
Improving quartet graph construction for scalable and accurate species tree estimation from gene trees
Abstract
Summary methods are widely used to estimate species trees from genome-scale data. However, they can fail to produce accurate
species trees when the input gene trees are highly discordant because of estimation error and biological processes, such as
incomplete lineage sorting. Here, we introduce TREE-QMC, a new summary method that offers accuracy and scalability under these
challenging scenarios. TREE-QMC builds upon weighted Quartet Max Cut, which takes weighted quartets as input and then constructs
a species tree in a divide-and-conquer fashion, at each step forming a graph and seeking its max cut. The wQMC method has
been successfully leveraged in the context of species tree estimation by weighting quartets by their frequencies in the gene
trees; we improve upon this approach in two ways. First, we address accuracy by normalizing the quartet weights to account
for “artificial taxa” introduced during the divide phase so subproblem solutions can be combined during the conquer phase.
Second, we address scalability by introducing an algorithm to construct the graph directly from the gene trees; this gives
TREE-QMC a time complexity of 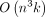 , where n is the number of species and k is the number of gene trees, assuming the subproblem decomposition is perfectly balanced. These contributions enable TREE-QMC
to be highly competitive in terms of species tree accuracy and empirical runtime with the leading quartet-based methods, even
outperforming them on some model conditions explored in our simulation study. We also present the application of these methods
to an avian phylogenomics data set.
, where n is the number of species and k is the number of gene trees, assuming the subproblem decomposition is perfectly balanced. These contributions enable TREE-QMC
to be highly competitive in terms of species tree accuracy and empirical runtime with the leading quartet-based methods, even
outperforming them on some model conditions explored in our simulation study. We also present the application of these methods
to an avian phylogenomics data set.
Estimating the evolutionary history for a collection of species is a fundamental problem in evolutionary biology. Increasingly, species trees are estimated from multilocus data sets, with molecular sequences partitioned into (recombination-free) regions of the genome (referred to as loci or genes). A popular approach to species tree estimation involves concatenating the alignments for individual loci together and then estimating a phylogeny under standard models of molecular sequence evolution, like the generalized time reversible (GTR) model (Tavaré 1986).
Such models assume the genes have a shared evolutionary history; however, this is not necessarily the case. The evolutionary histories of individual genes (referred to as gene trees) can differ from each other because of biological processes (Maddison 1997). Incomplete lineage sorting (ILS), one of the most well-studied sources of gene tree discordance, is an outcome of genes evolving within populations of individuals, as modeled by the multispecies coalescent (MSC) (Pamilo and Nei 1988; Rosenberg 2002; Degnan and Salter 2005). Concatenation-based approaches to species tree estimation can be statistically inconsistent under the MSC (Roch and Steel 2015). Moreover, simulation studies have shown concatenation can perform poorly when the amount of ILS is high (e.g., Kubatko and Degnan 2007). ILS is expected to impact many major groups, including birds (Jarvis et al. 2014), placental mammals (McCormack et al. 2012), and land plants (Wickett et al. 2014). Thus, species tree estimation methods that account for ILS, either explicitly or implicitly, are of interest.
An alternative to concatenation involves estimating gene trees (typically one per locus) and then applying a summary method. The most popular summary method to date, ASTRAL (Mirarab et al. 2014b), is a heuristic for the NP-hard maximum quartet support species tree (MQSST) problem (Lafond and Scornavacca 2019), which can be framed as weighting quartets (four-leaf trees) by their frequencies in the input gene trees and then seeking a species tree T that maximizes the total weight of the quartets displayed by T. The optimal solution to MQSST is a statistically consistent estimator of the (unrooted) species tree under the MSC model (Mirarab et al. 2014b), which is why heuristics for this problem are widely used in the context of multilocus species tree estimation. Proofs of consistency typically assume the input gene trees are error-free (Roch et al. 2018); however, this is unlikely in practice. An analysis of gene trees published for several recent systematic studies found low bootstrap support values on average (Table 1 in Molloy and Warnow 2018), suggesting that gene tree estimation error (GTEE) may be pervasive across modern phylogenomics data sets. GTEE can negatively impact the accuracy of summary methods, as shown by simulation (e.g., Xi et al. 2015) and systematic studies (e.g., Meiklejohn et al. 2016). Overall, GTEE and ILS present significant challenges to species tree estimation.
A third challenge is scalability. ASTRAL executes an exact (dynamic programming) algorithm for MQSST within a constrained version of the solution space constructed from the input gene trees. There have been many improvements to ASTRAL, with the latest version ASTRAL-III (Zhang et al. 2018) having a time complexity of O((nk)1.726x), where n is the number of species (also called taxa), k is the number of gene trees, and x = O(nk) is the size of the constrained solution space. Because x depends on the amount of gene tree heterogeneity, a recent method FASTRAL (Dibaeinia et al. 2021) runs ASTRAL-III in an aggressively constrained solution space to speed up species tree estimation.
The other popular quartet methods, wQMC (Avni et al. 2014) and wQFM (Mahbub et al. 2021), take weighted quartets as input and then execute a divide-and-conquer approach to phylogeny reconstruction. A recent study found wQFM to be more accurate than ASTRAL-III on challenging model conditions characterized by high ILS and high GTEE (Mahbub et al. 2021). In these analyses, wQFM was given Θ(n4) quartets as input, with each quartet weighted by the number of gene trees that displayed it. The related input processing step limits the scalability of this approach. Here, we enable improved accuracy and scalability by introducing TREE-QMC.
Results
Overview of TREE-QMC method
TREE-QMC builds upon the first widely used quartet method, wQMC, which reconstructs the species tree in a divide-and-conquer fashion. At each step in the divide phase, an internal branch in the output species tree is identified; this branch splits the taxa into two disjoint subsets (Fig. 1). The algorithm continues by recursion on the subproblems implied by the two subsets of taxa. “Artificial taxa” are introduced to represent the species on the opposite of the branch so that solutions to subproblems can be combined during the conquer phase. The recursion terminates when the subproblem has three or fewer taxa, as there is only one possible tree that can be returned. At each step in the conquer phase, trees for complementary subproblems are connected at the related artificial taxa, until there is a single tree on the original set of species (Supplemental Fig. S1).
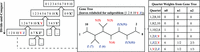
At each step in the divide phase, taxa are split into two disjoint subsets and then artificial taxa are introduced to represent the species on the other side of the split. To compute the quartet weights for a given subproblem, the leaves of each gene tree are relabeled by the artificial taxa. Without normalization (column n0), quartet 1, 2|Y, Z gets zero votes and the alternative quartets get six votes each (note: quartet 1, Y|2, Z gets six votes by taking either species 5, 3, or 4 for label Y and either species 0 or 9 for label Z). With normalization, each gene tree gets one vote for each subset of four labels, although this vote can be split across the three possible quartets. In the uniform normalization scheme (column n1), we simply divide column n0 by the total number of votes cast in the unnormalized case. In the nonuniform normalization scheme (column n2), we leverage that structure implied by the divide phase of the algorithm; the idea is that species should have lesser importance each time they are relabeled by artificial taxa.
Central to wQMC's approach is a graph built from weighted quartets. This graph is constructed in such a way that its max cut should correspond to a branch in the output species tree (Snir and Rao 2010, 2012; Avni et al. 2014). Our observation is that quartets on artificial taxa can have higher weights than quartets on only nonartificial taxa (called singletons) when looking at a single gene tree (Fig. 1). As we will show, normalizing the quartet weights so that each gene tree gets one vote for every subset of four species improves accuracy. The best performing normalization scheme (n2) weights quartets based on the subproblem decomposition; essentially, quartets are upweighted if their taxa are more closely related to the current subproblem (note: n1 denotes uniform normalization and n0 denotes no normalization). Moreover, we provide an algorithm to build the (normalized) quartet graph directly from the input gene trees, enabling TREE-QMC to have a time complexity of O(n3k) if the subproblem decomposition is perfectly balanced (Theorem 3 in the Supplemental Materials). This analysis is for a highly idealized setting and ignores large constant factors (Theorem 2 in the Supplemental Materials).
Beyond time complexity, methods can differ from each other in terms of data locality, code optimizations, and other theoretical guarantees (e.g., ASTRAL is guaranteed to find an optimal solution within its constrained solution space, whereas TREE-QMC has no such guarantee). Thus, in the remainder of this paper, we focus on evaluating the empirical performance of TREE-QMC (and its different normalization schemes) against the leading quartet-based summary methods on simulated and biological data.
Experimental evaluation
We now give an overview of our simulation study; details are provided in the Supplemental Materials.
Methods benchmarking
TREE-QMC is compared against four leading quartet methods: wQMC v1.3, wQFM v3.0, ASTRAL v5.5.7 (denoted ASTRAL-III or ASTRAL3), and FASTRAL. Two of these methods, wQMC and wQFM, take weighted quartets instead of gene trees as input (the input processing step is performed using the script distributed with wQFM). All methods are run in default mode. The current version of TREE-QMC requires binary gene trees as input so polytomies in the estimated gene trees are refined arbitrarily before running TREE-QMC (the same refinements are used in all runs of TREE-QMC to ensure a fair comparison across the normalization schemes).
Evaluation criteria
All methods are compared in terms of species tree error, quartet score, and runtime. Species tree error is the percent Robinson-Foulds (RF) error (i.e., normalized RF distance between the true and estimated species trees multiplied by 100). Because the true and estimated species trees are both binary, the RF error rate is equivalent to false negative error rate (i.e., the fraction of the internal branches in the true species tree that are missing from the estimated species tree). Two-sided Wilcoxon signed-rank tests are used to evaluate differences between TREE-QMC-n2 versus FASTRAL as well as TREE-QMC-n2 versus ASTRAL3 (TREE-QMC-n2 is also compared against wQFM when possible).
We report the difference in the quartet scores between the estimated and true species trees (scores are computed using the same set of gene trees). The quartet score is the number of quartets in the input gene trees that are displayed by the output species tree (divided by the total number of quartets in the gene trees). This quantity is simply the (normalized) MQSST objective function, so higher quartet scores imply a better solution to MQSST.
The runtime is the wall clock time, which for wQFM and wQMC includes the time to weight quartets based on the input gene trees (the fraction of time spent on input processing phase is reported in the Supplemental Materials). All methods are run on the same data set on the same compute node on our cluster; the maximum wall clock time is 18 h.
Simulated data sets
Our benchmarking study uses data simulated in prior studies, specifically the ASTRAL-II simulated data sets (Mirarab and Warnow 2015) as well as the avian and mammalian simulated data sets (Mirarab et al. 2014a). These data are generated by (1) taking a model species tree, (2) simulating gene trees within the species tree under the MSC, (3) simulating sequences down each gene tree under the GTR model, and (4) estimating a tree from set of gene sequences. Either the true gene trees from step 2 or the estimated gene trees from step 4 can be given as input to methods. This process is repeated for various parameter settings.
The ASTRAL-II data sets are generated from model species trees simulated under the Yule model given three parameters: species tree height, speciation rate, and number of taxa. The speciation rate is set so that speciation events are clustered near the root (deep) or near the tips (shallow) of the species tree. There are 50 replicates for each model condition (note that a new model species tree is simulated for each replicate data set). The avian and mammalian simulated data sets are generated from published species trees estimated for 48 birds (Jarvis et al. 2014) and 37 mammals (Song et al. 2012), respectively. The species tree branches are scaled to vary the amount of ILS, and the sequence length is changed to vary the amount of GTEE. There are 20 replicates for each model condition.
The data properties (ILS and GTEE levels) are summarized in Supplemental Tables S1 and S2. The ILS level is the percent RF error (between the true species tree and the true gene tree) averaged across all gene trees, and GTEE level is the percent RF error (between the true and estimated gene trees) averaged across all gene trees. Overall, these data sets cover a range of model conditions. The results are presented in four experiments looking at the impact of varying the number of taxa, the species tree scale/height (proxy for ILS), the sequence length (proxy for GTEE), and the number of genes.
Experimental results
Number of taxa
Figure 2, A and B, shows the impact of varying the number of taxa. The pipelines that need weighted quartets to be given as input (wQFM and wQMC) run on the order of seconds for 10 taxa, minutes for 50 taxa, and hours for 100 taxa. The runtime of these pipelines is dominated by the time to weight Θ(n4) quartets by their frequency in the input gene trees (Supplemental Table S3). This input processing step does not complete on our compute nodes within our maximum wall clock time of 18 h for most data sets with 200 taxa. Therefore, we could not run wQMC and wQFM on data sets with 200, 500, or 1000 taxa. In contrast, TREE-QMC implements a similar approach to wQMC but bypasses the input processing step, scaling to 1000 taxa and 1000 genes. For these data sets, FASTRAL, TREE-QMC-n2, and ASTRAL-III complete on average in 32 min, 64 min, and 5.3 h, respectively (note: ASTRAL-III fails to complete on 3/50 replicates within our maximum wall clock time of 18 h). Thus, TREE-QMC-n2 is much faster than ASTRAL-III and is not much slower than FASTRAL. TREE-QMC-n2 is significantly more accurate than either FASTRAL or ASTRAL-III when the number of taxa is 200 or greater. For these same conditions, quartet weight normalization, and especially the nonuniform (n2) scheme, improves TREE-QMC's accuracy. Results for methods given true gene trees as input or only 250 (out of 1000) gene trees as input are shown in Supplemental Figs. S7–S9).
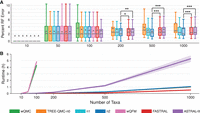
Impact of number of taxa. (A) Percent species tree error across replicates (bars represent medians; triangles represent means; outliers are not shown). The symbols *, **, and *** indicate significance at P < 0.05, 0.005, and 0.0005, respectively (all but one test survives Bonferroni multiple comparison correction; see Supplemental Table S4 for details). (B) Mean runtime across replicates (shaded region indicates standard error). All data sets have species tree height 1×, shallow speciation, and 1000 estimated gene trees. The ILS level is 17%–35%, and GTEE level is 19%–30%. Note: The input processing for wQMC and wQFM does not run within our maximum wall clock time of 18 h for data sets with 200 or more taxa.
Incomplete lineage sorting (ILS)
ASTRAL-II data (200 taxa, 1000 estimated gene trees)
Figure 3, A and B, shows the impact of varying the species tree height and thus the amount of ILS for the ASTRAL-II data sets. TREE-QMC-n2, FASTRAL, and ASTRAL-III produce highly accurate species trees, with median species tree error at or below 6% for all model conditions (note: the input processing for wQMC and wQFM does not run within our maximum wall clock time of 18 h for these 200-taxon data sets). For some conditions, TREE-QMC-n2 is significantly more accurate than FASTRAL or ASTRAL-III; otherwise, there are no significant differences between these pairs of methods. Quartet weight normalization improves the accuracy of TREE-QMC; this effect is most pronounced when the amount of ILS was very high (species tree height: 0.5×). On these same conditions, ASTRAL-III is much slower than the other methods, taking 73 min on average for the highest amount of ILS (species tree height: 0.5×) compared to 5 min on average for the lowest amount of ILS (species tree height: 5×). In contrast, both TREE-QMC-n2 and FASTRAL are quite fast, taking on average <3 min for model conditions with 200 or fewer taxa. Results for methods given true gene trees as input or only 250 (out of 1000) gene trees as input are shown in Supplemental Figs. S10–S12.
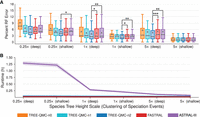
Impact of the amount of ILS. (A) Percent species tree error across replicates (bars represent medians; triangles represent means; outliers are not shown). The symbols *, **, and *** indicate significance at P < 0.05, 0.005, and 0.0005, respectively (three tests survive multiple comparison corrections; see Supplemental Table S5 for details). (B) Mean runtime across replicates (shaded region indicates standard error). All data sets have 200 taxa and 1000 estimated gene trees. One model condition with species tree height 1× and shallow speciation is repeated from Fig. 2. For species tree heights 0.5×, 1×, and 5×, the ILS level is 68%–69%, 34%, and 9%–21%, respectively, and the GTEE level is 44%, 27%–34%, and 21%–28%, respectively.
Avian simulated data (48 taxa, 1000 estimated gene trees)
Figure 4, A–C, shows the impact of varying the species tree scale and thus the amount of ILS on the avian simulated data sets. wQMC is the least accurate method and is even less accurate than TREE-QMC-n0 (no normalization). Normalization improves the performance of TREE-QMC for these data, enabling TREE-QMC-n2 to be among the most accurate methods when the amount of ILS is high (species tree scales: 0.5× and 1×). Testing for differences between TREE-QMC-n2 versus the other three leading methods (wQFM, FASTRAL, and ASTRAL-III) reveals that either TREE-QMC-n2 is significantly better or else there are no significant differences between these pairs of methods. All methods finish quickly: wQMC and wQFM complete in <13 min on average, ASTRAL-III completes in <4 min on average, and the other methods finish in <1 min on average.
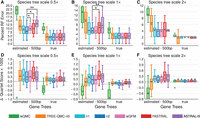
Impact of ILS and GTEE. (A), (B), and (C) Percent species tree error for the avian data set with 1000 estimated or true gene trees and species tree scales 0.5×, 1×, and 2×, respectively. Two-sided Wilcoxon signed-ranked tests were used to evaluate differences between TREE-QMC-n2 versus wQFM, FASTRAL, and ASTRAL3 (nine tests per subfigure). The symbols *, **, and *** indicate significance at P < 0.05, 0.005, and 0.0005, respectively (for 0.5× species tree scale with estimated gene trees, the difference between TREE-QMC-n2 and ASTRAL-II survives Bonferroni multiple comparison correction; see Supplemental Table S6 for details). (D), (E), and (F) show the difference in quartet score between the estimated and true species tree times 1000 for species tree scales 0.5×, 1×, and 2×, respectively (positive values indicate the estimated tree is a better solution to MQSST than the true tree). For species tree heights 0.5×, 1×, and 2×, the ILS level is 60%, 47%, and 35%, respectively, and the GTEE level is 60%, 60%, and 62%, respectively. Results for wQMC are cut off because otherwise the trends cannot be observed (see Supplemental Fig. S2 for full y-axes).
Figure 4, D–F, shows the difference in quartet score between estimated and true species trees. We find that most methods typically recover species trees with higher quartet scores than the true species tree, indicating that the true species tree is not the optimal solution to MQSST. Moreover, the relative performance of methods for quartet score is different than the relative performance of methods for species tree error for many model conditions. These two trends are especially pronounced when gene trees are estimated (mean error: 60%–62%).
Mammalian simulated data (37 taxa, 200 estimated gene trees)
All methods have similar performance for the mammalian data, although these data sets represent easier model conditions in terms of ILS and GTEE levels (Supplemental Fig. S3; Supplemental Table S7).
Gene tree estimation error (GTEE)
Avian simulated data (48 taxa, 1000 gene trees)
Figure 4, A–C, also shows the impact of GTEE for each species tree scale (ILS level). For each ILS level, methods are given true gene trees or estimated gene trees (mean error: 60%–62%). The trends for estimated gene trees are discussed above. For true gene trees, there are no significant differences between TREE-QMC-n2 versus the other leading methods (wQFM, FASTRAL, and ASTRAL-III), and all versions of TREE-QMC perform similarly so the utility of normalization is diminished. Moreover, these methods find species trees with similar quartet scores to the true species tree, unlike the case of estimated gene trees. Lastly, the performance of wQMC is in line with the other methods when there is very little gene tree heterogeneity because of ILS or GTEE (Fig. 4C).
Mammalian simulated data (37 taxa, 200 gene trees)
Similar trends between methods are observed for mammalian simulated data sets when varying the sequence lengths (Supplemental Fig. S4; Supplemental Table S8). TREE-QMC is significantly more accurate than FASTRAL and ASTRAL-III for the shortest sequence length (250 bp; GTEE level 43%); there are no differences in accuracy between these pairs of methods otherwise.
Number of genes
Similar trends between methods are observed when varying the number of genes (e.g., Supplemental Fig. S5; Supplemental Tables S9, S10).
Reanalysis of avian phylogenomics data set
We also reanalyze the avian data set from Jarvis et al. (2014) with 3679 ultraconserved elements (UCEs). This data set includes the best maximum likelihood tree and the set of 100 bootstrapped trees for each UCE. Although the true species tree is unknown, we discuss the presence and absence of strongly corroborated clades, such as Passerea and six of the magnificent seven clades excluding clade IV (Braun and Kimball 2021). We also compare methods to the published concatenation tree estimated by running RAxML (Stamatakis 2014) on UCEs only (Jarvis et al. 2014); thus the comparison between concatenation and the quartet-based summary methods is on the same data set. Branch support is computed for the estimated species trees using ASTRAL-III's local posterior probability (Sayyari and Mirarab 2016) as well as using multilocus bootstrapping (MLBS) (Seo 2008). We repeat this analysis (except MLBS) on the TENT data (14,446 gene trees), which includes gene trees estimated on UCEs as well as exons and introns. In this case, methods are compared to the published TENT concatenation tree estimated by running ExaML (Kozlov et al. 2015).
UCE data
For the UCE data (48 taxa, 3679 gene trees), ASTRAL-III completes in 65 min, making it the most time consuming method. All other methods run in less than a minute; however, the preprocessing step to weight quartets for wQFM takes 41 min.
Both FASTRAL and ASTRAL-III produce the same species tree (Fig. 5C), and both TREE-QMC-n2 and wQFM produce the same species tree (Fig. 5A). We compare these two trees to the published concatenation tree for UCEs (Fig. 5B). There are many similarities between these three trees, as all contain the magnificent seven clades. The TREE-QMC-n2 and FASTRAL trees differ from the concatenation tree by seven and nine branches, respectively, putting the TREE-QMC-n2 tree slightly closer to the concatenation tree than the FASTRAL tree. The TREE-QMC-n2 tree recovers Passerea and Afroaves and fails to recover Columbea, like the concatenation tree and unlike the ASTRAL-III tree (note that Passerea was considered to be strongly corroborated, after accounting for data type effects, by Braun and Kimball 2021). Overall, there are only five branches that differ between the TREE-QMC-n2 tree and the FASTRAL tree; all of these branches have nearly equal quartet support for their alternative resolutions so that both trees represent reasonable hypotheses.
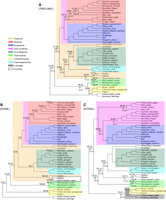
Avian UCE data. (A) Tree estimated from UCE gene trees using TREE-QMC-n2/wQFM. (B) Tree estimated from concatenated UCE alignment using RAxML. (C) Tree estimated from UCE gene trees using ASTRAL-III/FASTRAL. Above the branch, we show support values X, Y, where X is estimated using ASTRAL's local posterior probability (multiplied by 100) and Y is computed using RAxML's bootstrap support for B and using MLBS for A and C. Support values are only shown when X is <100. Below the branch, we show the quartet support (the two values below it correspond to quartet support for the two alternative resolutions of the branch). Taxa outside of Neoaves are not shown as all methods recovered the same topology outside of Neoaves.
TENT data
For the TENT data (48 taxa, 14,446 gene trees), TREE-QMC-n2 and FASTRAL complete in <3 min, whereas it takes 2.35 h to weight quartets. wQFM completes in less than a minute after this preprocessing phase. We do not run ASTRAL-III as this analysis was reported to take over 30 h (Dibaeinia et al. 2021).
All three methods produce a different tree, which is compared to the published concatenation tree for TENT data (Supplemental Fig. S6). None of the trees recover Passera, and only the concatenation and wQFM trees recover Afroaves, although this branch has very low local support (local posterior probability of 0.0) in the wQFM tree. Once again, the TREE-QMC-n2 and wQFM trees are closest to the concatenation tree, with the TREE-QMC-n2, wQFM, and FASTRAL trees differing from it by 8, 8, and 10 branches, respectively. There are five branches that differ between the wQFM tree and the TREE-QMC-n2 tree (two of these branches in the wQFM have very low support: local posterior probability of 0.03 and 0.0). There are only three branches that differ between the TREE-QMC-n2 tree and the FASTRAL tree; as with the UCE data, these branches are reasonable based on quartet support for their alternative resolutions.
Discussion
Our method TREE-QMC builds upon the algorithmic framework of wQMC (Avni et al. 2014) by introducing the normalized quartet graph and showing that it can be computed directly from gene trees. These contributions together enable our new method TREE-QMC to be highly competitive with the leading quartet-based summary methods in terms of species tree accuracy and empirical runtime, even outperforming them on some simulated data sets. Specifically, TREE-QMC (with nonuniform normalization) is competitive with wQFM in terms of species tree accuracy but scales to much larger data sets. Moreover, TREE-QMC is at least as accurate, and often more accurate, than the dominant method ASTRAL-III (and its improvement FASTRAL), while being highly competitive in terms of empirical runtime.
The model conditions where TREE-QMC outperforms ASTRAL-III are characterized by large numbers of species or high amounts of gene tree heterogeneity because of ILS and GTEE. For the latter scenario, the true species tree was typically not the optimal solution to MQSST (note: this observation is not out of line with the statistical theory because the proof of consistency assumes infinite error-free gene trees). Therefore, better heuristics to MQSST may not translate to more accurate species trees when GTEE is high.
A major goal is to develop summary methods that are robust to GTEE. One approach is weighting quartets not just by their frequency in the input gene trees. A new version of ASTRAL, dubbed weighted ASTRAL (Zhang and Mirarab 2022), which was published during our study, adjusts quartet weights based on the branch support and branch lengths in the estimated gene trees. TREE-QMC's nonuniform normalization scheme adjusts the quartet weights based on the subproblem division (i.e., quartets are upweighted if they are on species in more closely related subproblems, which ideally reflects closeness in the true species tree). In the future, it would be interesting to compare TREE-QMC to weighted ASTRAL as well as to implement other quartet weighting schemes within TREE-QMC.
There are several other opportunities for future work worth mentioning. First, the version of TREE-QMC presented here requires binary gene trees as input. Thus, TREE-QMC was given gene trees that are randomly refined in our experimental study, whereas all other methods were given gene trees with polytomies. This did not have a negative impact on TREE-QMC's performance relative to the other methods; however, it would be worth exploring this issue further. Ultimately, this inherent limitation of TREE-QMC could be addressed by devising an efficient algorithm for computing the “edges” in the quartet graph (see Methods section), although this would come at the cost of increased runtime. Second, the experimental study presented here only evaluates TREE-QMC in the context of multilocus species tree estimation where gene tree can be discordant with the species tree because of ILS and/or GTEE. Our study does not address the use of TREE-QMC as a more general quartet-based supertree method, and future work should explore whether quartet weight normalization is beneficial in this context. Lastly, TREE-QMC's algorithm operates on gene trees that are multilabeled because of artificial taxa, so the algorithms presented here can be applied to gene trees that are multilabeled because of other causes, such as multiple individuals being sampled per species (Rabiee et al. 2019) or genes evolving via duplications (Zhang et al. 2020; Legried et al. 2021; Yan et al. 2021; Smith et al. 2022). Future work should explore the effectiveness of TREE-QMC under these conditions as well as those characterized by missing data because of gene loss or other causes (Nute et al. 2018).
Methods
We now present the TREE-QMC method. To begin, we define some terminology for phylogenetic trees and the notation used through this section and the Supplemental Materials.
Terminology and notation
A phylogenetic tree T is a triplet 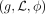 , where g is a connected acyclic graph, L is a set of labels (species), and ϕ maps leaves in g to labels in L. If ϕ is a bijection, we say that T is singly labeled; otherwise, we say T is multilabeled. Trees may be either unrooted or rooted. Edges in an unrooted tree are undirected, whereas edges in a rooted tree are directed away from the root, a special vertex
with in-degree 0 (all other vertices have in-degree 1). To transform an unrooted tree T into a rooted tree Tr, we select an edge in T, subdivide it with a new vertex r (the root), and then orient the edges of T away from the root. Conversely, we transform a rooted tree Tr into an unrooted tree T by undirecting its edges and then suppressing any vertex with degree 2.
, where g is a connected acyclic graph, L is a set of labels (species), and ϕ maps leaves in g to labels in L. If ϕ is a bijection, we say that T is singly labeled; otherwise, we say T is multilabeled. Trees may be either unrooted or rooted. Edges in an unrooted tree are undirected, whereas edges in a rooted tree are directed away from the root, a special vertex
with in-degree 0 (all other vertices have in-degree 1). To transform an unrooted tree T into a rooted tree Tr, we select an edge in T, subdivide it with a new vertex r (the root), and then orient the edges of T away from the root. Conversely, we transform a rooted tree Tr into an unrooted tree T by undirecting its edges and then suppressing any vertex with degree 2.
For a tree T, we denote its edge set as E(T), its internal vertex set as V(T), and its leaf set as L(T). Sometimes we consider a phylogenetic tree T restricted to a subset of its leaves R ⊆ L(T). Such a tree, denoted T|R, is created by deleting leaves in 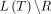 and suppressing any vertex with degree 2 (while updating branch lengths in the natural way). Henceforth, all trees are binary, meaning that nonleaf, nonroot vertices (referred to as internal vertices) have degree 3.
and suppressing any vertex with degree 2 (while updating branch lengths in the natural way). Henceforth, all trees are binary, meaning that nonleaf, nonroot vertices (referred to as internal vertices) have degree 3.
To present TREE-QMC, we need two additional concepts: bipartitions and quartets. A bipartition splits a set L of labels into two disjoint sets: E and 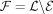 . Each edge in a (singly labeled, unrooted) tree T induces a bipartition because deleting an edge creates two rooted subtrees whose leaf labels form the bipartition
. Each edge in a (singly labeled, unrooted) tree T induces a bipartition because deleting an edge creates two rooted subtrees whose leaf labels form the bipartition 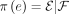 . A given bipartition is displayed by T if it is in the set {π(e):e ∈ E(T)}. The bipartition is trivial if
. A given bipartition is displayed by T if it is in the set {π(e):e ∈ E(T)}. The bipartition is trivial if 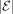 or
or 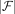 is 1; otherwise, it is nontrivial.
is 1; otherwise, it is nontrivial.
A quartet q is an unrooted, binary tree with four leaves a, b, c, d labeled by A, B, C, D, respectively. It is easy to see that there are three possible quartet trees given by their one nontrivial bipartition: (1) a,b|c,d; (2) a,c|b,d; and (3) a,d|b,c (note that we typically use lower case letters to denote leaf vertices and capital letters to denote leaf labels, although this distinction is only necessary when trees are multilabeled). A set of quartets can be defined by an unrooted tree T by restricting T to every possible subset of four leaves in L(T); the resulting set Q(T) is referred to as the quartet encoding of T. If T is multilabeled, then some of the quartets in Q(T) will have multiple leaves labeled by the same label. Lastly, we say that T displays a quartet q if q ∈ Q(T).
Review of wQMC
As previously mentioned, our new method TREE-QMC builds upon the divide-and-conquer method wQMC (Avni et al. 2014). To produce a bipartition on X, wQMC constructs a graph from Q, referred to as the quartet graph, and then seeks its maximum cut (Snir and Rao 2010, 2012; Avni et al. 2014). The quartet graph is formed from two complete graphs, B and G, both on vertex set V (i.e., there exists a bijection between V and X). All edges in B and G are initialized to weight zero. Then, each quartet 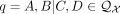 contributes its weight
contributes its weight 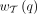 to two “bad” edges in B and four “good” edges in G, where
to two “bad” edges in B and four “good” edges in G, where 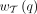 corresponds to the number of gene trees in the input set T that display q. The bad edges are based on sibling pairs: (A, B) and (C, D). The good edges are based on nonsibling pairs: (A, C), (A, D), (B, C), and (B, D). We do not want to cut bad edges because siblings should be on the same side of the bipartition; conversely, we want to
cut good edges because nonsiblings should be on different sides of the bipartition. Ultimately, we seek a cut C to maximize
corresponds to the number of gene trees in the input set T that display q. The bad edges are based on sibling pairs: (A, B) and (C, D). The good edges are based on nonsibling pairs: (A, C), (A, D), (B, C), and (B, D). We do not want to cut bad edges because siblings should be on the same side of the bipartition; conversely, we want to
cut good edges because nonsiblings should be on different sides of the bipartition. Ultimately, we seek a cut C to maximize
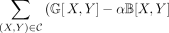 ), where α > 0 is a hyperparameter that can be optimized using binary search. Although MaxCut is NP-complete (Karp 1972), fast and accurate heuristics have been developed (Dunning et al. 2018). The cut gives a bipartition in the output species tree and the wQMC method proceeds by recursion on the two subsets of
species on each side of the bipartition. Artificial taxa are introduced to represent the species on the other side of the
bipartition.
), where α > 0 is a hyperparameter that can be optimized using binary search. Although MaxCut is NP-complete (Karp 1972), fast and accurate heuristics have been developed (Dunning et al. 2018). The cut gives a bipartition in the output species tree and the wQMC method proceeds by recursion on the two subsets of
species on each side of the bipartition. Artificial taxa are introduced to represent the species on the other side of the
bipartition.
TREE-QMC: quartet weight normalization
Our key observation in developing TREE-QMC is that artificial taxa change the quartet weights so that a single gene tree will
vote multiple times for quartets on artificial taxa and only once for quartets on only nonartificial taxa (called singletons).
As shown in Fig. 1, the weight of quartet M, N|O, P is
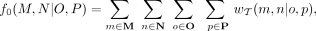 (1)
where
(1)
where  denotes the set of leaves (i.e., species) in T associated with label M (and similarly for N, O, P). When labels M, N, O, P are all singletons, each gene tree casts exactly one vote for one of the three possible quartets: M, N|O, P or M, O|N, P or M, P|N, O (assuming no missing data). Otherwise, each gene tree casts |M| · |N| · |O| · |P| votes (again assuming no missing data) and thus can vote for more than one topology.
denotes the set of leaves (i.e., species) in T associated with label M (and similarly for N, O, P). When labels M, N, O, P are all singletons, each gene tree casts exactly one vote for one of the three possible quartets: M, N|O, P or M, O|N, P or M, P|N, O (assuming no missing data). Otherwise, each gene tree casts |M| · |N| · |O| · |P| votes (again assuming no missing data) and thus can vote for more than one topology.
We propose to normalize the quartet weights so that each gene tree casts one vote for each subset of four labels, although
it may split its vote across the possible quartet topologies in the case of artificial taxa. To get this outcome, we can simply
divide by the number of votes cast so that the weight of M, N|O, P becomes
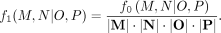 (2)
This can be implemented efficiently by assigning an importance value I(x) to each species
(2)
This can be implemented efficiently by assigning an importance value I(x) to each species 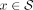 and then computing the weight as
and then computing the weight as
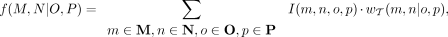 (3)
where I(m, n, o, p) = I(m) · I(n) · I(o) · I(p). Specifically, Equation 3 reduces to Equation 2 when I(m) = |M|−1 for all m ∈ M (and similarly for N, O, P). Because all species with the same label are assigned the same importance value, we refer to this approach as uniform normalization (n1). More broadly, the quartet weights will be normalized whenever Equation 3 corresponds to a weighted average, meaning that
(3)
where I(m, n, o, p) = I(m) · I(n) · I(o) · I(p). Specifically, Equation 3 reduces to Equation 2 when I(m) = |M|−1 for all m ∈ M (and similarly for N, O, P). Because all species with the same label are assigned the same importance value, we refer to this approach as uniform normalization (n1). More broadly, the quartet weights will be normalized whenever Equation 3 corresponds to a weighted average, meaning that
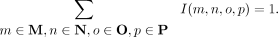 (4)
It is easy to see that this will be the case whenever
(4)
It is easy to see that this will be the case whenever 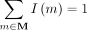 (and similarly for N, O, P). In unnormalized (n0) case, we assign all species an importance value of 1 so that Equation 3 reduces to Equation 1.
(and similarly for N, O, P). In unnormalized (n0) case, we assign all species an importance value of 1 so that Equation 3 reduces to Equation 1.
We now describe how to normalize quartet weights while leveraging the hierarchical structure implied by artificial taxa by
assigning importance values to species with the same label. The idea is that species should have lesser importance each time
they are relabeled by an artificial taxon. In Fig. 1, artificial taxon Z represents species Z = {0, 6, 7, 9} but species 0 and 9 were previously labeled by artificial taxon X. This relationship can be represented as the rooted “phylogenetic” tree TZ given by Newick string: (6, 7, (0, 9)X)Z. We use TZ to assign importance values to all species z ∈ Z, specifically
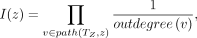 (5)
where outdegree(v) is the out-degree of vertex v and path(TZ, z) contains the vertices on the path in TZ from the root to the leaf labeled z, excluding the leaf. Continuing the example,
(5)
where outdegree(v) is the out-degree of vertex v and path(TZ, z) contains the vertices on the path in TZ from the root to the leaf labeled z, excluding the leaf. Continuing the example, 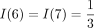 and
and 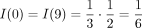 . By construction,
. By construction, 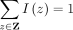 so this approach normalizes the quartet weights. Because different species with the same label can have different weights,
we refer to this approach as nonuniform normalization (n2). In our simulation study, normalizing the quartet weights in this fashion improved species tree accuracy for challenging
model conditions.
so this approach normalizes the quartet weights. Because different species with the same label can have different weights,
we refer to this approach as nonuniform normalization (n2). In our simulation study, normalizing the quartet weights in this fashion improved species tree accuracy for challenging
model conditions.
TREE-QMC: efficient quartet graph construction
Another key development in TREE-QMC is an efficient algorithm for constructing the quartet graph directly from k gene trees, each on n species. Our approach breaks down computing the weights for good and bad edges into three cases:
-
Case 1: taxa X, Y are both nonartificial taxa (called singletons).
-
Case 2: taxa X is a singleton and Y is an artificial taxon (or vice versa).
-
Case 3: taxa X, Y are both artificial taxa.
For one gene tree, G[X, Y] and B[X, Y] can be computed for all pairs X, Y in case 1, case 2, and case 3 in O(a2) time, O(abn) and O(b2n) time, respectively, where a is the number of singletons and b is the number of artificial taxa. Thus, for a subproblem with s = a + b taxa, the quartet graph can be constructed in O(s2nk) time by applying this algorithm to all gene trees (Theorem 1 in the Supplemental Materials).
After quartet graph construction, we seek a max cut. Our high-level approach is the same as wQMC. However, we differ in our binary search for α (interval and precision) and our max-cut heuristic, instead using the one proposed by Burer et al. (2002) and implemented in the open-source package MQLib by Dunning et al. (2018). Our approach for cutting the quartet graph runs in O(s2) time (Theorem 2 in the Supplemental Materials), so the time complexity for each subproblem is dominated by quartet graph construction. Overall, the divide-and-conquer algorithm runs in O(n3k) time (Theorem 3 in the Supplemental Materials) if the division into subproblems is perfectly balanced. We do not expect subproblem division to be perfectly balanced in practice, and moreover the time complexity analysis hides large constant factors (Theorem 2 in the Supplemental Materials). That being said, we find TREE-QMC is sufficiently fast, at least on the specific inputs in our study.
Idea behind efficient quartet graph construction
We now provide the idea behind our approach by considering the simplest case where there are no artificial taxa in gene tree
T with n leaves (i.e., T is singly labeled). For TREE-QMC, the weight of bad edges between taxa X and Y, denoted B[X, Y], is the number of quartets displayed by T with X, Y as siblings and similarly for G[X, Y] but nonsiblings. This means that we can easily compute the weight of the good edges if given the weight of the bad edges
by applying 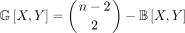 .
.
To compute B efficiently, we observe that there is exactly one leaf associated with label X (denoted x) and one leaf associated with label Y (denoted y), so there is a unique path connecting leaves x and y in T (Fig. 6A). Deleting the edges on this path (and their end points) produces a forest of K rooted subtrees, denoted {t1, t2, …, tK}. Let w and z be two leaves of subtrees ti and tj, respectively. Then, T displays quartet x, w|z, y for i < j, quartet x, y|w, z for i = j, and quartet x, z|w, y for i > j. To summarize, x, y are siblings if and only if leaves w, z are in the same subtree off the path from x to y. It follows that B[X, Y] can be computed by considering all ways of selecting two other leaves from the same subtree for all subtrees on the path from x to y.
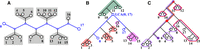
To count the quartets induced by T with 0 and 17 as siblings, we consider the path between them (shown in blue in panel A). The deletion of the path produces six rooted subtrees (highlighted in gray). Because 0 and 17 are siblings in a quartet
if and only if the other two taxa are drawn from the same subtree, the number of bad edges can be computed as 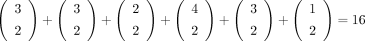 . Here we show how to compute the number of quartets induced by T with 0 and 17 as siblings after rooting T arbitrarily. Panel B shows that we need to consider the number of ways of selecting two taxa from the same subtree for three cases: (1) the subtree
above the lca(0, 17) (highlighted in green), (2) all subtrees off the path from the lca(0, 17) to the left taxon 0 (highlighted in red), and (3) all subtrees off the path from the lca(0, 17) to the right taxon 17 (highlighted in pink). Case 1 can be computed in constant time if we know the number of leaves
below the LCA, that is, A[0, 17] = 6 (Eq. 8). Cases 2 and 3 can also be computed in constant time as follows. Panel C shows the prefix of the left child of the lca(0, 17), denoted p[lca(0, 17).left]. This quantity is the number of ways of selecting two taxa from the same subtree for all subtrees off the path from the
root to this vertex (circled in red). Similarly, the prefix of taxon 0, denoted p[0], is the number of ways of selecting two taxa from the same subtree for all subtrees off the path from the root to 0 (circled
in purple). Therefore, the number of ways of selecting two taxa from all subtrees in case 2 (i.e., subtrees highlighted in
red in B) is L[0, 17] = p[0] − p[lca(0, 17).left] = 7 (Eq. 9). Case 3 can be computed in a similar fashion as R[0, 17] = p[17] − p[lca(0, 17).right] = 3 (Eq. 10). Putting this all together gives B[0, 17] = 16 (Eq. 6).
. Here we show how to compute the number of quartets induced by T with 0 and 17 as siblings after rooting T arbitrarily. Panel B shows that we need to consider the number of ways of selecting two taxa from the same subtree for three cases: (1) the subtree
above the lca(0, 17) (highlighted in green), (2) all subtrees off the path from the lca(0, 17) to the left taxon 0 (highlighted in red), and (3) all subtrees off the path from the lca(0, 17) to the right taxon 17 (highlighted in pink). Case 1 can be computed in constant time if we know the number of leaves
below the LCA, that is, A[0, 17] = 6 (Eq. 8). Cases 2 and 3 can also be computed in constant time as follows. Panel C shows the prefix of the left child of the lca(0, 17), denoted p[lca(0, 17).left]. This quantity is the number of ways of selecting two taxa from the same subtree for all subtrees off the path from the
root to this vertex (circled in red). Similarly, the prefix of taxon 0, denoted p[0], is the number of ways of selecting two taxa from the same subtree for all subtrees off the path from the root to 0 (circled
in purple). Therefore, the number of ways of selecting two taxa from all subtrees in case 2 (i.e., subtrees highlighted in
red in B) is L[0, 17] = p[0] − p[lca(0, 17).left] = 7 (Eq. 9). Case 3 can be computed in a similar fashion as R[0, 17] = p[17] − p[lca(0, 17).right] = 3 (Eq. 10). Putting this all together gives B[0, 17] = 16 (Eq. 6).
This observation can be used to count the quartets efficiently when gene trees are singly labeled. However, we need to be more careful when T is multilabeled, which is typically the case because of artificial taxa. Following our example, suppose that we want to count the number of bad edges between 0 and 17 contributed by the subtree with leaves 4, 5, and 6. However, if leaves 4 and 5 are both relabeled by artificial taxon M, the quartet on 0, 17|4, 5 corresponds to quartet 0, 17|M, M has no topological information and should not be counted. The other quartets 0, 17|4, 6 and 0, 17|5, 6 correspond to 0, 17|M, 6 and thus should be counted.
Computing the bad edges given a singly labeled gene tree
We now present an algorithm for computing the number of bad edges given a singly labeled gene tree T (later we will extend it to the more general case of a multilabeled gene tree). After rooting T arbitrarily, we again consider the path between x and y, which now goes through their lowest common ancestor, denoted lca(x, y) (Fig. 6B). This allows us to break the computation into three parts
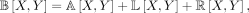 (6)
where A[X, Y] is the number of ways of selecting two leaves from the subtree above lca(x, y), L[X, Y] the number of ways of selecting two leaves from the same subtree for all subtrees off the path from lca(x, y) to leaf in its left subtree (say x), and R[X, Y] the number of ways of selecting two leaves from the same subtree for all subtrees off the path from lca(x, y) to the leaf in its right (say y). As we will show, each of these quantities can be computed in constant time, after an O(n) preprocessing phase, in which we compute two values for each vertex v in T. The first value c[v] is the number taxa below vertex v. The second value p[v], which we refer to as the “prefix” of v, is the number of ways to select two taxa from the same subtree for all subtrees off the path from the root to vertex v (Fig. 6C). It is easy to see that c can be computed in O(n) time via a postorder traversal. After which, p can be computed in O(n) via a preorder traversal, setting
(6)
where A[X, Y] is the number of ways of selecting two leaves from the subtree above lca(x, y), L[X, Y] the number of ways of selecting two leaves from the same subtree for all subtrees off the path from lca(x, y) to leaf in its left subtree (say x), and R[X, Y] the number of ways of selecting two leaves from the same subtree for all subtrees off the path from lca(x, y) to the leaf in its right (say y). As we will show, each of these quantities can be computed in constant time, after an O(n) preprocessing phase, in which we compute two values for each vertex v in T. The first value c[v] is the number taxa below vertex v. The second value p[v], which we refer to as the “prefix” of v, is the number of ways to select two taxa from the same subtree for all subtrees off the path from the root to vertex v (Fig. 6C). It is easy to see that c can be computed in O(n) time via a postorder traversal. After which, p can be computed in O(n) via a preorder traversal, setting
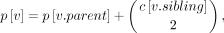 (7)
after initializing p[root] = 0. Now we can compute the quantities:
(7)
after initializing p[root] = 0. Now we can compute the quantities:
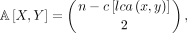 (8)
(8)
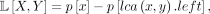 (9)
(9)
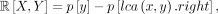 (10)
where v.left denotes the left child of v and v.right denotes the right child of v (see Fig. 6C). It is possible to access lca(x, y) in constant time after O(n) preprocessing step (Gusfield 1997), although we implemented this implicitly by computing the entries of B during a postorder traversal of T. Thus, we can compute B in O(n2) time, provided that T is singly labeled.
(10)
where v.left denotes the left child of v and v.right denotes the right child of v (see Fig. 6C). It is possible to access lca(x, y) in constant time after O(n) preprocessing step (Gusfield 1997), although we implemented this implicitly by computing the entries of B during a postorder traversal of T. Thus, we can compute B in O(n2) time, provided that T is singly labeled.
Computing the bad edges given a multilabeled gene tree
We now present an algorithm for computing the number of bad edges B[X, Y] given a multilabeled gene tree T. As previously mentioned, this breaks down into three cases. Case 1 (where taxa X, Y are both singletons) is presented below and cases 2 and 3 are presented in the Supplemental Materials.
As before, we focus on the number of ways to select two leaves w, z from a collection of subtrees; however, now that T is multilabeled, it is possible for two leaves w, z to have the same label. Therefore, we now need to count the number of ways to select two leaves z, w below vertex u so that they are uniquely labeled Z ≠ W (note that we use capital letters W and Z to denote the current labels of leaves w and z, respectively). This modified binomial is computed by revising the preprocessing phase. We now let c0[v] denote the number of leaves labeled by singletons below vertex v and let cD[v] denote the number of leaves labeled by artificial taxon D below vertex v. Thus, for each vertex v, we store a vector c[v] of length b + 1, where b is the number of artificial taxa in T. As before, we can compute c in O(bn) time via a postorder traversal. However, the number of ways to select two leaves with different labels is now broken into three cases:
-
the number of ways to select two singletons, which equals
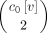 ,
,
-
the number of ways to select one singleton and one artificial taxa, which equals
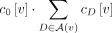 , where
, where 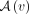 is the set of artificial taxa below vertex v, and
is the set of artificial taxa below vertex v, and
-
the number of ways to select two artificial taxa, which equals
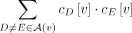 .
.
Putting this all together gives the modified binomial coefficient:
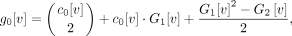 (11)
where
(11)
where 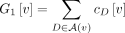 and
and 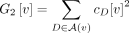 . At each vertex, the calculation of G1[v] and G2[v] takes O(b) time, after which we can compute g0[v] in constant time. Thus, g0 can be computed in O(bn) time. Note that we also need to compute modified binomial coefficient for the subtree “above” vertex v, denoted g0[v.above]. This can be computed in a similar fashion by noting that number of singletons above v is a − c0[v] and that the number of leaves above v labeled by each artificial taxon D is |D| − cD[v].
. At each vertex, the calculation of G1[v] and G2[v] takes O(b) time, after which we can compute g0[v] in constant time. Thus, g0 can be computed in O(bn) time. Note that we also need to compute modified binomial coefficient for the subtree “above” vertex v, denoted g0[v.above]. This can be computed in a similar fashion by noting that number of singletons above v is a − c0[v] and that the number of leaves above v labeled by each artificial taxon D is |D| − cD[v].
Using the modified binomial, we can apply our algorithm for singly labeled trees by redefining prefix sum:
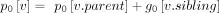 (12)
and then redefining the quantities from which we can compute B[x, y] in constant time, that is, A[X, Y] = g0[lca(x, y).above], and L[X, Y] = p0[x] − p0[lca(x, y).left], and R[X, Y] = p0[y] − p0[lca(x, y).right]. As there are a2 pairs of singletons in the subproblem, the total runtime is O(a2 + bn).
(12)
and then redefining the quantities from which we can compute B[x, y] in constant time, that is, A[X, Y] = g0[lca(x, y).above], and L[X, Y] = p0[x] − p0[lca(x, y).left], and R[X, Y] = p0[y] − p0[lca(x, y).right]. As there are a2 pairs of singletons in the subproblem, the total runtime is O(a2 + bn).
Normalizing quartet weights while computing the bad edges
To normalize the quartet weights, B[X, Y] becomes the weighted sum of quartets with X, Y are siblings, where each quartet x, y|z, w is weighted by I(x, y, z, w) = I(x)I(y)I(z)I(w), where I(x) is the importance value assigned to leaf x (which corresponds to a species in the singly labeled gene tree). When X, Y are singletons,
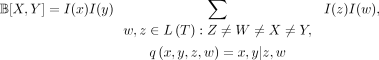 (13)
where the importance values of singletons are set to 1 so we know that I(x) = I(y) = 1. Note that all of the importance values are set to 1 in the unnormalized case.
(13)
where the importance values of singletons are set to 1 so we know that I(x) = I(y) = 1. Note that all of the importance values are set to 1 in the unnormalized case.
To compute the normalized version of B[X, Y] using the previous algorithm, we set cD[v] to be the sum of the importance values of the leaves below v that are labeled by D (i.e., 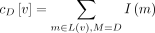 , where L(v) denotes the set of leaves below v). The proof of correctness follows from Lemma 1, in which we show that the total weight of selecting two uniquely labeled leaves below vertex u equals g0[u]. This is because all other quantities (p, A, L, R) are computed from g0[u].
, where L(v) denotes the set of leaves below v). The proof of correctness follows from Lemma 1, in which we show that the total weight of selecting two uniquely labeled leaves below vertex u equals g0[u]. This is because all other quantities (p, A, L, R) are computed from g0[u].
The total weight of all taxon pairs in the subtree rooted at internal vertex u
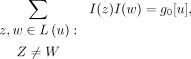 (14)
where L(u) is the set of leaves below vertex u.
(14)
where L(u) is the set of leaves below vertex u.
See Supplemental Materials for proof.
Lastly, we need to compute the good edges G[X, Y], which is the total weight of quartets in which X, Y are not siblings. This can be performed in constant time, following Lemma 2.
Let T be a multilabeled gene tree, and let X, Y be singletons. Then,
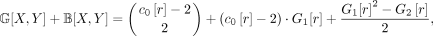 (15)
where r is the root vertex of T.
(15)
where r is the root vertex of T.
See Supplemental Materials for proof.
This concludes our treatment of case 1, in which X, Y are both singletons. To compute all entries of B and G, we also need to consider the other two cases. In case 2, X is a singleton and Y is an artificial taxon or vice versa (Supplemental Fig. S13), and in case 3, both X and Y are artificial taxa (Supplemental Fig. S14). These cases are more complicated because the naive approach would consider all paths in the tree between a leaf labeled X and a leaf labeled Y, which is not efficient. The algorithms and proofs for these cases are provided in the Supplemental Materials.
Software availability
TREE-QMC is available at GitHub (https://github.com/molloy-lab/TREE-QMC) and as Supplemental Code.
Acknowledgments
This research was funded by the State of Maryland. We thank the reviewers for feedback that greatly improved the quality of our work.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277629.122.
- Received December 23, 2022.
- Accepted May 4, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








