
Plant polymerase IV sensitizes chromatin through histone modifications to preclude spread of silencing into protein-coding domains
Abstract
Across eukaryotes, gene regulation is manifested via chromatin states roughly distinguished as heterochromatin and euchromatin. The establishment, maintenance, and modulation of the chromatin states is mediated using several factors including chromatin modifiers. However, factors that avoid the intrusion of silencing signals into protein-coding genes are poorly understood. Here we show that a plant specific paralog of RNA polymerase (Pol) II, named Pol IV, is involved in avoidance of facultative heterochromatic marks in protein-coding genes, in addition to its well-established functions in silencing repeats and transposons. In its absence, H3K27 trimethylation (me3) mark intruded the protein-coding genes, more profoundly in genes embedded with repeats. In a subset of genes, spurious transcriptional activity resulted in small(s) RNA production, leading to post-transcriptional gene silencing. We show that such effects are significantly pronounced in rice, a plant with a larger genome with distributed heterochromatin compared with Arabidopsis. Our results indicate the division of labor among plant-specific polymerases, not just in establishing effective silencing via sRNAs and DNA methylation but also in influencing chromatin boundaries.
The chromatin is decorated with modifications to DNA and histones constituting epigenetic modifications (Law and Jacobsen 2010; Feng and Michaels 2015). The proportion of genomic regions coding for proteins called euchromatin reduces with an increase in genome size across organisms, and such alterations and genome expansion are mainly owing to the proliferation of repeats and transposons (Pellicer et al. 2018). Plant genomes are especially enriched with latent transposons poised for transcription located proximal to the protein-coding genes (PCGs) in the euchromatic domains (Hirsch and Springer 2017).
The activity of RNA polymerase II (Pol II) on the euchromatic domains is regulated locally by transcription factors, chaperones, and chromatin remodeling enzymes that transduces the local epigenetic status (Hahn 2004; Gibney and Nolan 2010; Schier and Taatjes 2020). The evolution of plants with increased instances of gene-proximal repeats in the genome necessitates articulation of silencing states to the Pol II with precision. This locally silenced state is called facultative heterochromatin and is enriched with Polycomb Repressive Complex (PRC)–dependent H3K27me3 marks in contrast to the constitutive heterochromatin enriched with H3K9me2 modifications (Zhang et al. 2007). Studies in early land plant genomes like that of Marchantia polymorpha suggest that H3K27me3 evolved as a predominant silencing mark in plants (Montgomery et al. 2020), and H3K9me2 mark took over the constitutive silencing, leading to the observed dichotomy between facultative and constitutive heterochromatin (Déléris et al. 2021). Mutants of MET1, a major player in constitutive heterochromatin establishment, showed compensation by H3K27me3 marks at repeats and transposons (Soppe et al. 2002; Mathieu et al. 2005; Deleris et al. 2012; Rougée et al. 2021; Zhao et al. 2022), indicating that unknown players monitor heterochromatin states in specific domains to initiate compensatory marks in constitutive heterochromatic regions. Demarcation of facultative and constitutive heterochromatic boundaries is paramount in avoiding the intrusion of silencing states into neighboring PCGs, warranting the evolution of novel machineries curtailing silencing overshoot and simultaneously defending against genotoxic repeats. Although these mechanisms have been envisaged across eukaryotes, major upstream players involved in these beyond the obvious epigenetic readers, writers, and erasers are unknown.
Plants have evolved RNA silencing as an efficient mode of robust and targeted silencing of repeats and genes, at both transcriptional and post-transcriptional levels (Baulcombe 2004). Small RNAs (sRNAs), predominant effectors of plant RNA silencing, confer both specificity and amplification modality in silencing. The production of sRNAs associated with transcriptional silencing is primarily initiated by plant-specific RNA Pol II paralog RNA polymerase IV (Pol IV) and, in peculiar cases, by Pol II itself (Nuthikattu et al. 2013; Cuerda-Gil and Slotkin 2016). The Pol IV transcripts, majorly originating from the repeats and transposons, are acted upon by RNA-dependent RNA polymerase 2 (RDR2), whereas the Pol II aberrant transcripts are acted upon by RDR6, thereby converting them to double-stranded duplexes that become substrates for several Dicer-like proteins (DCLs) (Nuthikattu et al. 2013). The resultant short duplex sRNAs are picked up by specific Argonaute (AGO) effectors, specified by their length and by one of the strands possessing the preferred 5′-nucleotide (Mi et al. 2008). For post-transcriptional gene silencing (PTGS), majorly undertaken in plants by cleavage of target mRNA, 21- to 22-nt size class sRNAs with 5′-uracil (U) are loaded into the AGO1, leading to slicing of the target mRNA between positions 10 and 11 in the sRNA:mRNA duplex regions (Baumberger and Baulcombe 2005). In contrast, for transcriptional silencing, Pol IV–derived repeat-associated 24-nt sRNAs are loaded into AGO4 (Zilberman et al. 2003) with a 5′-adenosine (A) preference, and this ribonucleoprotein complex binds to the complementary region of the long scaffold transcript produced by RNA polymerase V (Pol V) (Wierzbicki et al. 2008). This triggers recruitment of DNA methyl transferases like Domains Rearranged Methyltransferase 2 (DRM2) and majorly results in asymmetric DNA methylation at the target locus (Matzke and Mosher 2014). Numerous noncanonical modalities in the concerted activity of these RNA polymerase variants have also been reported (Cuerda-Gil and Slotkin 2016).
Even though RNA silencing is an effective process to accurately delimit silencing, the loci over which different polymerases operate are dependent on the chromatin states. For instance, Pol IV is recruited by the readers of H3K9me2 marks named Sawadee Homeodomain Homolog 1 and 2 (SHH1 and -2) in coordination with CLASSY-type chromatin remodelers (Law et al. 2013; Zhang et al. 2013; Zhou et al. 2018). Pol V recruitment involves DNA methylation readers like SUVH2 and SUVH9 (Liu et al. 2014), a chromatin remodeling complex called the DDR complex (Law et al. 2010), and several other proteins that are modulated by chromatin states. The activity of these polymerases is tightly linked to the epigenetic status of the locus of interest and, hence, the RNA silencing. A few reports have probed the effects of the loss of DNA methylation machineries on the chromatin states in Arabidopsis and observed excessive reorganization of the chromatin domains. For example, in met1 mutants, chromatin is decondensed with a loss of clear heterochromatin and euchromatin boundaries, exemplifying the impact of the cross talk between these epigenetic states (Soppe et al. 2002; Mathieu et al. 2005; Zhong et al. 2021). Similarly, pol iv shows permanent loss of silencing at selected loci that are recalcitrant to rescue by complementation (CO) of Pol IV, and this phenomenon is owing to an associated loss of H3K9me2 marks (Li et al. 2020). Unrestricted to silencing, it is also evidenced that loss of Pol IV leads to transcriptional misregulation as observed by the accumulation of atypical nascent transcripts in maize (Erhard et al. 2015) and by increased Pol II activity at the 3′-ends of Pol II transcriptional units (McKinlay et al. 2018). The indirect facilitation of spurious transcription by virtue of loss of epigenetic players triggers cryptic transcription by Pol II in both genes and transposons (Le et al. 2020). The prolific presence of repeat fragments in the gene coding units like introns in rice has been shown to trigger transcriptional silencing-like features (Espinas et al. 2020). Taken together, the epigenetic landscape not only modulates the silencing of repeats directly but also encumbers the cryptic transcriptional activity.
Such counter-balancing reinforcement loops between epigenetic states must contribute to molecular and morphological phenotypes upon perturbation, especially in monocots with higher proportion of transposons. It has been conclusively documented that reproductive structures in rice, including gametes, undergo massive reprogramming in terms of sRNA production and DNA methylation in a locus-specific manner (Chenxin et al. 2020). Indeed, unlike Arabidopsis, loss of silencing players display exacerbated phenotypes in the monocot model rice in the cases of drm2, met1b, pol iv, pol v, and dcl3 (Moritoh et al. 2012; Wei et al. 2014; Yamauchi et al. 2014; Xu et al. 2020; Zheng et al. 2021). In agreement with this, grass family (Poaceae) members have evolved a specific neofunctionalized RNA polymerase paralog called RNA polymerase VI (Trujillo et al. 2018). These phenomena substantiate that a complex gene arrangement interleaved by repeats mandates a very robust mechanism of epigenetic silencing.
Pol IV is well known to be involved in sRNA biogenesis and silencing via DNA methylation. However, the roles of Pol IV in regulating other epigenetic layers such as chromatin marks, specifically in plants with complex genomes, are unknown. This study aims to explore the loss-of-function lines of Pol IV in rice and Arabidopsis, mainly focusing on the less well known roles of Pol IV in demarcating genome-wide silencing, chromatin boundaries, and aberrant transcription.
Results
Knockdown of RNA Pol IV induced pleiotropic phenotypic defects
The loss of function of the catalytically active component of the Pol IV complex, NRPD1, causes a spectrum of effects in different plant species, from delayed growth transition in Physcomitrella patens, delayed flowering in Arabidopsis, and reproductive defects in Brassica rapa, Capsella rubella, and Zea mays to increased tillering in Japonica rice (Erhard et al. 2009; Coruh et al. 2015; Grover et al. 2018; Xu et al. 2020; Wang et al. 2020b). To understand the multifaceted roles of Pol IV beyond RdDM in rice, we generated artificial microRNA (amiR)–mediated knockdown (kd) transgenic indica rice lines targeting the largest subunit of Pol IV (NRPD1). Since the rice genome encodes two isoforms of Pol IV, NRPD1a and NRPD1b, sharing 88.8% sequence identity, amiR was designed to target a conserved sequence in the N-terminal region of both the transcripts, incorporating optimal design parameters as identified earlier (Fig. 1A; Narjala et al. 2020). Previous attempts targeted both the rice NRPD1 isoforms using RNA interference with suboptimal targeting or the CRISPR-Cas9–mediated knockout (KO) that was embryonic lethal or was showing drastic reduction in fertility (Debladis et al. 2020; Xu et al. 2020; Zhang et al. 2020; Zheng et al. 2021). To obtain precise targeting and to avoid the lethality and sterility effects of KO, we resorted to the amiR technology. To prove that amiR is targeting only NRPD1, leading to the effects, we supertransformed the kd with a construct expressing a amiR-targeting-resistant version of NRPD1b CDS driven by its cognate promoter (Fig. 1A). The expression of amiR in kd lines was confirmed with northern hybridization, and a reduction in transcript abundance of both the isoforms was confirmed with RT-qPCR and RNA-seq. Restoration of these transcripts was observed in the CO lines (Fig. 1B,C; Supplemental Fig. S1A,B). Rice transformation was performed twice independently to score for consistent phenotypes and to eliminate T-DNA insertion effects. Independent transgenic events were identified by junction fragment Southern analyses (Supplemental Fig. S2A,B).
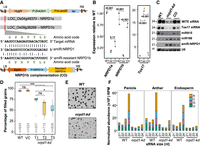
Knockdown of RNA polymerase IV (Pol IV) in rice results in pleiotropic phenotypes. (A) T-DNA map of amiR coding binary construct with the supertransformed NRPD1b complementation (CO) construct. The alignment depicts the amiR binding region and the modifications in the amiR-resistant CDS of NRPD1b that were driven by its cognate promoter (P:NRPD1b). Amino acids encoded by the original and amiR-resistant modifications were unchanged. (BlpR) Bialaphos resistance. The precursor-amiR (Pre-amiR) is driven by maize ubiquitin promoter (P:ZmUbi1). (ter) 35S-poly(A) signal, (HygR) hygromycin selection marker, (RB and LB) right and left border. (B) Plots representing relative abundance of NRPD1a, NRPD1b, and Tos17 transcripts in kd and CO with respect to WT, measured by RT-qPCRs. Student's t-test; P-values mentioned across comparisons. (C) Small RNA (sRNA) northern blots showing the accumulation of 24-nt siRNAs and miRNAs in WT, kd, and CO. U6 was used as loading control. (D) Boxplots showing the percentage of filled grains (n = number of grains). Dots represent the average of panicles in each plant. Tukey's test; (***) P-value of 0.001, (*) P-value of 0.01, (ns) nonsignificant. (VC) Vector control plants that lack amiR encoding region. (E) Representative images of pollen grains of dehisced anther stained with iodine. Scale: 50 μm. (F) Stacked bar plots showing the normalized abundance of sRNAs of different sizes with their 5′ nucleotides. The replicates were merged.
As observed previously (Xu et al. 2020), kd plants showed increased tiller number (Supplemental Fig. S1C). The percentage of viable filled grains was significantly low at 75% in T1 kd plants and dropped progressively over generations to reach 50% in T3 (Fig. 1D; Supplemental Fig. S1D). The unfilled florets showed structural deformities, and they were consistent among the distinct transgenic events, proving that the defects observed were not owing to transgenesis (Supplemental Fig. S2C–E). Specific lineages of the kd lines had extreme reproductive defects without viable grains (Supplemental Fig. S3A). These defects were not owing to zygosity of the T-DNA or the dosage of amiR, indicating a possible effect of induced epimutations (Supplemental Fig. S3B,C; Johannes and Schmitz 2019). Because NRPD1 is reported to influence pollen development (Wang et al. 2020b), we further investigated the pollen quality by measuring size, iodine staining potential, and ultrastructural morphology that revealed pollen defects in kd (Fig. 1E; Supplemental Fig. S1E–G). Taken together, amiR-mediated kd of the Pol IV complex in rice led to severe reproductive defects consistent across generations, unlike the mild abnormalities observed in Arabidopsis.
Rice Pol IV is required for biogenesis of repeat-associated sRNAs
To explore the molecular effects of the loss of Pol IV in rice and to discern the reasons for extreme reproductive defects upon NRPD1 kd, we performed sRNA profiling in three reproductive tissues that showed drastic defects: pre-emerged panicle, anther, and endosperm. As shown earlier in other species, kd plants showed evident reduction of 24-nt sRNAs in all the tissues (Fig. 1C,F; Supplemental Fig. S4A; Herr et al. 2005; Coruh et al. 2015; Grover et al. 2018; Wang et al. 2020b). As expected (Mi et al. 2008; Zhai et al. 2015), the maximum reduction in 24-nt sRNAs corresponded to 5′A-containing sRNAs in all the three tissues (Fig. 1F). On the contrary, bulk of the Pol II transcribed miRNAs and the sRNAs mapping to their precursors largely remained unchanged (Supplemental Fig. S4B,C). Further, as the Pol IV–derived sRNAs are known to be associated with transposons and repeats, we profiled the sRNAs from major annotated repeats in the rice genome. We observed a substantial loss of repeat-associated sRNAs, and this was also validated by northern blotting in different tissues (Fig. 1C; Supplemental Fig. S4D–F). The repeat-associated sRNAs were also restored to WT levels in the CO lines as proven by northern blots (Fig. 1C). In conclusion, RNA Pol IV is essential for production of repeat-associated sRNAs in different tissues of rice, and CO of one of the two isoforms of NRPD1 rescues the sRNA levels.
Given the severity of the defects found in the rice kd plants, we hypothesized that the loss of sRNA-mediated regulation of transposons and the concomitant misregulation of genes might be attributable to the defects. Because the 24-nt sRNAs are involved in establishing DNA methylation of the repeats, we profiled the DNA methylome at the annotated repeats in rice, which revealed a substantial decrease of methylation in kd compared with WT, especially in CHG and CHH contexts (Supplemental Fig. S5A). In addition, several of the transposon classes showed an increase in expression (Supplemental Fig. S6A–C). The up-regulation of repeats and loss of DNA methylation over transposons prompted us to question if they have gained proliferative potential upon Pol IV kd. This possibility was previously predicted to influence phenotypes and genomic integrity in a number of reports (Cui et al. 2013; Wei et al. 2014; Debladis et al. 2020). To verify such a possibility, we performed a PCR-based assay to detect the extrachromosomal circular DNA (ECC DNA) intermediates that are produced as by-products of transposition (Lanciano et al. 2017). We observed additional bands portraying increased proliferative potential of PopRice and Tos17 transposons (Supplemental Fig. S6D). These bands were observed even after the removal of linear DNA using a specific exonuclease (Supplemental Fig. S6E; Supplemental Methods). We examined if specific transposons proliferated by performing transposon display Southern hybridizations. Among the kd lines tested, a specific line showed a discernible copy number increase of the LINE-1 element across generations (Supplemental Fig. S6F). This observation is in line with a previous report in which silencing of NRPD1 caused an increase in transposon copy number (Debladis et al. 2020). The proliferation of transposons might be the reason for exacerbated reproductive defects in selected kd lines (Supplemental Fig. S3A). Ribosomal RNA (rRNA) precursor expression and the expression of several genes were also misregulated in the kd lines compared with WT (Supplemental Fig. S5B,C). The genes misregulated in anthers and panicles of the kd lines also showed a significant overlap with the DEGs seen in nrpd1-RNAi shoot bases (Xu et al. 2020), suggesting a conserved effect of loss of Pol IV in different tissues of rice.
In summary, the activity of RNA Pol IV and ensuing sRNAs are essential for regulating the expression of genes and repeats via DNA methylation, and loss of these processes results in the potentially genotoxic proliferation of transposons in the rice genome.
Activity of RNA Pol IV potentiates silencing by H3K27me3 and H3K9me2 marks and modulates their relative occupancy over genes and repeats
Several reports have suggested that the effective silencing is brought about by the cross talk between DNA methylation and histone modifications in a locus-specific manner in plants (Mathieu et al. 2005; Deleris et al. 2012; Gent et al. 2015; Zhou et al. 2016). These reports have found that loss of DNA methylation led to perturbation of post-translational modifications (PTMs) of histones at the respective loci, together influencing the status of transcriptional silencing (Jamge et al. 2022). Recent studies in Arabidopsis have suggested that the loss of Pol IV and associated sRNAs can influence the histone H3 PTMs levels at specific loci (Li et al. 2020; Parent et al. 2021). In particular, Rougée et al. (2021) have described H3K27me3 redistribution at a subset of ddm1 hypomethylated loci in Arabidopsis without being able to repress the hypomethylated loci. These observations in Arabidopsis, along with lack of in-depth epigenetic analysis in Arabidopsis nrpd1 mutants, prompted us to explore the multiple layers of epigenetic changes upon kd of Pol IV in rice.
To examine the polycomb-mediated H3K27me3 and constitutive heterochromatic mark H3K9me2 profiles, we performed nuclear immunostaining that revealed broader H3K27me3 occupancy in kd over the H3K9me2 regions, unlike the WT, corroborating observations in Arabidopsis DNA methylation mutants (Fig. 2A; Supplemental Figs. S8, S9). Unlike Arabidopsis nuclei with distinct chromocenters, rice nuclei showed a wide distribution of H3K9me2 signal, suggesting a broad distribution of chromatin states in monocots (Liu et al. 2017). Further, to examine the H3K27me3 marks and other epigenetic marks in a locus-specific manner, we performed ChIP-seq with H3K27me3-, H3K4me3-, and H3K9me2-specific antibodies in replicates that showed consistent enrichment profiles (Supplemental Fig. S7). We analyzed the profiles of these marks genome-wide, showing clear enrichment of H3K4me3 over PCGs, the absence of H3K9me2 at the genic loci, and the presence of H3K27me3 at both genic and nongenic regions (Methods) (Supplemental Fig. S10A). Differential enrichment analyses revealed 7686 loci possessing significantly higher H3K27me3 occupancy in kd compared with WT, validating the immunostaining observations (Supplemental Fig. S10B). We found that the loci that showed enhanced occupancy of H3K27me3 marks also showed loss of H3K9me2 marks (Fig. 2B). This observation points out that Pol IV activity is quintessential not only for maintaining the DNA methylation and sRNA profiles but also for buffering the relative locations of gene-specific H3K27me3 marks and repeat-specific H3K9me2 marks. In the absence of Pol IV, H3K27me3 marks intruded into new domains. Several sites showed reduced enrichment of H3K27me3 and misregulation of other marks (Supplemental Fig. S10B). On the other hand, H3K9me2 marks were reduced over the loci intruded by H3K27me3 marks, likely redistributing the silencing signatures (Fig. 2B).
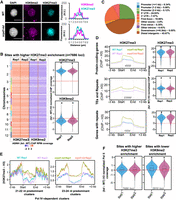
Pol IV maintains the genome-wide distribution of H3K27me3-mediated silencing states. (A) Immunostaining images of H3K27me3 and H3K9me2 marks in the nuclei extracted from WT and kd seedlings. DAPI-stained DNA. Scale bar: 5 µm. At least 30 nuclei from each genotype were examined. Fluorescence signals over the regions of interest are plotted. Regions of interest are also shown in Supplemental Figures S8 and S9. (B) Chromosome-resolved heatmaps showing the levels of difference in H3K9me2 enrichment at sites with higher H3K27me3 enrichment in kd compared with WT. Box-violin plots show the distribution of the differences in H3K9me2 and H3K27me3 levels. The y-axis is scaled to the inverse sine hyperbolic function of differences. (C) Pie chart showing the feature annotation of the sites shown in B. All the sites that do not fall in the ±3-kb windows of genes were categorized as distal intergenic regions. (D) Metaplots showing the levels of H3K27me3 enrichment (normalized to H3) over the annotated features overlapping with the identified H3K27me3 peaks. Numbers in gray depict the number of loci. Box-violin plots show the difference in enrichment in kd compared with the WT. The y-axis is scaled to the inverse sine hyperbolic function of H3 ChIP-normalized values. (E) Metaplots showing the abundance normalized enrichment of H3K27me3 marks w.r.t. H3 over the Pol IV–dependent sRNA clusters size categorized as 21- to 22-nt and 23- to 24-nt predominant clusters. (F) Box-violin plots showing the difference in Pol II coverage over the H3K27me3 higher-enriched sites and H3K9me2 lower-enriched sites in kd compared with the WT. Numbers in gray describe the number of loci taken for analyses. The y-axis is scaled to inverse sine hyperbolic function of enrichment values. (B,D,F) A Mann–Whitney U test was used to test statistical significance against WT Rep1. (*) P-value < 0.0001.
Furthermore, annotation of the H3K27me3 gained loci showed that almost 55% corresponded to coding genes, with ∼45% of the loci concentrated at the 5′ UTR (24.93%) and first exons (19.66%) of the PCGs (Fig. 2C). All the non-PCGs accounted for 44% of the H3K27me3 overenriched loci (Fig. 2C). Analysis of H3K27me3 peaks further revealed that it was globally overenriched at PCGs, TEs, and repeat loci, thus implicating the spread of this mark into both PCGs and non-PCGs in kd (Fig. 2D). Moreover, the activation mark H3K4me3 also showed differential enrichment and overabundance over the PCGs and non-PCGs in kd (Supplemental Fig. 10B,C). Because monocot genomes are relatively enriched with repeats where they can pronouncedly influence the proximal genes to a greater extent than dicots (Hirsch and Springer 2017), we selected peaks containing PCGs that have at least 10% of their length overlapping with repeats (“genes with repeats”) and explored the redistribution of histone marks. As expected, this subset of PCGs recapitulated the hyper-occupancy of the H3K27me3 marks over the gene bodies to a greater degree than PCGs (Fig. 2D). Moreover, the hallmark RdDM sites enriched in Pol IV–dependent sRNAs gained H3K27me3 marks (Fig. 2E). This is in line with the observation that, even though the silencing by H3K9me2 and H3K27me3 is compartmentalized, it can, to a certain degree, substitute and overlap in cases of perturbation of other silencing signatures (Deleris et al. 2012; Déléris et al. 2021).
In spite of the spreading of H3K27me3 silencing in kd, we observed enhanced transcription of transposons and repeats in kd lines (Supplemental Fig. S6), indicating that H3K27me3 overshoot and intrusion did not completely compensate for the loss of repressive signatures of H3K9me2, DNA methylation, and sRNAs, supporting earlier investigations in Arabidopsis (Rougée et al. 2021). To test if occupancy of H3K27me3 marks can effectively prevent transcription at the loci that lost H3K9me2 marks, we performed ChIP-seq with RNA Pol II antibody. This analysis revealed the overoccupancy of Pol II ChIP signals over the sites with increased H3K27me3 or reduced H3K9me2 occupancy (Fig. 2F), suggesting inefficient suppression of transcription by H3K27me3 as established in ddm1 mutants in Arabidopsis (Rougée et al. 2021). Taken together, we identify a novel effect of loss of Pol IV in buffering the occupancy of H3K9me2 and H3K27me3 marks over the PCGs and repeats. Failure to establish this balance led to a more permissive chromatin state, promoting misregulation of PCGs and repeats.
Aberrant transcription in kd lines gives rise to atypical sRNAs that are suppressed by Pol IV
Relaxed chromatin state and the reduction of silencing signatures in the kd lines might favor the enhanced transcription. Such observations were routinely reported in model dicot Arabidopsis and other organisms when mutants with altered chromatin state were analyzed (Zheng et al. 2009; McKinlay et al. 2018; Ishihara et al. 2021; Jamge et al. 2022). To address this possibility, we performed a MNase accessibility assay from the chromatin of WT and kd plants. This analysis indicated relaxed chromatin status in kd compared with WT as kd lines accumulated shorter digested fragments (Supplemental Fig. S11A). Also, we profiled the Pol II status over PCGs and genes with repeats using ChIP-seq. Even though we did not observe a significant difference of Pol II occupancy over all PCGs, we observed a significant hyperoccupancy over the genes with repeats (Supplemental Fig. S11B,C). Because the antibody captures all the states of Pol II, we postulated whether aberrant assembly, stalling, or elongation of the Pol II in kd might trigger the accumulation of misformed, potentially aberrant transcripts. We explored if these transcripts over the subset of PCGs might trigger production of sRNAs from these regions. As anticipated, sRNA analysis showed that the numerous ShortStack (Axtell 2013)-identified sRNA clusters (“clusters”) were up-regulated in kd in all the three tissues tested in both of the size classes (Supplemental Fig. S12A–C,E; Supplemental Methods). The sRNAs that are up-regulated in kd were denoted as Pol IV–suppressed sRNAs (“suppressed sRNAs”), and those that are reduced in kd were named as Pol IV–dependent sRNAs (“dependent sRNAs”; Supplemental Methods). Suppressed sRNAs did not show any first nucleotide bias (Supplemental Fig. S12D). Further analysis of the 21- to 24-nt sRNAs from the sRNA clusters revealed that these suppressed sRNA clusters had 21- to 22-nt predominant, 23- to 24-nt predominant, and mixed clusters of both of these size classes in almost equal proportions (Supplemental Fig. S13A; Supplemental Methods). The suppressed and dependent clusters had uniform length distribution (Supplemental Fig. S13B).
To have an unbiased estimation of the relative distribution of these sRNAs, we performed a genome window (100 bp)–based analysis (Supplemental Methods). Although dependent bins were higher in number in all the three Pol IV kd tissues, suppressed sRNA loci were fewer (Supplemental Fig. S14A–C). For exploring the size distribution of all inserts in the panicle sRNA data sets, we plotted size-density distribution of 16- to 45-nt sRNAs from suppressed and dependent bins (Fig. 3A). As expected, Pol IV–dependent sRNA bins accumulated sRNAs of the predominantly 24-nt size class, whereas suppressed sRNA bins accumulated wide fragment sizes ranging from 21–35 nt. We verified that suppressed sRNAs were not owing to oversampling of the residual sRNAs in the kd tissues and associated normalization artifacts by comparing the raw abundance of sRNAs in each library to the sum of miRNAs (Supplemental Fig. S15). The suppressed bins were concentrated at fewer selective loci (Supplemental Figs. S16A, S17A, S18A). Further, cumulative sum plots describing the relation between the abundance of sRNAs and the number of uniformly sized bins also indicated that suppressed sRNAs were abundantly concentrated in fewer bins (Supplemental Figs. S16B, S17B, S18B).
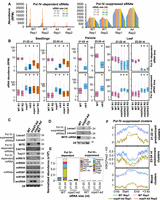
Pol IV complex suppresses sRNA production from several loci. (A) Bar plots showing the abundance of Pol IV–dependent and –suppressed sRNAs of 16- to 45-nt size range. (B) Boxplots showing the abundance of sRNAs from different mutants in rice. sRNAs were size-categorized into 21- to 22-nt and 23- to 24-nt and counted in 100-bp nonoverlapping windows. Plots depict the abundance of sRNAs in each size class over nrp(d/e)2 (leaf), nrpd1 (seedlings), and rdr2 (panicle) dependent and suppressed bins. The data sets were obtained from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE158709 (seedlings: nrpd1, nrpe1) and GSE130166 (seedlings and panicle: rdr2). The y-axis is scaled to inverse sine hyperbolic function of RPM values. Mann–Whitney U test was used to test statistical significance against WT R1. (*) P-value < 0.0001. (C) sRNA northern blots validating the presence of Pol IV–suppressed sRNAs, –dependent sRNAs, and –independent miRNAs. U6 was used as loading control. (D) sRNA northern blots showing the abundance of sRNAs in WT, kd, and NRPD1 CO lines. U6 was used as loading control. (E) Stacked bar plots showing abundance of Pol IV–dependent and –suppressed sRNAs from transposon categories. (F) Metaplots describing histone H3K27me3 occupancy normalized to total H3 signal over the Pol IV–suppressed sRNA clusters, categorized as 21- to 22-nt predominant clusters, 23- to 24-nt predominant clusters, and the mixed sized clusters.
Further, to validate if suppressed sRNAs in kd are associated with other Pol IV machineries, we analyzed the published sRNA data sets derived from rice nrpd1, rdr2, and nrpe1 seedlings (Zheng et al. 2021; Wang et al. 2022). We indeed observed the accumulation of suppressed sRNAs in nrpd1 and rdr2 but to a lesser degree in nrpe1 seedlings (Fig. 3B). Further, we observed that the nrp(d/e)2 sRNA data sets (Chakraborty et al. 2022) indeed retained the Pol IV–suppressed loci, suggesting only a minor role of Pol V in altering the chromatin state and the accumulation of these sRNAs (Fig. 3B). We also detected the Pol IV–suppressed sRNAs by sRNA northern hybridizations at two distinct loci (Fig. 3C; Supplemental Fig. S12E), proving the existence of the suppressed sRNAs in kd lines, and they were not observed upon NRPD1 CO (Fig. 3D). Furthermore, repeat features that are hallmarks of dependent sRNAs were completely devoid of suppressed sRNAs (Fig. 3E). Annotation of the differentially expressed sRNA bins from different tissues identified that nearly half of the Pol IV–suppressed sRNA bins mapped to coding regions, whereas dependent counterparts mapped to the noncoding regions (Supplemental Fig. S19A). Pol IV–suppressed sRNAs from the coding regions can be either generated from hairpin structures of aberrant transcripts or generated from duplex RNAs of aberrant transcripts by the activity of RDRs. Specific AGOs will stabilize the preferred sRNAs. To test if dsRNAs are generated by the activity of RDRs (which will not show strand bias with respect to the coding gene), we counted suppressed sRNAs from bins mapping to coding genes. Suppressed sRNAs uniformly mapped to both the same mRNA encoding strand and the opposite strand, suggesting activity of one of the RDRs (Supplemental Fig. S19B). Unlike dependent sRNAs, suppressed sRNAs were within the gene body PCGs (Supplemental Fig. S19C).
Furthermore, occupancy of H3K27me3 marks at the suppressed loci of all size classes were lower than the WT in kd lines, suggesting a chromatin misregulation associated with these loci (Fig. 3F). Moreover, genes overlapping with the suppressed sRNA loci showed reduced H3K27me3 and H3K9me2 levels and increased H3K4me3 levels (Supplemental Fig. S20). In summary, we revealed a population of novel sRNAs that were suppressed by Pol IV showing atypical genomic origins and molecular signatures.
Pol IV–suppressed sRNAs are conserved in Arabidopsis and are dependent on RDR6 and DDM1
We speculated that altered chromatin state owing to loss of Pol IV might be conserved across plants and might be leading to the accumulation of suppressed sRNAs. In agreement with this, we observed that loss of Arabidopsis Pol IV resulted in suppressed sRNAs, very similar to rice, but from fewer bins in inflorescence tissues (Supplemental Fig. S21A; Zhou et al. 2018). Unlike Pol IV–dependent bins, suppressed sRNAs in Arabidopsis showed nonuniform distribution like rice tissues (Supplemental Fig. S21B). The availability of various genetic mutants in Arabidopsis prompted us to check the mechanistic origins of these sRNAs and possible regulators of chromatin state when Pol IV is not functional. In this direction, we examined several siRNA biogenesis mutants, especially connected to RdDM, and found that the suppressed sRNAs were found mainly upon loss of Pol IV itself or associated proteins using northern blots with various mutants (Fig. 4A; Supplemental Fig. S22). The suppressed sRNAs were unique to nrpd1 and were not observed in sRNA-processing mutants such as dcl3, dcl234, or ago4 or the mutant of chromatin remodeler ddm1 (Fig. 4A). We also used sRNA data sets from the similar stage inflorescence tissues from additional set of mutants involved in RNA silencing (Zhai et al. 2015). This independent set of nrpd1 data sets also confirmed the increase of suppressed sRNAs compared with the WT mainly from nonrepeat regions (Supplemental Fig. S22A,B). The suppressed sRNAs were also seen in other nrpd1-associated mutants like rdr2, nrp(d/e)2, nrpd1dcl3, and rdr2dcl3 (Supplemental Fig. S22C). This indicates that the suppressed sRNAs are directly coupled to the absence of Pol IV complex (NRPD1 or RDR2). In addition, most of the suppressed sRNAs were brought back to the WT levels upon NRPD1 CO (Supplemental Fig. S22D).
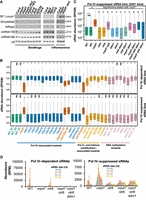
Pol IV–suppressed sRNAs are conserved in Arabidopsis. (A) sRNA northern blots showing the levels of Pol IV–suppressed sRNAs (SP. Locus1) and other sRNAs from Arabidopsis seedlings and inflorescence tissues. (B) Boxplots showing the sRNA abundance from different genotypes of Arabidopsis with counts from Pol IV–dependent and –suppressed 23- to 24-nt size class bins. The sRNA data sets were obtained from GEO GSE165574, GSE45368, and GSE99694. Analyses depicts sRNAs from inflorescence tissues. (C) Boxplots showing the sRNA abundance from different genotypes of Arabidopsis with counts from Pol IV–suppressed 23- to 24-nt size class bins depicted in B. The sRNA data sets were obtained from GEO GSE79780 (inflorescence tissues). The y-axis is scaled to the inverse sine hyperbolic function of RPKM values. Mann–Whitney U test was used to test statistical significance against WT R1, unless stated otherwise. (*) P-value < 0.0001. (D) Bar plots showing the abundance of Pol IV–dependent and –suppressed sRNAs of 16- to 35-nt size range.
To probe which part of the RdDM machinery is mainly involved in generation of Pol IV–suppressed sRNAs, we analyzed published sRNA data sets from inflorescence tissues of 28 different genotypes of Arabidopsis mutants, pertaining to three categories: Pol IV–associated mutants, Pol V and histone modification–associated mutants, and DNA methylation mutants (Supplemental Table S2). For the set of loci identified as suppressed and dependent loci, we evaluated the abundance of sRNAs across the mutants. We observed evident and drastic accumulation of suppressed sRNAs comparable to nrpd1 only in clsy1234 quadruple mutants and in the shh1clsy34 triple mutant (Fig. 4B). This observation points out that the suppressed sRNAs are the effect of ablation of Pol IV enzyme complex along with the machinery responsible for its assembly. On the contrary, the Pol IV–suppressed sRNAs are not initiated upon removal of either the Pol V arm of RdDM or the DNA methyltransferases (Fig. 4B).
To explore the molecular machinery involved in the biogenesis of suppressed sRNAs upon loss of nrpd1, we explored the abundance of sRNAs in combination mutants of nrpd1. The sRNA abundance in the Pol IV–suppressed bins (n = 3451) identified in previous analyses was counted from ddm1, rdr6, nrpd1 rdr6, and nrpd1 rdr6 ddm1 data sets (Panda et al. 2016). This revealed that the production of suppressed sRNAs in the nrpd1 mutant was partially dependent on RDR6 and DDM1, as the double or triple mutants of these genes in a nrpd1 background showed a reduction in suppressed sRNAs compared with the nrpd1 mutant (Fig. 4C; Supplemental Fig. S22E). Dependence on RDR6 and DDM1 was also observed across all the sRNA size classes that mapped to suppressed sRNA bins but not for the dependent sRNA bins (Fig. 4D). Importantly, the dependence of suppressed sRNAs on silencing machinery genes establishes an active mode of suppressed sRNA biogenesis in the absence of the Pol IV complex.
Probing the contributing molecular signatures associated with the suppressed sRNAs, we also tested the status of the DNA methylation in reproductive tissues over the suppressed sRNA clusters and found that methylation over the CG, CHG, or CHH contexts in both rice and Arabidopsis remains largely unchanged (Supplemental Figs. S23, S24). Pol II occupancy over the suppressed and dependent loci also remained unchanged, likely owing to the transient interaction by Pol II in generating these transcripts (Supplemental Fig. S25). On the other hand, reduction of H3K27me3 over specific genomic regions upon loss of Pol IV might have triggered production of suppressed sRNAs (Supplemental Fig. S26), which are conserved across monocots and dicots.
Pol IV–suppressed sRNAs are loaded into AGO1 to induce PTGS at protein-coding loci
Given that these abundant atypical sRNAs are from gene coding regions and they did not change the DNA methylation signature of the loci, we explored if they can target genes post-transcriptionally. Pol IV–suppressed sRNAs from PCGs are predominantly of the 21- or 24-nt size class unlike the broad size range of the bulk of suppressed sRNAs (Fig. 5A), suggesting processing by sRNA machinery. To test if AGO1-loaded suppressed sRNAs targeted genes post-transcriptionally, we performed degradome sequencing in WT and kd panicles. Among the genes that overlapped with the suppressed sRNAs (389 genes), target prediction tools identified 154 genes as potential targets of suppressed sRNAs (Supplemental Table S8; Supplemental Fig. S27B). The degradome tag density at the predicted target loci was substantially increased in kd lines, and nontargets did not accumulate degradome tags despite being sources of suppressed sRNAs (Fig. 5B; Supplemental Fig. S27C). In further agreement with the targeting process, the WT AGO1 IP was enriched with suppressed sRNAs that mediated slicing as seen in degradome analysis (Fig. 5C; Supplemental Fig. S27A; Wu et al. 2009). The transcript-to-degradome read depth ratio for targets and nontargets showed expected profiles (Supplemental Fig. S27D,E). It would be interesting to explore the loading distribution of suppressed sRNAs in kd lines into specific AGO1 isoforms (out of AGO1a, -b, -c, and -d). Many of the target RNAs had significantly reduced expression (∼40% mean), suggesting precise PTGS at these sites mediated by suppressed sRNAs (Fig. 5D–F; Supplemental Fig. S28). We also observed that many of the genes that underwent PTGS by suppressed sRNA-mediated targeting in rice were previously implicated in reproductive growth and development (Supplemental Table S8). For instance, OsMADS18 (APETALA1 homolog in rice), a member involved in floral architecture establishment (Wang et al. 2020a), a close homolog of fertility restorer (RF; Os10t0497366), a glycine-rich interaction partner of RF5 (Os12t0632000) (Hu et al. 2012), and a pollen-specific desiccation-associated protein (Os11t0167800) were targeted for degradation in kd lines (Supplemental Table S8). Targeting of aberrant transcripts originating from atypical loci specifically in kd, but not in WT, by suppressed sRNAs is an area for further investigation.
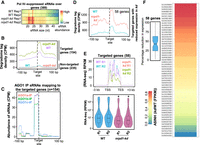
Pol IV–suppressed sRNAs get loaded into AGO1 to mediate PTGS. (A) Heatmaps showing size distribution of sRNAs from Pol IV–suppressed sRNA bins overlapping with genes (389 genes). (B) Metaplots showing the degradome tag density over the genes identified as targets in nrpd1-kd panicle degradome (green and purple; 154 genes) and the same for the genes that are not effectively targeted (category > 5) but produce Pol IV–suppressed sRNAs (cyan and orange; 235 genes). Targeted locations are centered, and 100 bp on either side is displayed. (C) Metaplots showing the abundance of AGO1 IP-enriched sRNAs over the identified targeted genes centered at the targeting site. (D) Metaplots showing the degradome tag density of the 58 genes that showed targeting and significant reduction in expression. (E) Metaplots and box-violin plots showing the expression status of 58 targeted genes described in D. (F) Heatmap showing the fold changes (scaled to inverse sine hyperbolic function) in expression (FPKM) of the 58 genes that showed reduction in expression compared with the WT. The inset boxplot shows the distribution of reduction in expression (FPKM) observed.
On the other hand, the suppressed sRNAs in Arabidopsis did not accumulate in the AGO1-enriched fractions in both Col-0 and nrpd1 plants more than the mock IP controls (Supplemental Fig. S29). On the contrary, Pol IV–dependent sRNAs were abundantly found in AGO4 IP data sets (Supplemental Fig. S29A; Zhai et al. 2015; Panda et al. 2020). AGO1 targeting is unlikely or is rare to be detected in bulk analyses in Arabidopsis in which suppressed sRNAs are lowly abundant nor loaded into AGO1 (Supplemental Fig. S29C). It is worth exploring the AGO1-mediated targets of suppressed sRNAs in other plant species in which perturbation of NRPD1 showed strong phenotypes (Grover et al. 2018; Wang et al. 2020b). Such a targeting mediated by suppressed sRNAs might act as strong deterrent for the loss of Pol IV activity in plants with complex genomes.
Discussion
The precise and dynamic regulation of epigenetic modifications in a coordinated manner hallmarks the center stage of regulation. Our investigation uncovers the role of plant-specific RNA polymerase, Pol IV, in control over locus-specific epigenetic marks and prevention of illegit transcription.
Evolutionary analyses suggest that the plant-specific RNA polymerases IV, V, and, in specific cases, VI are novel machineries evolved in conjunction with the complexity of plant genomes, and the degree of importance of these polymerases varies across plants. For instance, in early land plants like P. patens, the pathway is completely redundant with other sRNA-independent DNA methyltransferases introducing de novo DNA methylation (Yaari et al. 2019) as opposed to the widespread defects observed in rice (Fig. 1; Supplemental Figs. S1–S3; Xu et al. 2020; Zheng et al. 2021). The functions of these polymerases are pronounced in reproductive tissues as they are involved in faithful transmission of epigenetic information, antagonizing genome dosage aberrations, and hybridization (Zhang et al. 2016; Erdmann et al. 2017; Martinez et al. 2018; Satyaki and Gehring 2019). It is apparent that the green-lineage-specific RNA polymerases have neofunctionalized to perform additional roles with increasing genome complexity. The comparisons we present with respect to the redistribution of epigenetic marks between rice and Arabidopsis serve as strong evidence to these predictions.
Several investigations in the model plant Arabidopsis have established that Pol IV initiates biogenesis of sRNAs from repeats and transposons establishing de novo DNA methylation aiding against their genotoxic proliferation. Our studies in rice corroborate this function of Pol IV in multiple reproductive tissues (Figs. 1, 6A; Supplemental Fig. S4). Several reports have mentioned the synergistic effects and cross dependence of histone modifications and global CG DNA methylation in establishing epigenetic modalities in Arabidopsis (Soppe et al. 2002; Mathieu et al. 2005; Deleris et al. 2012; Li et al. 2018; Zhong et al. 2021; Zhao et al. 2022). These studies had used met1, ddm1, and other methyltransferase mutants, resulting in a loss of compaction at the pericentromeres and constitutive heterochromatin domains. On the other hand, the RdDM is not restricted to heterochromatin, and its purview extends into the gene-rich regions as well. Whether modulation of the sRNAs can directly perturb the chromatin states attributable to the gene expression was unclear. Especially, in monocots like rice, the interspersion of repeat fragments within genes mandates gene regulation not at extended length scales but in localized compartments even within the euchromatin (Espinas et al. 2020). Probing the effect of the loss of RdDM on the chromatin states over the genes in our study uncovered a distinct redistribution of H3K9me2 and H3K27me3 marks (Figs. 2, 6B). Predominantly, H3K9me2 marks, attributed with well-suppressed constitutively silenced loci and H3K27me3 associated with silenced gene coding units by action of PRC2 showed overoccupancy in kd over the transposons and genes (Fig. 6B). The authentic Pol IV transcribing loci that result in sRNAs showed significant H3K27me3 occupancy, likely an ectopic compensatory mode of silencing (Fig. 2E). This feature is evolutionarily conserved between plants and animals, and loss of transposon methylation can contribute to the redistribution of these marks into transposon territories (Deleris et al. 2012; Déléris et al. 2021). The proximity of the PCGs and transposons might be the trigger causing the intrusion of facultative silencing marks on the genes in species with larger genomes such as rice. This is well supported by the fact that upon kd of the Pol IV, genes that are dispersed with the repeat fragments showed an increased degree of H3K27me3 intrusion (Fig. 2E). PRC2-mediated H3K27me3 on genes impacts reproductive success in plants by controlling several imprinted genes that scale the genome dosage (Köhler et al. 2003; Roszak and Köhler 2011; Kradolfer et al. 2013; Jiang et al. 2017). Such indirect effects on the PCGs by unwarranted H3K27me3 marks, to a certain extent, might explain the defects in kd.
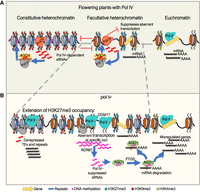
Pol IV complex precludes the spread of aberrant silencing. (A) Transcription by Pol IV produces sRNAs that guide silencing via DNA methylation at euchromatic TEs. Its activity also suppresses ungauged transcription by other polymerases and optimal distribution of H3K27me3 over coding genes. (B) Upon loss of Pol IV, distributions of H3K27me3, H3K9me2 marks are perturbed, and several transposons and genes are marked by H3K27me3. In addition, upon loss of Pol IV, ungauged aberrant transcription leads to production of Pol IV–suppressed sRNAs, partly dependent on RDR6 (post-transcriptionally) and DDM1 (transcriptionally). In rice, these transcripts are loaded into AGO1 and can target genes.
Loss of silencing signals and redistribution of H3K27me3 marks over the PCGs upon loss of RdDM in rice should result in differential accessibility of these loci to RNA polymerases. Ungauged transcription in the absence of Pol IV might feed to the sRNA pools via activity of DCLs that are devoid of Pol IV precursor load. Exploration of sRNA pools in the nrpd1 mutants of rice and Arabidopsis indeed showed resultant aberrant sRNAs, likely triggered by spurious transcripts. This pool of Pol IV–suppressed sRNAs is partly dependent on RDR6 and DDM1, as the double mutants of these genes in the nrpd1 background showed a reduction in their abundance. Such spuriously transcribed loci showed a reduction in H3K27me3 occupancy (Fig. 3F). This commonality of spurious sRNA transcripts in RdDM mutants was observed earlier (Zheng et al. 2021), where a similar notion of chromatin relaxation triggering Pol IV and Pol II transcription is promulgated. Similarly, studies in maize and Arabidopsis have suggested atypical transcripts in the nrpd1 or rdr2 mutation (Lu et al. 2006; Kasschau et al. 2007; Erhard et al. 2015; McKinlay et al. 2018). Our studies mechanistically delineate the causative chromatin features in the nrpd1 mutant. Not limiting to that, we find that the resultant Pol IV–suppressed sRNAs are capable of targeting genes post-transcriptionally by loading into specific AGOs (Fig. 6B). The ability of suppressed sRNAs to induce phased sRNAs and their activity in trans is worthy of investigation.
These analyses reveal that the atypical Pol IV–suppressed sRNAs in diverse plants such as rice and Arabidopsis were a result of misregulated chromatin states that are maintained by Pol IV. In rice, suppressed sRNAs were capable of targeting PCGs post-transcriptionally. We catalog reciprocity of the silencing between transcriptional and post-transcriptional modes, repurposing an existing RNA targeting machinery. We believe this acts as a second line of defense when the conventional transcriptional silencing via RdDM is impaired. This is in turn regulated by aberrant transcription and production of suppressed sRNAs capable of PTGS. Arabidopsis nrpd1-related mutants also showed production of these sRNAs; nevertheless, they did not get loaded into AGO1 for active targeting. The absence of efficient PTGS in Arabidopsis nrpd1 mutants might have alleviated the reproductive defects observed in rice. This difference between rice and Arabidopsis might be indicating stronger selection pressure for Pol IV function in complex genomes. Our results suggest a strong and multifaceted impact of Pol IV on the expression of PCGs, while enabling evolution of additional genomic complexities, architecture, and heterogeneity.
Methods
Plant material
Indica variety rice (Oryza sativa indica sp.) Pusa Basmati 1 (PB1) plants were grown in a growth chamber at 24°C/70% RH with a 16-h–8-h light–dark cycle for hardening before transferring to a greenhouse maintained at 28°C with a natural day–night cycle. Arabidopsis plants were grown in a growth chamber maintained at 22°C/70% RH with a 16-h–8-h light–dark cycle. The different mutants used are reported in Supplemental Table S4.
Binary vector construction and Agrobacterium mobilization
For the amiR construct, the artificial miRNA was designed using the WMD3 tool (Ossowski et al. 2008). The amiR was chosen so that it targets both of the NRPD1 isoforms (NCBI IDs: NRPD1a, XM_015781553.2; NRPD1b, XM_015756207.1). The mature amiR sequence (5′-UAUAGUGUUACUCUUGGACAU-3′) was embedded in the OsamiR528 precursor in the pNW55 plasmid (Supplemental Table S3; Ossowski et al. 2008; Warthmann et al. 2008). The precursor region was subcloned into a binary vector (pCAMBIA1300 backbone) between ZmUbiquitin1 promoter and 35S poly(A) signal and mobilized into Agrobacterium using the electroporation method.
For the CO construct, the NRPD1b promoter and 5′ UTR were amplified from the genomic DNA (Chr 9: 22,015,503–22,019,281) and fused to the amiR-resistant CDS of OsNRPD1b, and this was cloned into pCAMBIA3300 (with bialaphos selection, BlpR). The construct was supertransformed to the HygR kd calli.
Southern hybridization
Southern hybridization was performed as previously described (Ramanathan and Veluthambi 1995; G and Shivaprasad 2022). Total DNA (5 µg) was isolated and digested using 30 units of appropriate restriction enzyme. The digested DNA was electrophoresed on a 0.8% agarose gel in 1× TBE buffer and capillary-transferred to a Zeta probe nylon membrane (Bio-Rad). The probes (Supplemental Table S3) were labeled using [α-P32] dCTP (BRIT India) using a Rediprime labeling kit (GE Healthcare). The blots were exposed to phosphor screen and scanned using a Typhoon scanner (GE Healthcare).
sRNA northern hybridization
sRNA northern blots were performed as described earlier (Shivaprasad et al. 2012; Tirumalai et al. 2020). Around 15 µg of TRIzol extracted total RNA from different tissues was electrophoresed on a denaturing 15% acrylamide gel. The gel was electroblotted onto Hybond N + membrane (GE Healthcare) and UV cross-linked. The membrane was hybridized with the T4 PNK end-labeled oligonucleotides (with [γ-P32]-ATP) in Ultrahyb buffer (Invitrogen). The blots were washed, exposed to phosphor screen, and scanned using a Typhoon scanner (GE Healthcare). Post scanning, blots were stripped at 80°C in stripping buffer and proceeded with repeat hybridizations with subsequent probes.
Chromatin immunoprecipitation-sequencing (ChIP-seq) and analyses
ChIP was performed as described earlier (Saleh et al. 2008; Song et al. 2016). Around 1.2 g of pre-emerged panicle tissues was taken and cross-linked with 1% formaldehyde. The tissues were pulverized in liquid nitrogen, and nuclei were isolated. Equal numbers of nuclei were lysed and sheared using ultrasonication (Covaris) until fragments reached 150–350 bp in size. The sheared chromatin was incubated overnight with 50 µL of antibody (H3K4me3–Merck 07-473; H3K9me2–Abcam ab1220; H3K27me3–Active Motif 39155; H3–Merck 07-10254; Pol II–Abcam ab817) bound protein–G Dynabeads (Thermo Fisher Scientific) at 4°C. The purified IP products were taken for library preparation using NEBNext Ultra II DNA library prep kit with sample purification beads (NEB E7103L) as per the manufacturer's protocol. The libraries (with replicates) were sequenced on a Illumina HiSeq 2500 platform (Supplemental Table S1).
The obtained data sets were adapter-trimmed using cutadapt (Martin 2011) and aligned to the IRGSP 1.0 genome using the Bowtie 2 tool (Langmead and Salzberg 2012) with the following parameters: -v 1 -k 1 -y -a ‐‐best –strata. PCR duplicates were removed. The alignment files were converted to coverage files and compared (difference) to total H3 signal (for histone H3 PTMs) using bamcompare utility of deepTools (Ramírez et al. 2014). The average signals over the desired regions/ annotations were estimated using computematrix (deepTools), and the coverage signal metaplots were plotted using plotprofile that were modified using a custom R script (R Core Team 2021). The peak calling was performed using MACS2 (Zhang et al. 2008), with broad peak calling for all ChIP data sets except for H3K4me3. The peaks with enrichment above threefold compared with H3 ChIP were taken as valid peaks, and the peaksets were merged using BEDTools across genotypes and replicates. The composite peak sets were intersected with the annotated PCGs and repeats to get the peak overlapping features used for signal counting (deepTools). The replicate concordance was measured across the samples using deepTools plot correlation feature.
sRNA sequencing and analyses
sRNA sequencing was performed from pre-emerged panicle, anther, and endosperm of WT and kd plants. The size fractionation and library preparation were performed as previously described (Tirumalai et al. 2019). Reads were quality checked, adapter-removed, and size-selected using UEA sRNA workbench (Stocks et al. 2018). The reads are aligned after categorization into 21- to 22-nt or 23- to 24-nt sizes to IRGSP1.0 genome using Bowtie 2 (Langmead and Salzberg 2012) with the following parameters: -v 1 -m 100 -y -a ‐‐best –strata. Step-wise analyses for genomic annotations and 5′-nucleotide abundance were performed as previously described (Swetha et al. 2018). Only the mapped reads were used for further analyses, including differential expression analyses (Supplemental Table S5–S7). miRNA abundance was calculated using the miRProf tool (Stocks et al. 2018). Arabidopsis data sets were aligned to the TAIR10 genome.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE180457. All the necessary bioinformatics analyses scripts have been provided as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Prof. K. Veluthambi for Agrobacterium strains, PB1 seeds, and binary plasmids. We thank Prof. David Baulcombe for the Arabidopsis mutants. We thank CIFF, genomics, electron microscopy, IT, radiation, greenhouse, and laboratory-kitchen facilities at the NCBS. We acknowledge help from Anushree Narjala in bioinformatic analysis. We thank Dr. Aswin Sai Narain Seshasayee for his support. We thank Dr. Dimple Notani for sharing reagents. We thank Rahul Singh, M. Rajagopalan, and S. Goyal for subcloning. We acknowledge the R-based metaplotting script from the GitHub page of Jeffrey Grover (https://github.com/groverj3). We thank Nitish Dua and Mohammad Shariq for the help with immunostaining. We thank all the laboratory members for discussions and comments. This work was supported by NCBS-TIFR core funding, Department of Atomic Energy, Government of India, project identification no. RTI 4006 (1303/3/2019/R&D-II/DAE/4749 dated 16.7.2020) and grants (BT/IN/Swiss/47/JGK/2018-19; BT/PR25767/GET/ 119/151/2017) from Department of Biotechnology (DBT), Government of India. C.S. and K.P. acknowledge fellowship from DBT, India. These funding agencies did not participate in the designing of experiments, analysis, or interpretation of data or in writing the manuscript.
Author contributions: P.V.S. and V.H.S. designed all experiments, discussed results, and wrote the manuscript. V.H.S. performed most of the experiments and bioinformatics analyses. C.S. performed bioinformatics analysis. D.B. generated transgenic lines. K.P. performed electron microscopy and micro-CT scanning. S.R. performed confocal imaging. T.C. and R.A.M. generated and analyzed the nrp(d/e)2 sRNA data sets. All authors have read and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277353.122.
-
Freely available online through the Genome Research Open Access option.
- Received September 22, 2022.
- Accepted April 16, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








