version 0.4.5; https://github.com/FelixKrueger/TrimGalore) to remove unwanted methylation bias arising from the library preparation. Adapter contamination was also removed using Trim Galore! using the ‐‐illumina option. Trimmed reads were aligned to the human reference genome (hg19) using Bismark (version 0.19.1) (Krueger and Andrews 2011) with the ‐‐bowtie1 option (Langmead et al. 2009). Bismark (deduplicate_bismark) was used to remove PCR duplicates. Methpipe (version 3.4.3) (Song et al. 2013) was used to determine the methylation status of individual CpG sites. The hg19 reference genome contains about 56.4 million CpG loci (hg19) or 28.2 million CpG dyads. Methylation levels were calculated for individual CpG dyads and are henceforth referred to as CpG sites. A comparison of TE content in hg19 and hg38 is included in the Supplemental Material.
LEp DNA methylation profiles were compared with published WGBS data of ER+ luminal, ER− luminal, and basal/MEp cells isolated from RM breast tissue (noncultured cells) to rule out any culture-induced artifacts (Pellacani et al. 2016). Our LEp DNA methylation profiles correlated very well with ER− luminal cells (Pearson correlation coefficient r ≥ 0.78 for young LEps and r ≥ 0.76 for old LEps). At promoters, the same trend was observed with correlation coefficients being higher (r ≥ 0.9 for LEps compared with ER− luminal) (Supplemental Fig. S8).
ATAC-seq and analysis
Fifty thousand live FACS sorted young (n = 4) and old (n = 4) LEps were used for Tn5 transposition in duplicates using the Omni-ATAC method (Corces et al. 2017). Libraries were sequenced to a depth of 40 million paired reads each on an Illumina HiSeq 2500 system. Raw sequencing reads were trimmed using Trim Galore! (version 0.4.5; https://github.com/FelixKrueger/TrimGalore) to remove adapters. Trimmed reads were aligned to the human reference genome (hg19) using Bowtie (Langmead et al. 2009) with the ‐‐best -k 1 -X 2000 ‐‐mm ‐‐chunkmbs 1024 parameters. Unmapped reads, mates, and low-quality reads and secondary alignments were removed using SAMtools (Danecek et al. 2021). PCR duplicates were identified using Picard and removed using SAMtools. The reads were shifted by four bases on the sense strand and five bases on the antisense strand to account for Tn5 bias. Reads from technical replicates were pooled to generate pseudoreplicates. Peaks were called using MACS2 version 2.1.1.20160309 (Zhang et al. 2008) using -p 0.01 ‐‐nomodel ‐‐shift -37 ‐‐extsize 73 ‐‐SPMR ‐‐keep-dup all ‐‐call-summits as parameters. Peak calls from technical replicates and pseudoreplicates were compared using IDR to get the most conservative set of peaks for each sample.
Enrichment of TE subfamilies at open chromatin sites, DMRs, and variably methylated regions
The extent of overlap of open chromatin regions (ATAC-seq peaks), DMRs, and variable regions at repeat subfamilies (RepeatMasker annotation) (Smit et al. 2023) was calculated using BEDTools intersect (Quinlan and Hall 2010). Randomly shuffled control regions of the same length were generated 1000 times, and the expected number of overlaps was calculated using BEDTools intersect. Enrichment values were calculated by dividing the number of DMR overlaps (observed) by the average number of random overlaps (expected). P-values were calculated by dividing the number of extreme values (at either tail of the distribution) from random overlaps by 1000. Significantly enriched repeat subfamilies had P < 0.05, enrichment greater than 1.5-fold, and more than 10 copies overlapping DMRs or variable regions.
Motif analysis
Motif enrichment analyses at specific TE subfamilies were performed using AME available in the MEME suite using the HOCOMOCO v11 human TF motif database (Bailey et al. 2015). The enrichment of motifs in given sequences was calculated over shuffled control sequences. Motif scanning was performed using FIMO (Grant et al. 2011), available in the MEME suite (Bailey et al. 2015) with default parameters. Only binding sites with TFs that were expressed (TPM > 1) in LEps or MEps were used for further analyses. For ATAC-seq peaks and DMRs, only the sequence overlapping a TE was used for motif analysis.
DMR identification
DMRfinder (Gaspar and Hart 2017) was used to identify DMRs. Briefly, CpG sites with at least 4 × coverage in at least six out of 10 samples were determined. Genomic regions with at least four CpG sites within a distance of 100 bp were identified. The maximum length of regions was set to 500 bp. Regions with coverage of fewer than 20 reads were discarded. The final set of regions was then used to calculate DMRs with >20% change (P-value < 0.05) between pooled young and old samples. Motif enrichment analyses at DMRs were performed using findMotifsGenome.pl in HOMER (Heinz et al. 2010). For each set of genomic regions, background sequences with matched GC content were selected. P-values for motif enrichment were calculated using the cumulative binomial distributions.
RNA-seq analysis
RNA-seq data from normal and DCIS samples were obtained from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) (accession number GSE69240) (Abba et al. 2015). We used this data set as it was paired-end stranded data, which performs relatively better with TE quantification tools than unstranded and single-end data. FASTQ reads were quality- and adapter-trimmed using Trim Galore! with the ‐‐paired option. Quality-trimmed reads were aligned to the hg19 genome using the squire Map function of the SQuIRE pipeline (Yang et al. 2019). Subsequently, squire count and call functions were used to measure gene and TE expression changes in DCIS compared with normal tissues. TEs with increased expression in DCIS were first identified (log2FC > 0.5, Padj < 0.05). The SQuIRE call output contains information about the expressed strand. TEs with reads in the opposite strand as the TE itself were filtered out as those reads are likely from the host gene for intragenic TEs. Genes that were within ±500 kb of variably methylated TEs were then identified, and gene–TE pair lists were created and annotated based on whether the TE was intragenic (inside the gene) or intergenic (outside the gene). Intragenic TEs were further annotated based on whether the reads from the TE were on the same or opposite strand as that of the host gene. TEs that showed reads on the same strand as its host gene were discarded from the TE–gene pair list. Spearman's correlation values were calculated for log-transformed counts of genes and TEs from each gene–TE pair. Significantly correlated gene-TE pairs have adjusted P-values < 0.05 (Benjamini–Hochberg method).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE153696 (WGBS and ATAC-seq).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank other members of the Schones laboratory for helpful discussions and comments and the continued involvement of patient advocates Susan Samson and Sandy Preto. The results shown and referenced here are based in part upon data generated by the TCGA Research Network (https://www.cancer.gov/tcga) and the ENCODE Consortium (https://www.encodeproject.org/). This work was supported by the National Institutes of Health, grants R01DK112041, R01CA220693 (D.E.S.), and K01DK104993 (A.L.). P.S. was supported by a Mentored Research award from the Neuroendocrine Tumor Research Foundation. The research reported in this publication included work performed in the Integrative Genomics and Analytical Cytometry Cores supported by the National Cancer Institute (NCI) of the National Institutes of Health under award number P30CA033572, as well as a Department of Defense/Army Breast Cancer Era of Hope Scholar Award BC141351 and Expansion Award BC181737, the Conrad N. Hilton Foundation, the City of Hope Center for Cancer and Aging (M.A.L.); the NCI Cancer Metabolism Training Program Postdoctoral Fellowship T32CA221709 (R.W.S.), and the City of Hope Program in Molecular and Cellular Biology pilot award (D.E.S. and M.A.L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding bodies had no role in the design of the study, data collection, analysis, interpretation, the decision to publish data, or the writing of the manuscript.
Author contributions: P.S., M.A.L., and D.E.S. designed the study. P.S. and M.M. performed experiments. P.S., R.W.S., M.B., and A.L. analyzed and interpreted the data. P.S., M.A.L., and D.E.S. prepared the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277511.122.
-
Freely available online through the Genome Research Open Access option.
- Received November 15, 2022.
- Accepted June 23, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








