
Highly complete long-read genomes reveal pangenomic variation underlying yeast phenotypic diversity
- Cory A. Weller1,8,10,
- Ilya Andreev1,8,11,
- Michael J. Chambers1,8,
- Morgan Park2,
- NISC Comparative Sequencing Program2,9,
- Joshua S. Bloom3,4,5,6,7 and
- Meru J. Sadhu1
- 1Computational and Statistical Genomics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA;
- 2NIH Intramural Sequencing Center, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA;
- 3Department of Human Genetics, University of California, Los Angeles, Los Angeles, California 90095, USA;
- 4Department of Biological Chemistry, University of California, Los Angeles, Los Angeles, California 90095, USA;
- 5Howard Hughes Medical Institute, University of California, Los Angeles, Los Angeles, California 90095, USA;
- 6Institute for Quantitative and Computational Biology, University of California, Los Angeles, Los Angeles, California 90095, USA;
- 7Department of Computational Medicine, University of California, Los Angeles, Los Angeles, California 90095, USA;
- 12NIH Intramural Sequencing Center, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA
-
↵8 These authors contributed equally to this work.
Abstract
Understanding the genetic causes of trait variation is a primary goal of genetic research. One way that individuals can vary genetically is through variable pangenomic genes: genes that are only present in some individuals in a population. The presence or absence of entire genes could have large effects on trait variation. However, variable pangenomic genes can be missed in standard genotyping workflows, owing to reliance on aligning short-read sequencing to reference genomes. A popular method for studying the genetic basis of trait variation is linkage mapping, which identifies quantitative trait loci (QTLs), regions of the genome that harbor causative genetic variants. Large-scale linkage mapping in the budding yeast Saccharomyces cerevisiae has found thousands of QTLs affecting myriad yeast phenotypes. To enable the resolution of QTLs caused by variable pangenomic genes, we used long-read sequencing to generate highly complete de novo genome assemblies of 16 diverse yeast isolates. With these assemblies, we resolved QTLs for growth on maltose, sucrose, raffinose, and oxidative stress to specific genes that are absent from the reference genome but present in the broader yeast population at appreciable frequency. Copies of genes also duplicate onto chromosomes where they are absent in the reference genome, and we found that these copies generate additional QTLs whose resolution requires pangenome characterization. Our findings show the need for highly complete genome assemblies to identify the genetic basis of trait variation.
The individual members of a species will have myriad trait differences, including in disease phenotypes, agricultural traits, traits that are subject to natural or sexual selection, and fundamental biological processes. Trait variation is often caused by genetic differences between individuals, and understanding how genetic variation is tied to phenotypic variation has been a central goal of genetics research since its inception under Gregor Mendel. The budding yeast Saccharomyces cerevisiae is a popular model organism in genetics research. It was the first eukaryotic species to have a sequenced reference genome (Goffeau et al. 1996), and, to date, most genes found in the reference genome have been functionally characterized (Engel et al. 2022). This was aided by the remarkable ease of testing the effects of genetic changes in yeast (Giaever and Nislow 2014), which has accelerated even further with CRISPR technology. Beyond the reference genome, S. cerevisiae presents substantial genetic and phenotypic diversity, making yeast a premier model for understanding the genetic basis of trait differences between diverse individuals. S. cerevisiae has been isolated from around the world and from diverse environments (Peter et al. 2018), including domestic fermentative environments, patient samples, and wild forests (Lee et al. 2022). Whole-genome sequencing of diverse yeast isolates has revealed that many strains’ genomes contain genes not seen in the reference genome. Although the reference genome contains approximately 6000 genes, the “pangenome” composed of all genes in the global yeast population is thought to contain approximately 8000 genes, 3000 of which are variably present across strains (Peter et al. 2018). The presence or absence of variable genes could have large effects on yeast traits. For instance, some strains have extra nonreference genes that allow them to metabolize xylose or melibiose (Naumov et al. 1990; Wenger et al. 2010). Highly diverged alleles of reference genes, as seen for the GAL genes, or copy number variation, as seen for CUP1, can also dramatically affect phenotypes (Zhao et al. 2014; Boocock et al. 2021). But pangenomic variation in strains’ genomes is not well annotated, as it is generally found in repetitive subtelomeric regions that recombine frequently (Louis et al. 1994) and are difficult to genotype (Yue et al. 2017). Thus, the nature of yeast pangenomic variation is still being understood.
Of the thousands of genetic differences spread throughout genomes, including single-nucleotide polymorphisms (SNPs) and variable pangenomic genes, only some affect a given trait of interest. In yeast, an effective strategy to locate relevant genetic differences is linkage mapping on a biparental cross (Ehrenreich et al. 2009). Two haploid yeast strains are mated to produce a diploid strain, which undergoes meiosis to produce haploid progeny strains known as segregants. Researchers determine genotypes and phenotypes for hundreds to thousands of segregants and then identify genomic loci in which segregants with one parental genotype have a significantly different phenotype from those with the other genotype. These mapped loci, known as quantitative trait loci (QTLs), are usually several kilobases in size and encompass multiple genes owing to genetic linkage between causal variants and neighboring variants, requiring further study to identify the causal variants. QTLs can be mapped even if the parental genome sequences are not completely known, if genetic variants linked to the causal variant are genotyped. Complete genome sequences are desired, however, for confidently identifying QTLs’ causal genetic variants. To date, yeast genome sequences have predominantly been assembled from high-throughput short-read DNA sequencing, using millions of reads <600 bp long (Peter et al. 2018). The resulting inferred genome sequences are often fragmented and incomplete, with particular assembly challenges arising from repetitive sequences and structural variants (SVs) such as translocations, copy number variants, and pangenomic insertions and deletions. In contrast, the advent of long-read sequencing, which generates reads of ≥10 kb (Koren and Phillippy 2015), allows for highly complete yeast genome assembly, as repetitive or diverged DNA sequences are generally short enough that a 10-kb-long read can span them completely (Istace et al. 2017; Yue et al. 2017; Bendixsen et al. 2021; Lee et al. 2022; O'Donnell et al. 2022; Saada et al. 2022). Highly complete genome sequences can significantly aid the resolution of QTLs to their true underlying causal genetic factors, especially those caused by pangenomic, repetitive, or structural variation, as seen for humans, rice, and the tomato (Qin et al. 2021; Wu et al. 2022; Zhou et al. 2022).
A recent study performed linkage mapping on crosses between 16 diverse yeast strains (Bloom et al. 2019). Each parent strain was crossed to two other parent strains, for 16 total crosses. Approximately 900 segregants were isolated for each cross, and the segregants’ growth was measured in 38 environments, including different nutritional sources, treatment with toxic compounds, and temperature and pH stress. Across the 16 biparental crosses and 38 traits, Bloom et al. (2019) mapped 7751 QTLs with a median size of 31.7 kb (12.7 QTLs per cross per trait), which were responsible for most of the genetic component of the phenotypic variance. Here, we investigated the contribution of pangenomic variation to growth on maltose, oxidative stress, sucrose, and raffinose using highly complete genome sequences generated with long-read sequencing technology for the 16 parental strains.
Results
Long-read genome sequences
To determine their pangenome content, we generated de novo genome assemblies for the 16 QTL-mapped strains using Pacific Biosciences (PacBio) HiFi technology, which produces reads ∼10–20 kb in length with very high sequence accuracy (Wenger et al. 2019). The final assemblies all contained precisely 17 contigs that corresponded to 16 nuclear chromosomes and the mitochondrial DNA (Supplemental Table S1). For 11 strains, every chromosome sequence started and ended with telomeric TG1–3 repeats, and across the 16 strains, 505 of 512 nonmitochondrial contig ends had either TG1–3 repeats or telomere-associated Y′ or X elements (Fig. 1A; Wellinger and Zakian 2012), indicating that the genome assemblies were largely or entirely complete.
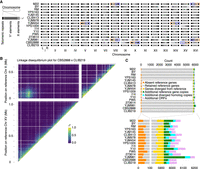
Long-read genomes. (A) Detection of telomeric TG1–3 repeats and telomere-associated Y′ or X element sequences at ends of assembled contigs. TG1–3 repeats were detected as more than 20 TG dinucleotides in the final 100 bp of a contig, whereas Y′ and X elements were detected by Liftoff. Dark, medium, and light gray shadings indicate the presence of TG1–3 repeats, Y′ elements, or X elements on a contig end, respectively, whereas white indicates the feature was not detected. Orange and purple highlighting mark chromosome arms affected by reciprocal translocations. (B) Linkage analysis of markers on reference Chr X and XV in the CBS2888 × CLIB219 cross. The existence of strong cross-chromosome correlation between Chr X and XV markers is consistent with a translocation between these chromosomes in strain CBS2888. (C) Gene and ORF content in assembled genomes. Genes and gene copies with >30% sequence identity to reference genes were classified as retained reference genes or diverged homologs using a threshold of 95% sequence identity to the reference gene.
Genome assemblies of four strains had major chromosomal rearrangements relative to the reference genome (Fig. 1A; Supplemental Fig. S1). Three of these strains’ reciprocal translocations were previously known: Chromosomes (Chr) VIII and XVI in strains M22 and YJM981 and Chr V and XIV in strain YJM454 (Hou et al. 2014). As seen by Hou et al. (2014), the translocation between Chr VIII and XVI led to unequal transmission of alleles to segregants (Supplemental Fig. S2). We also detected a reciprocal translocation between Chr X and XV in strain CBS2888, which has not been described previously. We checked for evidence of this translocation in the genotypes of the biparental cross between CBS2888 and CLIB219 generated by Bloom et al. (2019), which resulted in 943 recombinant haploid progeny. In these segregants, we observed linkage between genetic markers corresponding to reference Chr X and XV, confirming the observed translocation in CBS2888 (Fig. 1B) and indicating our assembled genomes reflect the true chromosomal architecture. We annotated SVs >500 bp in the 16 genomes using MUMandCo (O'Donnell and Fischer 2020). The most common SVs were deletion and insertion of repetitive sequences (Supplemental Fig. S1). Of interest, we detected the recently described recurrent inversion of 24 kb on Chr XIV (Salzberg et al. 2022); two strains had the reference orientation, and 14 strains had the inversion. We also detected a novel tandem repeat specific to strain YJM454 that amplified the uncharacterized open reading frame (ORF) YGR201C to 27 copies (Supplemental Fig. S3). YGR201C is predicted to be the sole eEF1Bγ gene family member without a C-terminal domain in the S. cerevisiae reference genome, but up to 15 have been found in other fungal species’ genomes (Renou et al. 2022); their function remains unknown. We detected an enrichment of SVs at chromosome ends (Supplemental Fig. S4).
We annotated the genes present in the assembled genomes using a combined strategy of finding genes that have homology with existing gene annotations in the S288C reference genome and of predicting ORFs de novo. The annotation tool Liftoff (Shumate and Salzberg 2021) was used to annotate genes that have sequence homology with known genes in the reference genome. To annotate any genes missed by Liftoff, we used ORFfinder (Sayers et al. 2011) to annotate ORFs longer than 100 codons. Gene identity and content varied widely among the 16 strains. Up to 96 reference genes per strain were replaced by diverged homologs, which could reflect introgressions from other species or balancing selection within S. cerevisiae (D'Angiolo et al. 2020; Boocock et al. 2021), and up to 84 reference genes were absent without replacement (Fig. 1C). We found additional copies of reference genes, with 190 in strain YJM981 owing to its highly repetitive subtelomeres, which are thought to reflect a deficiency in telomere maintenance (Bergström et al. 2014). We found up to 47 additional distant homologs of reference genes and 73 ORFs that did not have homology with reference genes per strain. Variable genes were strongly enriched at subtelomeres (Supplemental Fig. S4), as seen previously (Yue et al. 2017). Encouragingly, the strain BY had very few differences from the reference genome, as expected owing to its close relation to S288C, the reference genome strain (Brachmann et al. 1998). The most major difference arose from the BY genome assembly containing nine copies of the CUP1 tandem repeat locus (Supplemental Fig. S3), which is represented in condensed form in the reference genome with two copies (Zhao et al. 2014). One hundred sixty-six nonreference genes from the yeast pangenome determined from 1011 strains (Peter et al. 2018) were present in our 16 genomes (Supplemental Fig. S5), representing ∼10% of the nonreference pangenome with <2% of the strains.
To determine how differences in pangenome content between strains contribute to trait variation, we analyzed our annotated genomes in conjunction with previously mapped growth QTLs (Bloom et al. 2019). As variable pangenomic genes were predominantly localized at subtelomeres (Supplemental Fig. S4), we noted with interest that growth on maltose, paraquat, sucrose, and raffinose each had multiple large-effect subtelomeric QTLs. Below, we investigated how pangenomic variation contributes to these traits.
Highly variable maltose loci
Maltose is a disaccharide composed of two glucose molecules. It is a major component of brewer's wort (He et al. 2014) and is fermented by yeast to produce beer. Metabolism of maltose in S. cerevisiae involves genes from the MALR, MALT, and MALS gene families, which encode transcription factors, maltose transporters, and maltase enzymes, respectively (Brown et al. 2010). MALR transcription factors can activate MALT and MALS expression upon detection of maltose. Genes from these families are typically found clustered together in the genome to form MAL loci, often alongside IMA genes that encode enzymes that digest isomaltose (Teste et al. 2010). Previous analysis has found MAL loci variably present on five chromosomes (Charron et al. 1989); for instance, the reference genome encodes MAL loci on Chr II and VII.
Bloom et al. (2019) mapped QTLs for maltose growth in the 16-isolate panel. A recurrent QTL at the right end of Chr VII was the primary contributor to variable maltose growth (Fig. 2A), appearing in 12 of the 16 crosses with LOD scores from 38 to 173. As this coincides with the position of a MAL locus in the reference genome, we hypothesized that these QTLs could result from variation in MALR, MALT, or MALS. We examined the MAL genes on Chr VII across our 16 high-quality genomes. In contrast to the reference genome, which has a single copy of each gene, the size of the Chr VII MAL locus varied from two to 10 MAL and IMA genes, with zero to four MALRs, one to three MALTs, zero or one MALSs, and zero to two IMAs (Fig. 2B). This variation was not because of recent gene duplication events, as homologs had low sequence identity to each other. For instance, no pair of Chr VII MALRs in a strain had >85% DNA identity (Fig. 2C). Whereas diverse MALR genes have been found previously on different chromosomes (Charron et al. 1989), we found that Chr VII alone harbored homologs of all previously identified MALR genes except MAL33 and YPR196W (Fig. 2C). We checked the placement of these genes on Chr VII using linkage mapping genotype data. We observed that segregant genotypes on the right arm of Chr VII were strongly correlated with the presence of short reads matching variable MALRs, confirming they reside on Chr VII (Supplemental Figs. S6–S8). Similar to MALR diversity, we observe up to three distinct MALT genes in a single MAL cluster, which correspond to three distinct maltose transporters that have been previously characterized in S. cerevisiae (Han et al. 1995; Dietvorst et al. 2005).
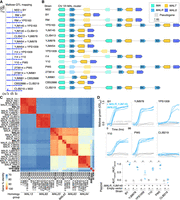
Diverse, highly complex MAL loci on Chr VII. (A) QTL mapping of maltose growth in 16 biparental S. cerevisiae crosses (Bloom et al. 2019). Each plot shows linkage (calculated logarithm of the odds [LOD] score), in one biparental cross, between segregant genotype and growth on 2% maltose, plotted against genomic coordinates. Twelve of 16 crosses have a QTL with LOD score greater than 35 at the right end of Chr VII, highlighted in peach; plots are labeled with which parent provided the Chr VII allele associated with superior maltose growth, if any. (B) Diagram of the Chr VII MAL locus in each strain. MALR genes are labeled by homology group, as determined in panel C. (C) DNA percentage identity between MALR genes found on Chr VII (McWilliam et al. 2013). Previously identified MALR genes are included for reference, in bold (Gibson et al. 1997). MALRs with >97% sequence identity are sorted into homology groups, labeled at the bottom. As seen by Song et al. (2015), we observed a homology group of MALRs with low sequence identity to any named MALR, which we labeled MAL83 (assigned systematic name YSC0072 at the Saccharomyces Genome Database [SGD; https://www.yeastgenome.org/]). (D, top) Maltose growth for maltose-poor strains BY, YJM978, YPS1009, Y10, PW5, and CLIB219 with plasmids carrying MALRYJM145 (blue) or empty vector (gray). n = 5 biological replicates, except for YJM978 + empty vector, which had four biological replicates. (Bottom) Growth was quantified as the area under the growth curve (AUC) for maltose normalized to the AUC for glucose (Supplemental Fig. S10). Maltose growth was significantly higher with MALRYJM145 than empty vector for strains BY, YJM978, YPS1009, Y10, and PW5. (*) P < 0.005, Tukey's HSD following one-way ANOVA.
Previous studies on diverse MALRs found they differ in which sugars they sense, and only some support growth on maltose. MAL23, MAL43, and MAL63 support growth on maltose, whereas MAL13, MAL33, MAL64, and YPR196W do not (Gibson et al. 1997; Brown et al. 2010). In contrast, the three characterized S. cerevisiae MALT genes all transport maltose (Han et al. 1995; Dietvorst et al. 2005). To test whether Chr VII MAL genotypes associated with poor maltose growth lacked a maltose-proficient MALR, we introduced MALRYJM145, which was the sole Chr VII MALR in maltose-proficient strain YJM145 and has 99% protein identity with the known maltose-proficient Mal63, into six maltose-poor strains using a CEN plasmid. MALRYJM145 significantly improved maltose growth for five strains (Fig. 2D). For one of these strains, YPS1009, the improved maltose growth was still slow, likely because its sole maltose transporter is a homolog of MTY1 (Supplemental Fig. S9), which has low maltose affinity (Salema-Oom et al. 2005). The remaining strain, CLIB219, lacked MALS (Fig. 2B); segregants of the CLIB219 × M22 cross grew on maltose if they inherited the MALS gene on M22's Chr II (Supplemental Fig. S9). Thus, variation in all three MAL genes affected strains’ ability to grow on maltose, with a major contribution from the presence or absence of maltose-proficient MALRs.
The RM and CLIB413 genome assemblies contained MAL genes in the subtelomeric region on the right end of Chr XI (Fig. 3A,B), which we confirmed by linkage analysis (Supplemental Figs. S11, S12). We hypothesized these MAL loci would benefit maltose growth, as the RM and CLIB413 Chr XI MALR genes encoded chimeras between Mal23 and Mal43 (Fig. 3C), which are maltose-proficient (Gibson et al. 1997). Indeed, all crosses involving RM and CLIB413 had maltose QTLs at the right end of Chr XI, with segregants that inherited MALR on Chr XI having superior maltose growth (Figs. 2A, 3D,E), as previously observed for the BY × RM cross (Bloom et al. 2013). As maltose-proficient MALR was sufficient for BY and YJM978 maltose growth (Fig. 2D), we concluded the extra MALRs caused the Chr XI maltose QTLs. Notably, our high-quality genome assemblies allowed us to dissect QTLs to causal genes that are entirely absent from Chr XI in the reference genome.
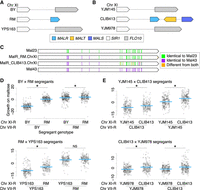
Maltose QTLs on Chr XI are caused by nonreference MAL loci. (A,B) Diagrams of MAL loci on Chr XI of RM and CLIB413, respectively, along with the MAL-less loci in the strains they were crossed to for QTL mapping. (C) Amino acid sequence comparison of Chr XI MALRs from RM and CLIB413 to Mal23 and Mal43. (D,E) Scatterplots of maltose growth of segregants from crosses involving strains RM and CLIB413, respectively, partitioned by segregant genotype at Chr XI and VII. Colony radiuses were measured by Bloom et al. (2019) after 48-h growth on maltose plates, normalized to colony radiuses on glucose plates, and then scaled to have a mean of zero and a variance of one. The mean phenotype of each genotypic grouping of segregants is shown with a blue line. (NS) Not significant, (*) P < 0.05, one-tailed Student's t-test, with the alternative hypothesis that the extra MAL genes improve maltose growth.
SGE1 copy-number variation affects paraquat resistance
Paraquat, a widely used herbicide, is a cationic small molecule that catalyzes the formation of reactive oxygen species, especially superoxide, from cellular electron donors (Fukushima et al. 2002). To explore natural variation in oxidative stress tolerance, Bloom et al. (2019) performed QTL mapping for paraquat resistance in the 16 strains. Eleven crosses had a paraquat-resistance QTL at the right end of Chr XVI with LOD score greater than 10, which was the strongest paraquat QTL for six crosses (Fig. 4A). The Chr XVI subtelomeric region contains SGE1, encoding a transporter that extrudes cationic compounds. Natural variation in SGE1 underlies variation in tolerance of ionic liquids used in biofuel production (Higgins et al. 2018). Genome-wide screens have found that SGE1 overexpression confers paraquat resistance (Österberg et al. 2006) and SGE1 deletion causes paraquat sensitivity (Helsen et al. 2020). In our genome sequences, we observed that paraquat-sensitive Chr XVI loci had one of three SGE1 alleles: SGE1-P282S/L284S, SGE1-D429E, or SGE1-E332Q/K467E (Fig. 4B). We tested the effect of these three SGE1 alleles on paraquat resistance in BY sge1Δ, along with the ancestral allele (Fig. 4C). Ancestral SGE1 provided the most paraquat resistance, SGE1-D429E provided intermediate resistance, and SGE1-P282S/L284S and SGE1-E332Q/K467E were indistinguishable from empty vector (Fig. 4C). Thus, we concluded that single-nucleotide variation in SGE1 is a major contributor to differences in paraquat resistance.
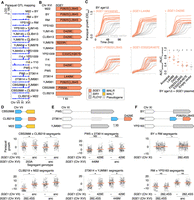
Diversity in SGE1 underlies widespread variation in paraquat tolerance. (A) QTL mapping of growth on 0.75 mM paraquat (Bloom et al. 2019), as in Figure 2A. The site of a recurrent QTL on Chr XVI is highlighted in peach. (B) Diagram of the SGE1 alleles on Chr XVI, with derived nonsynonymous variants labeled. (C) Growth over time in liquid YPD media with 3.5 mM paraquat for BY sge1Δ with plasmids carrying SGE1 from strains BY, YJM145, YPS1009, I14, or 273614 (orange) or empty vector (gray). n = 10 biological replicates. The last panel quantifies strain growth as the AUC for growth in paraquat normalized to the AUC for growth without paraquat (Supplemental Fig. S13). SGE1 alleles from strains BY, YJM145, and YPS1009 provided significantly less protection than the ancestral allele from strain I14. (*) P < 0.005, Tukey's HSD following one-way ANOVA. In addition, SGE1 alleles from strains YJM145, I14, and 273614 provided significantly greater protection than empty vector ([†] P < 0.001, Tukey's HSD), whereas alleles from strains BY and YPS1009 did not. (D–F) Additional SGE1 genes were found in strains CLIB219, 273614, and RM, respectively. Scatterplots below show growth on paraquat for segregants in crosses involving those strains, partitioned by SGE1 genotype. Plots are shown as in Figure 3, with normalization to growth on plates without paraquat. Mean phenotypes are shown with orange lines. (*) P < 0.05, one-tailed Student's t-test, with the alternative hypothesis that the extra SGE1 copy improves growth on paraquat.
Though the reference genome only has SGE1 on Chr XVI, we found extra copies of SGE1 on other chromosomes in three strains: RM, 273614, and CLIB219 (Fig. 4D–F). Linkage analysis indicated these SGE1 copies were correctly placed (Supplemental Figs. S14–S16). We examined the paraquat growth of segregants in crosses involving these strains. Segregants with the extra ancestral SGE1 on Chr VII from CLIB219 had substantially improved paraquat growth (Fig. 4D). Segregants with the extra SGE1-D429E on Chr XI from strain 273614 had moderately improved growth, especially if their Chr XVI locus had the inferior SGE1-P282S/L284S allele from YJM981 (Fig. 4E). The extra SGE1-P282S/L284S on Chr XI from strain RM corresponded to slightly improved growth in the BY × RM cross and no detectable benefit in the RM × YPS163 cross (Fig. 4F). These results mirrored our SGE1 allele replacement results, in which the ancestral SGE1 was best, SGE1-D429E was moderate, and SGE1-P282S/L284S was poor. Thus, we concluded that, through duplication, SGE1 causes additional QTLs on chromosomes other than where it is normally known to reside. As with the maltose Chr XI QTLs, resolving these QTLs would not have been possible without characterization of pangenomic variation. Furthermore, long-read sequencing facilitated correct genotyping of SGE1 SNPs, a prerequisite for QTL resolution, in strains with SGE1 copies on multiple chromosomes.
SUC gene content affects sucrose and raffinose growth
Sucrose, a disaccharide of glucose and fructose, is a common plant sugar found in fruits and tree sap (Liu et al. 2006; Fink et al. 2018). In yeast, sucrose is hydrolyzed by the enzyme invertase (Marques et al. 2016) to produce glucose and fructose, which then enter standard carbon metabolism pathways. Invertase is encoded in the S. cerevisiae reference genome by SUC2 on Chr IX. Some strains carry additional nonreference subtelomeric SUC loci; nine have been identified (Carlson et al. 1985; Naumov and Naumova 2010a,b). We found subtelomeric SUC genes on eight chromosomes (Fig. 5A), including three novel SUC loci, supported by linkage data (Supplemental Figs. S17–S19). The subtelomeric SUC genes had high sequence identity to each other and low sequence identity to SUC2 (Fig. 5B; Naumova et al. 2014), indicating they recently spread across yeast chromosomes through inter-telomeric recombination (Carlson et al. 1985) long after diverging from SUC2. There was likely recent recombination between subtelomeric SUC genes and SUC2, as the predominant subtelomeric SUC allele had alternating regions of low and high sequence identity to SUC2 (Fig. 5C), and some strains had chimeric SUC genes between the two (Fig. 5B,D). SUC2 and the subtelomeric SUC progenitor may have split before S. cerevisiae’s speciation, as their highest diverged regions are more dissimilar than SUC2 from S. cerevisiae and its sister species S. paradoxus (Fig. 5C), although subtelomeric SUC genes have not been found in S. paradoxus (Bozdag and Greig 2014; Naumova et al. 2014; Yue et al. 2017).
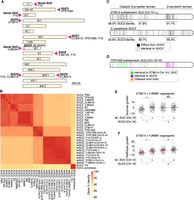
Subtelomeric copies of genes encoding invertase. (A) Invertase genes in the 16 genomes, with subtelomeric SUC loci and the SUC2 locus marked with wedges and a vertical bar, respectively. The SUC9 locus on Chr XIV (Naumov and Naumova 2010b) was not mapped to a specific arm, so we labeled both arms as SUC9. We did not observe SUC genes on Chr VIII, XIII, or XVI (SUC7, SUC4, and SUC10, respectively) (Naumova et al. 2014). (B) Gene percentage identity between SUC genes. (C) Gene sequence comparison of SUC2 from strain BY to the representative subtelomeric SUC gene from strain 273614 (top) and SUC2 from S. paradoxus (bottom). (D) Gene sequence comparison of the Chr VII SUC gene from strain YPS1009 to the subtelomeric SUC gene from strain 273614 and SUC2 from strain BY. (E,F) Scatterplots of growth on 2% sucrose or raffinose, respectively, of segregants of the 273614 × YJM981 cross, partitioned by SUC genotype at Chr IV and IX, as in Figure 3. Mean phenotypes are shown with magenta lines. (*) P < 0.05, one-tailed Student's t-test, with the alternative hypothesis that the extra SUC copy improves growth.
Previous studies have found that subtelomeric SUC genes allow strains lacking SUC2 to grow on sucrose (Carlson and Botstein 1983), which we also observe. Strain YJM981 has a natural frameshift loss-of-function mutation in SUC2, whereas strain 273614 has both a functional SUC2 and a subtelomeric SUC on Chr IV. In the 273614 × YJM981 cross, segregants with YJM981's suc2-fs allele grew substantially better on sucrose if they inherited the Chr IV subtelomeric SUC from strain 273614 (Fig. 5E). In contrast, segregants with the functional SUC2 allele from strain 273614 had no detectable boost to their sucrose growth from the Chr IV subtelomeric SUC. This was intriguing, as it suggested that although subtelomeric SUCs have maintained invertase function, growth on sucrose may not select for that function for the majority of the yeast population carrying functional SUC2.
Invertase also digests other sugars, which we conjectured could have selected for subtelomeric SUC gene invertase activity. Raffinose is a trisaccharide composed of galactose, glucose, and fructose. Invertase hydrolyzes raffinose to produce fructose and melibiose, a disaccharide of glucose and galactose. Invertase levels are more likely to be limiting for growth in raffinose than sucrose: Invertase has lower activity on raffinose (Gascón et al. 1968; Sainz-Polo et al. 2013), and raffinose hydrolysis produces less energy than sucrose, as most yeast strains can only use the resulting fructose and not the melibiose. Indeed, in contrast to sucrose, segregants with functional SUC2 in the 273614 × YJM981 cross grew significantly better on raffinose with the additional subtelomeric SUC gene (Fig. 5F). Thus, the extra invertase activity provided by subtelomeric SUC genes may be particularly beneficial in complex sugar environments.
Discussion
There has been increasing interest in describing species genetically not only through a single reference genome but with a population-wide pangenome that accounts for genes variably present in individuals. We used long-read sequencing to generate highly complete genome assemblies for 16 diverse strains of S. cerevisiae and to annotate pangenomic variation. To ascertain the effects of the uncovered genetic variation, we combined our long-read-derived genome sequences with linkage mapping data and identified QTLs caused by variable pangenomic genes. By leveraging linkage mapping data, we were also able to validate the long-read genome sequences, confirming a translocation and the placement of variable pangenomic genes through linkage to nearby genetic markers. Thus, combining linkage mapping and long-read sequencing strengthens our understanding of both the sequenced genomes and mapped QTLs.
In budding yeast, variable pangenomic genes are mostly found at subtelomeric locations (Yue et al. 2017). This could be related to yeast subtelomeres recombining between chromosomes. Inter-chromosomal recombination can lead to addition or loss of genes in the daughter cells following cell division (Louis et al. 1994). The genes we examined here, MALR, SGE1, and SUC, are all subtelomeric. Consistent with the subtelomeres experiencing inter-chromosomal recombination, copies of all three genes were found at the end of more than one chromosome. In some cases, we found identical gene copies on different chromosomes, suggesting their recombination events occurred recently. For SGE1, three distinct alleles seen on Chr XVI are also seen on other chromosomes, implying three independent recombination events, further underscoring the frequency of inter-chromosomal recombination.
In addition to subtelomeres, pangenomic variation is occasionally found at interior chromosomal regions in S. cerevisiae. We searched genome-wide for recurrent large-effect QTLs whose confidence intervals contained structural variation >250 bp (Supplemental Table S2). Although most recovered loci were subtelomeric, we also identified previously known copy number variants in CUP1 and the ENA locus, affecting copper and lithium sensitivity, respectively (Dunn et al. 2012; Zhao et al. 2014). Thus, interior loci can also harbor recurrent and significant trait-affecting pangenomic variation.
Increased gene dosage of limiting gene products can increase fitness, and we observed that increasing from one functional gene to two improved fitness through MALR for maltose, SGE1 for paraquat, and SUC for raffinose. The variable presence of highly identical sequences on different chromosomes is particularly challenging to detect using reference genomes and short-read genotyping. Correctly determining the genomic locations of variable genes is especially important for resolving the genetic basis of mapped QTLs, which requires precise knowledge of the sequence differences in the QTL. QTLs were caused by genes in locations absent from the reference genome for all three traits examined here.
We predict that variable pangenomic genes have many more effects on yeast phenotypic diversity than the traits we studied. The variable component of the yeast pangenome comprises thousands of genes (Peter et al. 2018), and each of those genes may contribute to very particular traits, such as metabolism of specific nutrients or growth in specific environments. For instance, several homologs of MALR were widespread in our 16 genomes but did not support growth on maltose, including a previously uncharacterized MALR we termed MAL83, raising the possibility that they function in a different sugar environment. With the variable pangenomic content annotated here for a large, genotyped mapping population, studying a wide variety of phenotypes in the panel's segregants could reveal the phenotypic effects of many other pangenomic genes in these genomes.
The design of the linkage mapping population, in which all individuals were direct progeny of 16 parental strains, meant that generating high-quality genome assemblies for just the parental strains was sufficient for resolving QTLs to specific pangenomic variation. This strategy of generating high-quality genome assemblies of select individuals can be used for linkage mapping studies on any group of closely related individuals, such as family studies in humans. As the cost and analytical constraints of generating long-read genome assemblies continues to decline (Jarvis et al. 2022), it will become possible to generate high-confidence de novo whole-genome sequences of every individual in a mapping study, including genome-wide association studies of unrelated humans. We expect that assembly of highly complete genomes using long-read sequencing will be vital for us to fully understand the genetic sources of trait variation.
Methods
Genome sequencing and assembly
DNA was isolated from the 16 parental strains using the Masterpure yeast DNA purification kit (Lucigen) and sequenced on a Sequel II sequencer (PacBio) to produce circular consensus sequence (CCS) reads, as detailed in the Supplemental Methods.
CCS reads from each sample were assembled using Canu v. 2.2 (parameters genomeSize = 12mb -pacbio-hifi) (Koren et al. 2017). When read coverage was greater than 50×, reads were down-sampled to 50× coverage using seqtk v. 1.2 before assembly (https://github.com/lh3/seqtk). Redundant contigs were identified from each initial draft assembly using purge_dups v. 1.0.1 (Guan et al. 2020). Mitochondrial contig sequences were visualized and manually circularized using Gepard v. 1.30 (Krumsiek et al. 2007). Further refinement of the contig sequences involved comparing the draft contigs against the S. cerevisiae S288C reference genome (ASM205763v1) using BLASTN (v. 2.2.24) to identify contigs representing telomeric ends that were not incorporated into the corresponding chromosome and overlapping chromosome contigs, particularly the two contigs representing Chr XII that were split at the rDNA cluster. Overlapping contigs were visualized using Gepard to identify the overlapping regions and manually joined using exported trimmed sequences. The structure of the rDNA cluster in the joined Chr XII contigs represents a collapsed version of this extended repeat region.
Genome annotation
All code and input data for performing genome annotation are hosted at Zenodo (http://doi.org/10.5281/zenodo.7846378). Genes were annotated in the strain FASTA files using a combined strategy of lifting over gene annotations from the reference S. cerevisiae genome and annotating ORFs de novo. For lifting over reference annotations, we used Liftoff v. 1.6.1 (Shumate and Salzberg 2021) with the parameters -p 4 -infer_transcripts -copies -s 0.3 -sc 0.3. We chose Liftoff owing to its process of annotating the products of a gene duplication as either original or copy genes. Reference annotations were downloaded from https://downloads.yeastgenome.org/latest/saccharomyces_cerevisiae.gff.gz on December 1, 2021.
To annotate ORFs de novo, we used ORFfinder v. 0.4.3 with the parameters -outfmt = 0 -ml 210. ORFs were checked for homology with reference genes using BLASTN. We removed highly repetitive ORFs, identified with TRF v. 4.09.1 (Benson 1999). Using custom R scripts (R. v. 4.1.2) (R Core Team 2023), ORFs were required to be >300 bp long, to begin with ATG, and to not overlap other genomic features (genes, transposable elements, centromeres, tRNAs, subtelomeric X and Y′ elements, and autonomously replicating sequences), which were annotated using Liftoff, and other ORFs.
Validation of assemblies using genetic maps
For a given cross, we used MUMmer (v. 4.0.0) (Marçais et al. 2018) to generate an alignment between chromosomes of interest from the two parent genome sequences and to call SNPs. Using custom scripts, we generated a final reference sequence that only included sites of SNPs at least 20 bp from an alignment mismatch, with invariant or poorly aligned regions replaced with Ns. Illumina short reads of the segregants, obtained from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (study SRP201965), were aligned with BWA-MEM (v. 0.7.17) (Li and Durbin 2009), and SNPs called with BCFtools (v. 1.16) (Li 2011). We called segregant genotypes in 1-kb windows, assigning parentage if at least 90% of SNPs agreed on parentage (otherwise, assigned N). Linkage disequilibrium and association mapping were performed with PLINK (v. 1.9) (Chang et al. 2015).
To test whether gene sequences of interest were assembled onto correct chromosomes, we performed linkage mapping in segregants of biparental crosses predicted to be heterozygous for the sequence. For each segregant, we quantified Illumina reads per million (RPM) matching the sequence using BWA with strict matching parameters: -B 40 -O 60 -E 10 -L 100. Linkage mapping was performed using segregant RPM as a quantitative phenotype.
Yeast growth assays
Strains, plasmids, and oligonucleotides are listed in Supplemental Tables S3, S4, and S5, respectively, with construction detailed in the Supplemental Methods. Plasmids carrying the MALR and SGE1 alleles were generated by Gibson assembly (New England Biolabs [NEB]) (Gibson et al. 2009). A lithium acetate transformation protocol (Becker and Lundblad 2001) was used to transform yeast strains with plasmids. Strain growth curves were measured in 96-well plates (Corning 3370) over 3–5 d on a SPECTROstar Omega microplate reader equipped with a plate stacker (BMG Labtech) at room temperature with 30 s of shaking at 600 RPM before each plate reading. Sample growth was quantified as the ratio of the AUC in maltose or paraquat media to the AUC in glucose media. Statistical analysis was performed using one-way ANOVA followed by Tukey's post-hoc HSD test.
Data access
The genome assembly data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA779940 (see Supplemental Table S1). Sequences of MAL83 generated in this study have been submitted to the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers OR078935–OR078941. The code used to generate our analyses is available as Supplemental Code, and code and data are available as a snapshot of our Git repository hosted on Zenodo (http://doi.org/10.5281/zenodo.7846378).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Arang Rhie, Sergey Koren, Adam Phillippy, Bill Pavan, David Bodine, Frank Albert, and all members of the Sadhu laboratory for helpful discussion. We thank Trey Sato, Leonid Kruglyak, and Fred Cross for strains and plasmids. This work used the computational resources of the NIH High Performance Computing Biowulf cluster. This work was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (1ZIAHG200401, 1ZIBHG000196). J.S.B. was supported by the Howard Hughes Medical Institute.
Author contributions: M.J.S. conceived and supervised the project. J.S.B. and M.J.S. provided resources. I.A., C.A.W., M.J.C., and M.J.S. performed investigation and formal analysis. M.P. and the NISC Comparative Sequencing Program sequenced and assembled yeast genomes. M.J.S. wrote the original draft; all authors reviewed and edited the manuscript and approved the final version.
Footnotes
-
↵9 A complete list of NISC Comparative Sequencing Program members appears at the end of this paper.
-
↵Present addresses: 10Center for Alzheimer's and Related Dementias, National Institutes of Health, Bethesda, MD 20892, USA;
-
↵11 Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277515.122.
- Received November 16, 2022.
- Accepted April 26, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








