
Comparing genomic and epigenomic features across species using the WashU Comparative Epigenome Browser
- Xiaoyu Zhuo1,2,
- Silas Hsu1,2,
- Deepak Purushotham1,2,
- Prashant Kumar Kuntala1,2,
- Jessica K. Harrison1,2,
- Alan Y. Du1,2,
- Samuel Chen1,2,
- Daofeng Li1,2 and
- Ting Wang1,2,3
- 1Department of Genetics, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 2The Edison Family Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 3McDonnell Genome Institute, Washington University School of Medicine, St. Louis, Missouri 63110, USA
Abstract
Genome browsers have become an intuitive and critical tool to visualize and analyze genomic features and data. Conventional genome browsers display data/annotations on a single reference genome/assembly; there are also genomic alignment viewer/browsers that help users visualize alignment, mismatch, and rearrangement between syntenic regions. However, there is a growing need for a comparative epigenome browser that can display genomic and epigenomic data sets across different species and enable users to compare them between syntenic regions. Here, we present the WashU Comparative Epigenome Browser. It allows users to load functional genomic data sets/annotations mapped to different genomes and display them over syntenic regions simultaneously. The browser also displays genetic differences between the genomes from single-nucleotide variants (SNVs) to structural variants (SVs) to visualize the association between epigenomic differences and genetic differences. Instead of anchoring all data sets to the reference genome coordinates, it creates independent coordinates of different genome assemblies to faithfully present features and data mapped to different genomes. It uses a simple, intuitive genome-align track to illustrate the syntenic relationship between different species. It extends the widely used WashU Epigenome Browser infrastructure and can be expanded to support multiple species. This new browser function will greatly facilitate comparative genomic/epigenomic research, as well as support the recent growing needs to directly compare and benchmark the T2T CHM13 assembly and other human genome assemblies.
To meet the need to visualize genomic sequences and features at different scales in the genomic era, scientists developed genome browser/viewers to help interpret genomes. The UCSC Genome Browser, equipped with comprehensive annotations and intuitive navigation, gained widespread popularity in the community (Kent et al. 2002; Lee et al. 2022). In addition to the UCSC Genome Browser, there are multiple other tools available to visualize genomes, each with its own advantages and focuses (e.g., Ensembl [Fernández-Suárez and Schuster 2010; Cunningham et al. 2022], GBrowse [Stein et al. 2002], WashU Epigenome Browser [Zhou et al. 2011; Li et al. 2019, 2022], the Integrative Genomics Viewer [IGV] [Robinson et al. 2011, 2023], and JBrowse [Buels et al. 2016; Diesh et al. 2023]).
With sharply decreasing sequencing cost, many more genomes of different species have become available, and there is an increased effort around the world to systematically sequence a wide variety of organisms (Teeling et al. 2018; Feng et al. 2020; Rhie et al. 2021). The advancement in sequencing technology has also promoted many functional genomic assays, which has enabled functional annotation of genomic regions (The ENCODE Project Consortium 2012; Roadmap Epigenomics Consortium et al. 2015; Bujold et al. 2016; Dekker et al. 2017). Based on whole-genome alignment between species, orthologous regions can be directly compared, and insights on the conservation and adaptation of genomic features can be drawn. Comparative genomics thus has become an important tool to decipher genomic code (Alföldi and Lindblad-Toh 2013). Comparative epigenomics, which compares the epigenomic features of orthologous regions of multiple species, is also gaining popularity (Xiao et al. 2012; Prescott et al. 2015; Zhou et al. 2017; Modzelewski et al. 2021).
Starting from Miropeats, various visualization tools have been developed to display regional orthologous relationship between species (Parsons 1995; Guy et al. 2010; Sullivan et al. 2011; https://github.com/daewoooo/SVbyEye; Goel and Schneeberger 2022; https://github.com/mrvollger/SafFire [DOI:10.5281/ZENODO.6376287]). These tools provide a variety of comparative features. The gEVAL Browser was designed for genome assembly quality evaluation and can be used to visualize and compare genome assemblies (Chow et al. 2016). Nguyen et al. (2014) developed comparative assembly hubs using UCSC Genome Browser's framework. It uses snake track to show multiple query assemblies aligned to a target assembly, and annotations mapped to query assemblies can also be displayed with an automatic “liftOver.” JBrowse2 implemented linear synteny view to support cross-species comparison since v1.6.4 (Buels et al. 2016; Diesh et al. 2023). CEpBrowser was developed to compare epigenomic data sets between human, mouse, and pig based on the UCSC Genome Browser framework in a gene-centric manner (Cao and Zhong 2013). It organizes linear representation of different species in different windows parallelly. By displaying different species in different windows, CEpBrowser can be implemented relatively easily without breaking the continuity of each genome. However, it only marks syntenic regions using the same color scheme but does not connect syntenic regions from different species or display any genetic differences. In addition, only comparisons between human (hg19), mouse (mm9), and pig (susScr2) are supported. Despite being the first comparative epigenome browser, it has not been widely used by the scientific community.
The WashU Epigenome Browser was developed in 2010 to host and display massive epigenomics data sets (Zhou et al. 2011; Li et al. 2019, 2022). It hosts data sets generated from the Roadmap Epigenomics Project (Roadmap Epigenomics Consortium et al. 2015), Encyclopedia of DNA Elements (ENCODE) (The ENCODE Project Consortium 2012), International Human Epigenome Consortium (IHEC) (Bujold et al. 2016), The Cancer Genome Atlas (TCGA) (Hutter and Zenklusen 2018), Toxicant Exposures and Responses by Genomic and Epigenomic Regulators of Transcription (TaRGET) (Wang et al. 2018), and 4D Nucleome Project (4DN) (Dekker et al. 2017). We recently refactored the browser and vastly improved its performance (Li et al. 2019). We further developed new features to display 3D genome structure, dynamic tracks, and imaging data associated with genomic coordinates (Li et al. 2022).
All the WashU Epigenome Browser features described before are still anchored on a single reference genome. Building upon the WashU Epigenome Browser, we extended the support to multiple species and developed the WashU Comparative Epigenome Browser based on four principles: (1) each assembly uses its own coordinates to anchor annotation and data sets mapped to it; (2) orthologous relationship and genetic variations between assemblies are intuitively illustrated; (3) it is adaptable to display any whole-genome alignment at different scales and resolution; (4) it inherits all features of modern genome browsers to facilitate user experience.
The WashU Comparative Epigenome Browser is a versatile tool that can be used at different phases during research. By providing a graphical representation of genomic data, the browser allows researchers to easily explore and understand the complex relationships between genes and other genomic features. The browser's cross-species comparison feature allows researchers to compare different epigenomic data sets across different species and identify similar or different epigenomic features and genomic structural boundaries, providing a powerful tool for validating discoveries made from genome-wide analysis. The browser also allows the integration and comparison of data from different sources, which can be used to generate new hypotheses about the evolution of epigenomic mechanisms across different organisms.
Here we present the WashU Comparative Epigenome Browser to address the needs to navigate multiple genomes at once and visualize comparative genomics/epigenomics data.
Results
The genome-align track connects syntenic regions of two genome assemblies
The foundation that enables comparative genome browsing is the alignment between genome assemblies. We developed a new track type called “genome-align track,” which contains genome-wide syntenic relationship between the reference (target) genome and the secondary (query) genome at base-pair resolution. The genome-align track file can be constructed from standard chained alignment AXT files, and we obtained whole-genome pairwise alignment AXT files from the UCSC Genome Browser (Schwartz et al. 2003; Lee et al. 2022). We developed a customized script to convert AXT files to genome-align track files for browser display (Methods).
We created a comparative epigenome gateway to help organize and facilitate the selection and display of curated genome-align tracks (http://comparativegateway.wustl.edu/). The gateway works in all modern web browsers on PC, MAC, and mobile platforms. We provided examples, video tutorials, and documentation on the website. As of this writing, 13 species were available as either the reference or the secondary genome, most of which were mammals because of data availability. We actively develop new browser functions and will support more species, and we welcome new genome and alignment requests on our browser GitHub repository (https://github.com/lidaof/eg-react/issues). Users can access the species selection interface by clicking “select genomes.” Within the species selection interface, users first select the reference assembly. When one reference genome is selected, all the available genome-align tracks will be populated as a list of secondary genomes (Fig. 1). Then the user can select one or more genome-align tracks anchored to the reference genome, save the selection, and open a new WashU Epigenome Browser window with all the selected genome-align tracks. With genome-align tracks loaded, the user can then use the browser's web interface to load available annotations (Tracks → Annotation Tracks), public data (Tracks → Public Data Hubs), or user's own data (Tracks → Remote/Local Tracks) on the browser mapped to either the reference genome or any of the loaded secondary genomes (Fig. 1).

The web user interface of the WashU Comparative Epigenome Browser. The genome-align track selector web interface is shown on the left. After selecting desired alignment tracks, the user will be redirected to the main WashU Epigenome Browser with the genome-align track loaded. Last, the user can load data and annotations to either the reference or secondary genomes on the browser.
The genome-align track supports comprehensive, multiresolution genome alignment display. At the finest resolution, orthologous coordinates from query genomes are vertically aligned and anchored to the reference genome. Detailed whole-genome alignment at the single-nucleotide resolution is displayed in the genome-align track, enabling users to navigate and examine the genetic differences between the query genome and the reference genome. It is straightforward to visualize single-nucleotide variations (SNVs) and short insertions/deletions (indels) between the two genome assemblies (Fig. 2A).
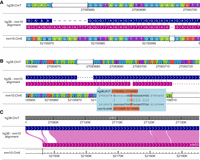
Displaying genome alignments using the WashU Comparative Epigenome Browser. (A) Displaying hg38-mm10 BLASTZ alignment at the nucleotide level with more than 10 pixels per nucleotide. The sequence strand in the alignment is illustrated using arrows. Syntenic nucleotides from hg38 and mm10 are vertically aligned with gaps inserted. Matching nucleotides are connected using a short vertical line in the alignment track. (B) Displaying hg38-mm10 alignment between 0.1 pixels per nucleotide and 10 pixels per nucleotide. The alignment is organized the same as in panel A without displaying nucleotides within the alignment. Alignments at nucleotide resolution are visible in the cursor tip hover box, and the nucleotide alignment under the cursor is highlighted in orange (G–T). (C) Displaying alignment with >10 nt per pixel. Both hg38 and mm10 genomes are continuously displayed without breaks. Syntenic regions are connected using pink Bezier curves.
Users can pan and zoom on the genome-align track using the tools bar on top of the displayed window in a similar fashion as they operate on any other browser track types. When the number of nucleotides within a browser window exceeds the available pixels to display each nucleotide clearly (10 pixels per nucleotide), the browser stops displaying individual nucleotides within the alignment. Instead, it would display a 20-bp alignment in a floating box next to the cursor when the user mouses over the genome-align track (Fig. 2B). This feature helps users to visualize a larger aligned region without missing the base-pair resolution information in the alignment.
Vertically aligning and anchoring query genomes to the reference genome is a straightforward and convenient way to display SNVs and small indels between query and reference genomes. However, it is insufficient to show any large, more complexed structural variations (SVs) between species. The WashU Comparative Epigenome Browser displays both the reference and query genomes in a linear manner and connects syntenic regions using Bezier curves if the browser window contains a long genomic alignment (more than 10 bases per pixel) (Fig. 2C). By doing so, large-scale genetic variations can be directly visualized in the browser. Because both genomes are continuously and colinearly displayed, epigenomic features are also displayed in full without sudden truncation.
Using the WashU Comparative Epigenome Browser to compare conserved epigenomic features between species
The genome-align track is more than just a visualization tool to display pairwise whole-genome alignments. After loading the genome-align track onto the browser, users can load annotations and data sets mapped to the secondary genome in the browser and compare them with those mapped to the reference genome. With this feature, the browser connects annotations and data sets from different genomes together using their syntenic relationship in the same window. While users navigate the reference genome, the browser retrieves syntenic coordinates from other genomes and fetches all the loaded tracks.
We can use the browser to characterize deeply conserved epigenomic marks. In Figure 3A the browser displays deeply conserved CpG methylation in the liver between mouse and zebrafish using methylC tracks (Yue et al. 2014; Zhou et al. 2014; Yang et al. 2020). By displaying the Hoxc gene cluster from both the mouse and zebrafish reference genomes and their syntenic relationship, we can appreciate that only a small fraction of their genomic sequences can be aligned with each other after hundreds of million years of independent evolution, recapitulating the discovery made by Zhang et al. (2016). Even conserved CpG islands between these two species are sparse. However, except for a few species-specific transposable elements, the displayed regions are hypomethylated, with an average methylation of 0.04 and 0.08 in the zebrafish and mouse, respectively. Our browser showed that despite limited sequence conservation, the overall hypomethylation of this region is conserved in both the mouse and zebrafish.
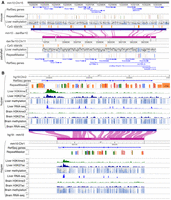
Comparing conserved epigenomic features between species. (A) The DNA methylation status of the Hoxc gene cluster is conserved between the mouse and zebrafish. Mouse and zebrafish DNA methylomes were characterized by Zhang et al. (2016). Mouse and zebrafish reference genomes (mm10 and danRer7) are shown back-to-back anchored by the mouse–zebrafish genome-align track along with their gene, repeat, and CpG island annotations. Liver DNA methylome data are from Zhang et al. (2016) with enhanced reduced representation bisulfite sequencing (ERRBS) displayed. (B) H3K4me3 and H3K27ac ChIP-seq, WGBS, and RNA-seq of brain and liver samples from both human and mouse of the SPP2/Spp2 gene are displayed using the WashU Comparative Epigenome Browser. Both DNA methylation level and read depth are illustrated in the methylC track. Both methylation percentage and read coverage of each CpG site were annotated within the methylC tracks. All CpG sites are marked by gray, with methylation percentage annotated by the blue bar in the foreground (0% methylated CpGs are displayed as full gray bars, whereas 100% methylated CpGs are displayed as full blue bars). The read coverage over CpG sites across the region is represented by the black line in the background.
Epigenomic modifications underlie tissue specificity. It has been shown before that the tissue-specific epigenomic patterns are often conserved between species (Zhou et al. 2017). The comparative browser makes it intuitive to examine the conservation pattern of tissue-specific gene activities. Figure 3B illustrates the conserved liver-specific expression and epigenome landscape of gene secreted phosphoprotein 2 (SPP2) between human and mouse. Epigenomic data, including whole-genome bisulfite sequencing (WGBS), H3K4me3 ChIP-seq, H3K27ac ChIP-seq, and RNA-seq data of liver and brain from human and mouse ENCODE, are displayed on the respective reference genomes in the comparative browser (The ENCODE Project Consortium 2012; Yue et al. 2014), spanning the syntenic region around human SPP2 gene and its orthologous mouse Spp2 gene (Fig. 3B). Both species share the pattern of liver-specific active histone marks, low DNA methylation in promoter, and high RNA expression, as well as a lack of active histone/expression and high DNA methylation in promoter in the brain, indicating tissue-specific epigenetic conservation. To aid in visual interpretation (showing a hypomethylated promoter region in the liver of both species), we have tabulated the methylation data presented in Figure 3B as Table 1.
Quantification of methylation data of Figure 3B
Visualizing species-specific feature
In addition to showcasing conserved features, the browser is equally effective at visualizing lineage-specific epigenomic features. Figure 4A displays H3K27ac and transcription factor NR2F1 ChIP-seq data from iPSC-derived cranial neural crest cells (CNCCs) of both human and chimpanzee (Prescott et al. 2015). This region has been identified as a putative human-biased enhancer previously, defined by the differential H3K27ac ChIP-seq peak between human and chimpanzee in the intron of the SMAP2 gene (Prescott et al. 2015). The epigenomic signature suggests that this is either a human gain or chimpanzee loss of a putative CNCC enhancer. Zooming in to examine the alignment at base level, we identified a single-nucleotide difference between human and chimpanzee that maps to a high information content position in the NR2F motif, potentially explaining the difference in both H3K27ac and NR2F1 ChIP-seq signal. This example shows that our browser can be used to associate epigenomic differences between species with their genetic differences.
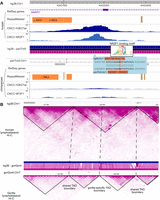
Highlighting species-specific features using the comparative browser. (A) Lineage-specific epigenomic innovation. H3K27ac, NR2F1 ChIP-seq data from both human and chimpanzee CNCCs in SMAP2 gene (annotated in human only) regions were plotted in the WashU Comparative Epigenome Browser. A human-specific NR2F1 and H3K27ac peak suggests a putative human-specific enhancer in this region. The putative enhancer is associated with a human-specific NR2F1 binding motif. (B) 3D genome structure differences between species. A human lymphoblastoid Hi-C contact map mapped to hg38 and gorilla lymphoblastoid Hi-C data mapped to gorGor4 were compared by anchoring to the human–gorilla alignment track.
The comparative browser also supports visualization and comparison of long-range chromatin interaction data across different genomes, thus facilitating the studies of 3D genome evolution (Vietri Rudan et al. 2015). Figure 4B directly compares the 3D genome structure between human and gorilla in the human Chr1q42.13 region. Hi-C data from lymphoblastoid cells of human and gorilla reveal several conserved TADs. However, one TAD in human is split into two different TADs in the gorilla. This observation using the comparative browser recapitulated insights from Yang et al. (2019).
Visualizing the relationship between genomic variation and epigenomic variation
There has been a growing interest in understanding the relationship between genetic variation and epigenetic variation. We have already shown using the browser to display the association between epigenomic changes with a SNP (Fig. 4A). Recently, we characterized SVs between human and chimpanzee and their impact on the epigenome (Zhuo et al. 2020). Figure 5A illustrates an interesting case of human-specific TE-derived putative enhancer we identified previously. In this comparative browser view, investigators can easily and intuitively compare a species-specific TE insertion and its associated epigenomic modification. Here, a human-specific retrotransposon SVA-F appears in the intron of the DNMBP gene. The sequence of this SVA-F element is highly repetitive; thus, it shows low mappability scores (average 50-bp score <0.05), indicating that short sequencing reads derived from this element may not be uniquely mapped back (Derrien et al. 2012). Indeed, a CNCC H3K27ac ChIP-seq data set (sequenced using 50-bp reads) does not contain signal within the SVA-F element but reveals a peak at the 3′ boundary of the element. Further analysis suggests that this boundary peak reflects enhancer signals from within this SVA-F element (Zhuo et al. 2020). In contrast, an iPSC H3K9me3 ChIP-seq data set (sequenced using 100-bp paired-end reads) is able to uniquely reveal an enrichment peak over this SVA-F element, indicating the deployment of repressive chromatin onto this newly inserted retrotransposon in iPSCs (Zhuo et al. 2020). The parallelly displayed chimpanzee genome and corresponding epigenomic data sets illustrate the lack of this specific SVA-F insertion and absence of respective epigenomic marks. This direct visual comparison of the retrotransposon insertion and epigenomic changes between the two species recapitulates the discovery of a tissue-specific enhancer derived from a human-specific retrotransposon insertion.
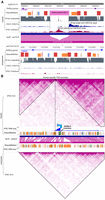
Connecting epigenomic changes with genomic changes using the WashU Comparative Epigenome Browser. (A) RefSeq genes, RepeatMasker, and 50-bp mappability annotations along with H3K27ac ChIP-seq data from cranial neural crest cells (CNCCs) and H3K9me3 ChIP-seq data from iPSCs in both human and chimpanzee were plotted in DNMBP gene region. The H3K9me3 peak in the human-specific SVA insertion indicates epigenomic repression of this element in iPSCs, and the human-specific H3K27ac peak indicates the creation of a putative new CNCC enhancer in the human lineage. (B) Human-specific HERV-H expression is correlated with a new TAD boundary in iPSCs in the human genome compared with the marmoset genome. The iPSC Hi-C contact map, RNA expression, and repeat annotations from human (hg19) and marmoset (calJac3) are compared vertically. HERV-H insertion and expression in the human lineage are associated with a human-specific TAD boundary.
Zhang et al. (2019) showed that the expression of HERV-H is associated with new TAD boundaries in primates. This association can be easily appreciated in a comparative browser view. In Figure 5B, Hi-C maps and RNA expression of human iPSC and marmoset iPSC can be directly compared in the context of their genome alignment. In the human genome, an HERV-H insertion is associated with a human-specific TAD boundary and human-specific RNA expression, both absent in the marmoset genome (Fig. 5B). Our browser allows examining the association between the Hi-C contact map and the HERV-H insertion and expression at a higher resolution, making it easier to appreciate that the TAD boundary is ∼20 kb away from the HERV-H insertion in the human genome. Our observation is consistent with the investigators’ hypothesis that it is the expression of instead of the presence of the HERV-H that contributes to the TAD boundary (Zhang et al. 2019). These examples show that the WashU Comparative Epigenome Browser can be used to directly compare genomic data sets across species and visualize the association with genetic changes.
Displaying genome annotations and data sets from multiple species using the WashU Comparative Epigenome Browser
A natural extension of the pairwise comparison function is to support comparison among multiple species. Conceptually, this extension is equivalent to visualizing genomic data aligned to a multiple genome alignment across species. Practically, we use multiple genome-align tracks to anchor the visualization to the same reference genome, thus enabling an intuitive comparison of genomic data across orthologous regions of multiple species.
We use CTCF turnover events characterized by Schmidt et al. (2012) and Choudhary et al. (2020) to illustrate the comparative analysis across multiple genomes. Schmidt et al. (2012) characterized the CTCF binding sites of six mammalian species (human, macaque, mouse, rat, dog, and opossum) and identified thousands of conserved as well as lineage-specific, retrotransposon-derived CTCF binding sites. We display both CTCF ChIP-seq data and called CTCF binding peaks of the six species from this study using the WashU Comparative Epigenome Browser, anchored on the human reference genome hg19 (Fig. 6). This allows direct comparison of CTCF binding across species along with genetic changes in each species.
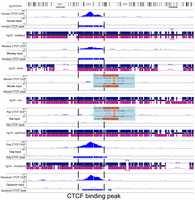
Using the WashU Comparative Epigenome Browser to visualize and compare the CTCF binding sites from six mammals. CTCF ChIP-seq and input from human (hg19), rhesus macaque (rheMac2), mouse (mm9), rat (rn4), dog (canFam2), and opossum (monDom5) were displayed on the browser. Human reference genome hg19 was used as the reference genome, and all the other species were anchored to their orthologous region from hg19 using whole-genome alignments. The region hg19:Chr4:23,456,625–23,458,090 shows a conserved CTCF binding peak in the orthologous loci in all mammal genomes except the two rodents, indicating a rodent-specific loss of a conserved CTCF binding site. The loss of CTCF binding also coincided with a rodent-specific 6-bp insertion.
Figure 6 highlights the loss of a conserved CTCF binding site in rodents (Fig. 6). In the zoomed-in view (<10 bp per pixel), base pair–level alignment is available to the user. To ensure multiple genome-align tracks at this view are vertically aligned, we introduced extra gaps in the pairwise alignment tracks when necessary. In contrast to the other four genomes, the mouse and rat do not display a CTCF binding peak in this region, and this event is associated with a rodent-specific 6-bp insertion in the ortholog site of the CTCF site conserved in the other four species. Again, the WashU Comparative Epigenome Browser makes it intuitive to display and identify associations between genetic changes and epigenomic changes across multiple species.
Extending comparative genomic analysis to nonmodel organisms and new assemblies
The WashU Comparative Epigenome Browser is built on an actively maintained and expandable platform. New genomes are routinely added to the browser to serve scientists around the world. The browser engineers respond to new comments and feature requests (including requests for new genomes) on the browser GitHub repository frequently (https://github.com/lidaof/eg-react/issues). We also documented how to add new genomes to the browser for a local environment for advanced users with a JavaScript background (https://epigenomegateway.readthedocs.io/en/latest/add.html).
Using this flexible framework, we created multiple nonmodel organism reference genomes in our browser. For example, we created reference cattle genome UMD3.1.1/bosTau8, and generated a bosTau8-mm10 genome-align track using bosTau8 as the reference genome. Figure 7A displays a direct comparison of DNA methylation patterns between cattle and mouse across the heart, lung, and liver (Liu et al. 2020; Zhou et al. 2020). We display the methylation pattern of the liver-specific gene Spp2 promoter in the comparative browser, and we can see the tissue-specific methylation pattern is conserved between the mouse and cow (Fig. 7A). Thus, the application of the WashU Comparative Epigenome Browser can easily extend beyond traditional model organisms.
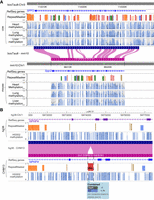
Applying comparative genomic analysis to nonmodel organisms and new genome assemblies. (A) Creating a cattle–mouse comparative browser view and using it to compare DNA methylation in the heart, lung, and liver between the cow and mouse. RefSeq genes and RepeatMasker tracks along with DNA methylation status of the heart, lung, and liver tissues from both the cow and mouse were plotted on the Comparative Epigenome Browser. (B) Using the browser to compare the difference between hg38 and CHM13 and how it may affect genomic analysis. The same HG002 WGBS data were mapped to hg38 and CHM13, respectively. The DNA methylation difference by either genome is a minimum across most of the genomic region, but an Alu insertion is only present in the CHM13 reference, and the hypermethylation of this Alu element can only be assessed using the CHM13 reference. Both methylation percentage and read coverage of each CpG site were annotated within the methylC tracks. All CpG sites are marked by gray, with methylation percentage annotated by the blue bar in the foreground (0% methylated CpGs are displayed as full gray bars, whereas 100% methylated CpGs are displayed as full blue bars). The read coverage over CpG sites across the region is represented by the black line in the background.
Finally, the comparative browser also fulfills a growing need in the field to compare and benchmark the performance of different human genome assemblies (Aganezov et al. 2022). The recent release of the T2T CHM13 genome assembly, as well as multiple alternative human genome assemblies from the Human Pangenome Reference Consortium (Cheng et al. 2021; Ebert et al. 2021; Garg et al. 2021; Porubsky et al. 2021; Jarvis et al. 2022; Wang et al. 2022), represents a major improvement for genomics, but the impact of analyzing functional genomics data using different genome assemblies remains to be evaluated. Our browser supports direct visualization of such evaluations. We mapped the public HG002 WGBS data (Gershman et al. 2022) to both the hg38 and CHM13 reference genomes, and in Figure 7B, we illustrate an Alu insertion present in CHM13 but absent in hg38. In this case, the presence and hypermethylation of the Alu in HG002 are only visible when the reads were mapped to the CHM13 reference genome (Foox et al. 2021; Nurk et al. 2022). Therefore, the WashU Comparative Epigenome Browser provides a near-term, conventional visualization of differential mapping results before the maturation of pangenome graph mapping and subsequent visualization (Miga and Wang 2021; Guarracino et al. 2022; Wang et al. 2022; Hickey et al. 2023; Liao et al. 2023).
Discussion
Here we present the WashU Comparative Epigenome Browser to visualize comparative genomic/epigenomic features. The browser functions may help scientists interested in comparative genomics/epigenomics to examine their regions of interest and produce publication-quality browser views to showcase their findings. In addition to a growing number of genomes, genome-align tracks, and genomics data sets we currently host, users can build and host their own comparative browser with customized species and genome builds. It enables scientists, especially those working on nonmodel organisms, to visualize and compare genomic and epigenomic features of different species.
The comparative browser is made possible by genome alignment tools developed by the community (Schwartz et al. 2003; Li 2018). On the other hand, the comparative function is also limited by the alignment algorithm. Therefore, the comparison between distantly related organisms may not be feasible beyond some ultraconserved genes. The comparative features are fundamentally enabled by the genome-align track, a pairwise genomic alignment track derived from the AXT format (Schwartz et al. 2003). Comparison across multiple genomes is achieved by using multiple genome-align tracks anchoring to the same reference genome. Although it is possible to generalize the comparative functions based on a multigenome alignment, the pairwise comparison is more technically practical and intuitive on a two-dimensional computer screen. Our browser supports all modern web browsers on a MAC, PC, and mobile platform. Displaying multiple tracks, especially Hi-C tracks from more than two species, is resource intensive and could affect the responsiveness depending on local machine configuration and internet speed. We envision continued exploration of advanced web technologies to further enhance the performance of multigenome comparison (Paten et al. 2011).
Methods
Genome-align track
The genome-align track was developed based on the widely used and available pairwise genome alignment AXT format (Schwartz et al. 2003). AXT pairwise genome alignment files were obtained from the UCSC Genome Browser, and they can also be converted from the minimap2 alignment PAF files (Kent et al. 2002; Li 2018, 2021; Lee et al. 2022). We use the paftools.js script from the minimap2 package to convert PAF files to MAF files and use the maf-convert tool from LAST alignment to convert MAF files to AXT files (Kiełbasa et al. 2011; Li 2018, 2021). We use a customized Python script to convert AXT files to genome-align track files. The script is available at GitHub (https://github.com/lidaof/eg-react/blob/master/backend/scripts/axt2align.py). The track file is a BED-like file, where the first three columns represent the reference genome coordinates, and the fourth column contains a JSON string that includes secondary genome coordinates along with the sequences from both assemblies.
The genome-align format is described in https://eg.readthedocs.io/en/latest/tracks.html#genome-align-track, and the JavaScript visualization is implemented by genomeAlignTrack.tsx, available at GitHub (https://github.com/lidaof/eg-react/blob/master/frontend/src/components/trackVis/GenomeAlignTrack.tsx).
Assembly selection widget
We developed an assembly selection widget to streamline the genome-align tracks loading. The widget is available at https://comparativegateway.wustl.edu/start/. It allows users to select one reference genome with one or more secondary genomes and launch a new WashU Epigenome Browser window with desired genome-align tracks loaded. The widget is available at GitHub (https://github.com/debugpoint136/comparative-selection-widget).
Figure data files
The zebrafish liver CpG methylation BED files in Figure 3A were downloaded the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE134055. Mouse liver CpG methylation BED files were downloaded from ENCODE experiment ENCSR733ZTZ. All download methylation BED files from ENCODE were converted to the methylC track files (Yue et al. 2014; Zhou et al. 2014).
All ChIP-seq, RNA-seq, and methylation data in Figure 3B were downloaded from The ENCODE Project (The ENCODE Project Consortium 2012; Yue et al. 2014).
Human and chimpanzee ChIP-seq data from Figures 4A and 5A were downloaded from GEO repository GSE70751 (Prescott et al. 2015). The reads were aligned to human genome hg38 and chimpanzee genome panTro5 using BWA with default parameters as described by Li (2013) and Zhuo et al. (2020).
Human and gorilla Hi-C files in Figure 4B were shared by the courtesy of Jian Ma and Yang Yang from Carnegie Mellon University (Yang et al. 2019).
Hi-C and RNA-seq data of human and marmoset in Figure 5B were downloaded from GEO repository GSE116862 (Zhang et al. 2019).
CTCF ChIP-seq and annotated binding regions in Figure 6 were downloaded from ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) under accession numbers E-MTAB-437 and E-MTAB-424 (Schmidt et al. 2012).
Cow methylation data in Figure 7A were downloaded from GEO repository GSE147087 (Liu et al. 2020; Zhou et al. 2020). The matching mouse methylation data were downloaded from the mouse ENCODE Project (Yue et al. 2014).
HG002 WGBS data were downloaded from open data repository EPI2ME Labs (https://labs.epi2me.io/gm24385-5mc) as described by (Gershman et al. 2022).
All bigWig files and Hi-C files were displayed on the browser directly. Methylation data were converted to the methylC track described by Zhou et al. (2014) for browser visualization.
All figure panels from Figures 3⇑⇑⇑–7, including all the process data files, were organized as browser sessions and saved as JSON format session files (https://comparativegateway.wustl.edu/showcases/). Session files were available for download, and they can be directly loaded into the browser (https://eg.readthedocs.io/en/latest/url.html).
Software availability
Repository
The WashU Comparative Epigenome Browser is available at https://comparativegateway.wustl.edu/. The browser source code is available in the Supplemental Material (Supplemental Code S1). It is also open source available at GitHub (https://github.com/lidaof/eg-react).
We welcome suggestions and requests, including but not limit to bug reports, new genome support suggestions, and new cross-species comparison suggestions. To make new suggestions, please start a new issue in the browser repository (https://github.com/lidaof/eg-react/issues).
Tutorial
The Browser tutorial is available at https://eg.readthedocs.io/en/latest/comparativeBrowser.html, and an accompanied video tutorial is at https://comparativegateway.wustl.edu/tutorials/. To set up a datahub and load multiple tracks together, please follow the datahub tutorial page (https://eg.readthedocs.io/en/latest/datahub.html). A datahub file of each of our showcases is also available for download in the browser showcases page (https://comparativegateway.wustl.edu/showcases/).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Jian Ma and Yang Yang from Carnegie Mellon University for providing the human and gorilla Hi-C data set. This work was funded by National Institutes of Health grant numbers R01HG007175, U01CA200060, U01HG009391, U41HG010972, U24HG012070, UM1HG011585, and UM1MH130994. Both A.Y.D. and J.K.H. were supported by National Human Genome Research Institute training grant T32 HG000045.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277550.122.
-
Freely available online through the Genome Research Open Access option.
- Received November 29, 2022.
- Accepted May 3, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








