
Genome-wide evaluation of the effect of short tandem repeat variation on local DNA methylation
Abstract
Short tandem repeats (STRs) contribute significantly to genetic diversity in humans, including disease-causing variation. Although the effect of STR variation on gene expression has been extensively assessed, their impact on epigenetics has been poorly studied and limited to specific genomic regions. Here, we investigated the hypothesis that some STRs act as independent regulators of local DNA methylation in the human genome and modify risk of common human traits. To address these questions, we first analyzed two independent data sets comprising PCR-free whole-genome sequencing (WGS) and genome-wide DNA methylation levels derived from whole-blood samples in 245 (discovery cohort) and 484 individuals (replication cohort). Using genotypes for 131,635 polymorphic STRs derived from WGS using HipSTR, we identified 11,870 STRs that associated with DNA methylation levels (mSTRs) of 11,774 CpGs (Bonferroni P < 0.001) in our discovery cohort, with 90% successfully replicating in our second cohort. Subsequently, through fine-mapping using CAVIAR we defined 585 of these mSTRs as the likely causal variants underlying the observed associations (fm-mSTRs) and linked a fraction of these to previously reported genome-wide association study signals, providing insights into the mechanisms underlying complex human traits. Furthermore, by integrating gene expression data, we observed that 12.5% of the tested fm-mSTRs also modulate expression levels of nearby genes, reinforcing their regulatory potential. Overall, our findings expand the catalog of functional sequence variants that affect genome regulation, highlighting the importance of incorporating STRs in future genetic association analysis and epigenetics data for the interpretation of trait-associated variants.
One of the main challenges in the genomics era is to associate genetic variation to human traits and disease. Over the past two decades, genome-wide association studies (GWAS) have successfully identified many susceptibility loci for a wide range of human phenotypes, revolutionizing the field of human genetics (Tam et al. 2019). Despite these successes, the interpretation of GWAS signals represents a challenging task as they are often located within noncoding regions of the genome and manifest as complex linkage disequilibrium (LD) blocks where identification of the causal variant(s) can be difficult. Because of these shortcomings, GWAS require complementary studies that go beyond the statistical output, helping to aid the biological interpretation of the identified signals and, eventually, identification of the genuine causal variant(s). In this sense, the discovery of quantitative trait loci (QTLs) often allows characterization of the functional consequences of sequence variation on gene expression (eQTLs) (Dixon et al. 2007; Göring et al. 2007; Stranger et al. 2007), DNA methylation (mQTLs) (Martin-Trujillo et al. 2020; Villicaña and Bell 2021), histone modifications (McVicker et al. 2013), or chromatin accessibility (Degner et al. 2012), thus providing insights into the underlying molecular mechanism of a given trait or disease. However, because of either statistical power or technical limitations, QTL studies have been mainly focused on common single-nucleotide variants (SNVs), ignoring low-frequency SNVs or other relevant sources of genetic variation such as tandem repeats (TRs).
Short tandem repeats (STRs), also known as microsatellites, are stretches of repeated units of DNA with motifs 2–6 bp in size that are distributed broadly across the genome and, in total, make up ∼3% of the human genome (Hannan 2018). Because of their repetitive nature, many STRs show some of the highest spontaneous mutation rates in the genome (Sun et al. 2012), which is typically much higher than that of SNVs. As a consequence of this instability, STRs show a high degree of length polymorphism within and across human populations (O'Dushlaine and Shields 2008; McIver et al. 2011; Willems et al. 2014; Mallick et al. 2016), representing an important source of human genetic variation that, in some cases, has been implicated in human disease (Hannan 2018). Multiple studies have shown transcriptional and epigenetic effects associated with STR length polymorphism, indicating that they can act as modulators of genome function (Gymrek et al. 2016; Quilez et al. 2016; Fotsing et al. 2019; Garg et al. 2020). Although the functional consequences of polymorphic STRs have been extensively studied in relation to gene expression (Gymrek et al. 2016; Quilez et al. 2016; Fotsing et al. 2019), there are few studies that have assessed their impact on epigenetic mechanisms such as DNA methylation. One of these studies has shown that the length of some STRs can modulate methylation levels at local CpG sites (CpGs; henceforth termed mSTRs) that, in turn, regulate the expression levels of nearby genes (Quilez et al. 2016). However, this study only assessed STRs located within the promoter regions of genes and thus overlooked the effect of STRs that map within introns or other functional regions such as enhancers. Supporting the likely widespread functional role of STRs on DNA methylation, we recently showed that many large TRs (those with motif lengths ≥10 bp, also known as variable number of tandem repeats [VNTRs]) act to regulate local DNA methylation (Garg et al. 2021). Given that DNA methylation plays a role in multiple biological processes such as the regulation of tissue-specific gene expression, X Chromosome inactivation, and genomic imprinting, and has been involved in a wide variety of human diseases ranging from developmental disorders to different types of cancer (Portela and Esteller 2010; Smith and Meissner 2013; Monk et al. 2019), the comprehensive study of the mechanisms influencing DNA methylation will provide new insights into the biological pathways and molecular mechanisms underlying human traits and diseases and will possibly implicate STRs in additional traits/disorders.
Here, we hypothesized that STRs genome-wide can act as functional elements of the human genome by modulating local DNA methylation profiles. To test our hypothesis, we performed a QTL analysis of variation in DNA methylation in relation to STR length using STR genotypes derived from short-read whole-genome sequencing (WGS) and DNA methylation levels generated from the lllumina Infinium MethylationEPIC BeadChip (henceforth termed the Illumina 850K array) in two independent cohorts of 245 and 484 individuals. Subsequently, we determined the set of likely causal mSTRs through fine-mapping analysis, determined their co-occurrence with eQTL, examined the population stratification, and, finally, by integrating GWAS signals, examined their contribution to human phenotypes, thereby providing insights into potential mechanisms that could underlie a fraction of human traits.
Results
Identification of cis-acting mQTLs
As a part of our pipeline to define a set of STRs with putative regulatory roles in DNA methylation (Fig. 1), we first performed a genome-wide QTL analysis between STR genotypes and DNA methylation levels of nearby CpGs in a discovery cohort of 245 samples collected by the Pediatric Cardiac Genomics Consortium (PCGC). This analysis identified a total of 19,129 pairwise associations at Bonferroni-corrected P < 0.001, comprising 11,870 and 11,774 unique mSTRs and CpGs, respectively (Supplemental Table S1). Overall, we did not observe any significant bias in the direction of the effect; that is, longer STR alleles were equally likely to associate with gains or losses of methylation at nearby CpGs.
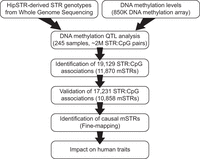
Overview of DNA methylation QTL analysis study design. Schematic summarizing the main steps used to identify functional STRs that modulate local DNA methylation profiles. Briefly, we performed (1) pairwise association analysis between STR length and DNA methylation levels within ±50 kb in a discovery cohort, (2) replication analysis in a second independent cohort, (3) fine-mapping analysis to define a set of causal mSTRs that act as DNA methylation regulators in cis, and finally (4) evaluation of the contribution of fine-mapped mSTRs to human traits by integration of tag SNVs with published GWAS signals.
We observed that the vast majority of mSTRs are located within either intronic (n = 5960) or intergenic (n = 5577) regions, with only a minority located within coding (n = 28) or 5′ and 3′ untranslated regions (UTRs; n = 305). Despite being abundant, intergenic STRs are underrepresented in the set of mSTRs (OR = 0.74, Fisher's exact test P < 0.0001). Conversely, we observed relative enrichments of mSTRs within introns (OR = 1.27, P < 0.0001), coding regions (OR = 2.57, P < 0.0001), enhancers (OR = 2.52, P < 0.0001), and promoter regions (defined as TSS ± 2 kb, OR = 2.57, P < 0.0001) compared with all STRs tested. Although these results are consistent with a functional role of mSTRs, we cannot rule out that they might be confounded owing to the design of the Illumina 850K array, which is biased for functional elements (Moran et al. 2016).
Although 70.8% (n = 8416) of the mSTRs associated with methylation levels of a single CpG, the remaining fraction of mSTRs associated with multiple CpGs. These mSTRs are frequently located outside gene promoter (n = 343) or enhancer regions (n = 290), with the vast majority being located in intronic (n = 1778) and intergenic (n = 1556) regions (for summary, see Supplemental Table S2). Within this set of mSTRs, we observed interesting associations. For example, we observed a positive association affecting multiple CpGs that involves a CCCCG repeat (Chr 5: 170,861,820–170,861,870; hg38) present in the promoter region of RANBP17 [MIM: 606141] (Fig. 2A), a gene that encodes a nuclear transporter receptor involved in the trafficking of molecules across the nuclear pore complexes. Similarly, we also identified a negative association between the length of an intronic AT repeat (Chr 15: 26,593,982–26,594,004; hg38) and methylation levels of multiple CpGs located in the promoter region of GABRB3 [MIM: 137192] (Fig. 2B), which has been suggested to play a role in neurotransmission and be involved in developmental and epileptic encephalopathy (Allen et al. 2013). Moreover, we also identified a functional GT repeat located at an intronic enhancer element of SRGAP1 [MIM: 606523], a gene involved in nonmedullary thyroid cancer and congenital anomalies of kidney and urinary tract (He et al. 2013; Hwang et al. 2015), which regulates the methylation levels of two CpGs present within the same enhancer (Fig. 2C). Additional examples of associations are included in Supplemental Figure S1.
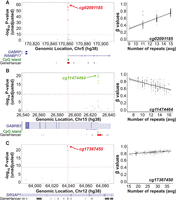
Examples of cis associations between STR length and local CpG methylation levels. Each Manhattan plot shows the association signals with CpG methylation ±50 kb of a genotyped STR: (A) Chr 5: 170,861,820–170,861,870, (B) Chr 15: 26,593,982–26,594,004, and (C) Chr 12: 64,039,629–64,039,660. Each circle represents a CpG, with significant associations shown either in red (positive correlations) or green (negative correlations). Panels A and B show a consistent positive and negative correlation between the STR length and β-values of multiple CpGs that map in the promoter region of RANBP17 and GABRB3, respectively. Panel C shows a consistent positive correlation between the STR length and β-values of two CpGs located at an enhancer. The location of the STR is shown by the vertical red line. The horizontal gray dashed line indicates the genome-wide significance threshold (Bonferroni-adjusted P < 0.001). Manhattan plots are annotated with genes (blue), CpG islands (green), enhancers (gray bars), and promoters (red bars) using NCBI RefSeq, cpgIslandExt, and GeneHancer tracks from the UCSC Genome Browser, respectively. In each case, the right panel shows a scatter plot of the average diploid STR genotypes (x-axis) and methylation β-values (y-axis) of the strongest associated (by P-value).
To further characterize mSTRs, we investigated the properties of their motifs according to both length and sequence content. In line with previous findings (Quilez et al. 2016), we observed a positive trend between motif length and functionality of the mSTRs (R2 = 0.74, P = 0.063) (Supplemental Table S3), with dinucleotide repeats being the least likely class of STRs to act as mQTLs (odds ratio 0.76, Fisher's exact test P < 0.0001) (Supplemental Fig. S2A). To investigate whether any motif was over- or underrepresented among the mSTRs, we first grouped motifs that were the same upon circular permutation or reverse complementation (e.g., AC, CA, GT, and TG were considered as a single motif) and then calculated the relative enrichment for each. After Bonferroni correction for the number of unique motifs tested, three were significantly underrepresented (OR < 0.9, Bonferroni-corrected P < 0.05) and 10 motifs were significantly overrepresented (OR > 1.1, Bonferroni-corrected P < 0.05) across the set of mSTRs (Supplemental Fig. S2B). Among the enriched motifs, we observed several with high GC content, such as CG, CCG, ACGC, and CCCC. Conversely, those motifs with low GC content (e.g., AC and AT) were depleted from the set of mSTRs. This finding is consistent with the strong influence of local CpG density on DNA methylation status.
Finally, we also determined the separation between each mSTR and its associated CpG. In line with previous mQTL studies of SNVs, STRs, and larger TRs (Kerkel et al. 2008; Gutierrez-Arcelus et al. 2013; Banovich et al. 2014; Smith et al. 2014; Quilez et al. 2016; Garg et al. 2021), we observed that mSTRs tended to occur close to their target CpGs (Supplemental Fig. S3), with an average separation of 645 bp.
Replication analysis of methylation STRs
We next performed a replication analysis using 484 quality-filtered samples collected from the Parkinson's Progression Markers Initiative (PPMI) cohort (https://www.ppmi-info.org/). Following the same statistical method as previously applied in our discovery cohort, we identified 53,908 significant STR:CpG pairwise associations (27,449 unique STRs and 28,375 CpGs) at Bonferroni-adjusted P < 0.001. This represents an approximately threefold increase in the number of associations observed in our initial analysis using 245 samples from the PCGC cohort, which is most likely owing to enhanced statistical power as a result of the larger sample size of the PPMI cohort (n = 484) compared with PCGC (n = 245).
Overall, we replicated 17,231 (89.7%, including 10,858 STRs and 10,714 unique CpGs) of the STR:CpG associations identified in the PCGC discovery cohort (Fig. 3; Supplemental Table S1), showing the robustness of our findings. Furthermore, 99.9% of these signals showed the same direction of effect between the two cohorts. Only replicated STR:CpG associations were retained for further analysis.
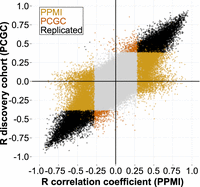
Replication of STR:CpG associations. Scatter plot showing the comparison between the correlation coefficient (r) obtained from the association analysis in our discovery (PCGC, n = 245; y-axis) and replication cohort (PPMI, n = 484; x-axis). Each dot represents a single association test (STR:CpG). Dots are color-coded according to their significance (Bonferroni-adjusted P < 0.001). Light gray dots represent nonsignificant STR:CpG associations identified in our discovery and replication analysis; black dots represent significant associations present in both cohorts. Light and dark orange dots represent significant associations observed solely in either our replication or discovery cohort, respectively.
Given that the PPMI cohort includes samples collected from patients with Parkinson's disease and healthy individuals, in order to investigate whether diagnosis status influenced our findings, we repeated the association analysis separately in patients (n = 362) and controls (n = 117) and observed that 99.9% of the validated STR:CpG associations retained the same directionality (Supplemental Fig. S4) in both groups. This result indicates that the effect of STR variation on DNA methylation is independent of disease status. Similarly, we further confirmed that our results were not influenced by sex by repeating our association analysis using only male or female individuals in both the PPMI (females = 159; males = 325) and PCGC (females = 105; males = 140) cohorts and by comparing the directionality of the resulting associations (PCGC: Spearman's rank correlation = 0.875, P < 2.2 × 10−16; PPMI: Spearman's rank correlation = 0.960, P < 2.2 × 10−16) (Supplemental Fig. S5).
Finally, to ensure the robustness of the STR genotypes included in our association analysis, we compared the genotypes obtained for 4167 mSTRs for which genotypes were obtained from both Illumina WGS and Pacific Biosciences (PacBio) HiFi long-read sequencing data. We observed a very high concordance between genotypes obtained from the two sequencing technologies (Spearman's rank correlation coefficient = 0.989, P < 0.0001) (Supplemental Fig. S6), indicating that HipSTR provides highly accurate genotypes that can be used in QTL analyses.
Identification of mSTRs with putative regulatory effects
As variation in DNA methylation can also be influenced by other genetic variants besides STRs (Kerkel et al. 2008; Gutierrez-Arcelus et al. 2013; Banovich et al. 2014; Smith et al. 2014), we next attempted to determine whether candidate mSTRs are the causal modulators of local DNA methylation or whether, instead, they simply occur in LD with the true causal variants that lie elsewhere within the region. To address this question, we performed a conditional analysis adjusting each STR:CpG association based on the genotype of the SNV most strongly associated with methylation levels of the associated CpG. Upon conditioning, we considered an mSTR as the likely causal regulator of DNA methylation levels when the direction of the STR:CpG association remained the same and with P < 0.05. To identify the lead associated SNV for each CpG, we first extracted SNV genotypes present within the locus (CpG ± 250 kb) and then tested each SNV for association with DNA methylation levels of the target CpG (median of 1168 SNVs tested per locus). After selecting the lead SNV for each CpG associated with a candidate mSTR, we repeated the STR:CpG association analysis using only those samples that were homozygous for the major allele of the corresponding lead SNV. Doing so, we observed that 2480 mSTRs out of the 17,231 (14.4%) replicated mSTRs still retained significant associations, indicating that these act to independently modulate DNA methylation (Fig. 4A).
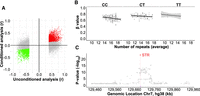
Genetic fine-mapping of mSTRs. (A) Comparison between results obtained with and without conditioning association tests for the lead associated SNV. Red (positive) and green (negative) dots represent individual STR:CpG associations that retain the same directionality and remain significant (P < 0.05) upon conditioning for the lead SNV. Conversely, gray dots represent STR:CpG associations that either became nonsignificant or show opposite directionality after conditioning, suggesting that the STR is not the causal variant at the locus. (B) An example of a conditional analysis for the STR located at the intronic region of KLHDC10 (Chr 7: 130,090,768–130,090,795). Scatter plots showing resulting associations between the methylation levels (β-values) of the mSTR-target CpG (probe ID cg09813917) and STR allele size for samples that were homozygous for the major allele (left) or were heterozygous (middle), and homozygous samples for the minor allele (right) of the lead SNV (rs4731662, Chr 7: 130,142,537). Upon conditioning for this SNV, the association of STR with methylation level retains the same directionality and remains significant (P < 0.05), suggesting that the STR acts as an independent regulator of methylation. (C) Regional Manhattan plot showing genetic associations (−log10 P-values; y-axis) against chromosome position (x-axis) for the Chr 7: 130,090,768–130,090,795 mSTR (red) and SNVs located within ±250 kb with methylation levels of the cg09813917 CpG probe. Across all the tested variants, the mSTR shows the strongest association and is predicted by CAVIAR to be the causal variant with 99% probability.
To provide further evidence of causality for mSTRs, we also performed fine-mapping of the putative responsible variant at each mQTL locus (associated CpG ± 250 kb). Here, we used the candidate mSTR, together with the top 300 associated local SNVs based on the resulting P-values from SNV:CpG association tests. Subsequently, we estimated the causal probability for each of the tested variants using CAVIAR (Hormozdiari et al. 2014). Using these probabilities, we identified 715 mSTR:CpG pairwise associations (585 and 685 unique STRs and CpGs, respectively) in which the mSTR showed the highest causal probability among the tested variants and had causal probability >0.3 (Supplemental Fig. S7), suggesting that the mSTR is most likely the causal variant underlying these associations.
We considered these 585 mSTRs as fine-mapped mSTRs (fm-mSTRs) (Supplemental Table S4). Notably, of these, our conditional analysis suggested that 398 of them (68%) act as independent modulators of DNA methylation (Supplemental Fig. S8). Given that the results from conditional analysis can be influenced by the allele frequency of the lead SNV, we decided to follow a conservative approach and consider mSTR as the causal variant (fm-mSTRs) according to our CAVIAR results.
We next explored the genomic features associated with fm-mSTRs. Similar to mSTRs, fm-mSTRs are frequently located at intergenic (n = 268) or intronic (n = 273) regions, with 29.6% of them overlapping functional genomic regions such as gene promoter (n = 118) and enhancer (n = 55) regions. Despite their relatively low abundance, fm-mSTRs showed 7.6-fold (Fisher's exact test P < 0.0001) and threefold enrichment (Fisher's exact test P < 0.0001) for gene promoter and enhancer elements, respectively, compared with the remaining tested STRs (n = 131,050). Genomic location and feature enrichment for the set of fm-mSTRs are summarized in Supplemental Table S5.
An example of an fm-mSTR is shown in Figure 4, where the conditional analysis for the lead SNV (Fig. 4B) did not affect the association between the fm-mSTR (Chr 7: 130,090,768–130,090,795, hg38) located intronic within KLHDC10 [MIM: 615152] and its target CpGs (Fig. 4C). Subsequent CAVIAR analysis confirmed that this STR was the likely causal genetic driving the observed methylation variation. Altogether these data suggest that a subset of our mSTRs represent the major genetic modulator of local DNA methylation profiles.
Additional replication of fm-mSTRs using DNA methylation data derived from Oxford Nanopore technology sequencing reads
We further validated a subset of our set of fm-mSTRs using DNA methylation values extracted from long-read Oxford Nanopore technology (ONT) WGS data. Despite using different techniques to measure DNA methylation (Illumina 850K array vs. ONT WGS) in different sample types (whole-blood vs. immortalized lymphoblastoid cells) and with a minimal sample size for the ONT data derived from individuals with highly variable genetic ancestry, we observed that 68.1% of the 600 STR:CpG associations generated using the ONT data had the same directionality as observed in the PCGC cohort (Supplemental Fig. S9). This result not only provides additional evidence about the robustness of our associations and suitability of DNA methylation measurements generated by the Illumina 850K array but also shows that our findings are generalizable across DNA methylation detection methods.
Co-occurrence of mSTRs with eQTLs
Given that DNA methylation can regulate the transcriptional activity of target genes, we used the set of polymorphic STRs to perform eQTL mapping using gene expression data from 405 individuals collected by the PPMI consortium. After multiple testing, we identified a total of 5203 significant STR:transcript associations (Bonferroni-adjusted P < 0.001), comprising 3856 and 1652 unique STRs (eSTRs) and transcripts, respectively. Similar to mSTRs, eSTRs tend to map in close proximity to the associated genes (Supplemental Fig. S10).
We observed that 12.6% (n = 68) of the fm-mSTRs tested for eQTLs (n = 540) are also able to regulate the expression levels of nearby genes (Supplemental Table S6). Among these repeats, we observed an interesting association involving an AG repeat (Chr 6: 42,959,499−42,959,540, hg38) located at the promoter region (TSS ± 2 kb) of GNMT [MIM: 606628], where the length of this STR showed both a strong negative correlation with the expression levels of GNMT and a strong positive correlation with DNA methylation levels of CpGs located within its promoter region (Fig. 5), respectively. This association is consistent with the inverse correlation between DNA methylation and transcriptional activity at promoter regions. Additional examples are included in Supplemental Figure S11.
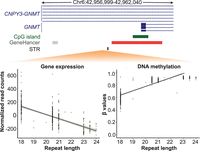
STR-directed DNA methylation leads to changes in gene expression levels of target genes. Scatter plots showing opposite correlation between the length of an AG repeat (Chr 6: 42,959,499–42,959,540) with the expression (left plot) and promoter DNA methylation (right plot) of the GNMT gene. Each dot represents data from one individual of the PPMI cohort, whereas the diagonal line represents the best-fit for all data points. Genomic locations of the STR (black), gene (blue), CpG islands (dark green), promoter (red), and enhancers (gray) are shown above the plots for the interval Chr 6: 42,956,999–42,962,040 (hg38).
Overall, these findings strengthen the regulatory role of a subset of our set of fm-mSTRs, providing insights into the functional consequences of the STR-directed DNA methylation.
Population stratification of fine-mapped mSTRs
We next attempted to explore the population structure of our set of polymorphic STRs in samples from The 1000 Genomes Project (1KGP) using the VST index and to detect loci with differences in allele size across continental populations as a result of possible selection. Using filtered HipSTR genotypes, we computed VST for 126,171 STRs that were represented across all the tested ancestries (100 or more individuals/ancestry). We observed 750 STRs (0.6%) showing evidence of population differentiation (VST > 0.3 and P < 0.01) in at least one of the tested superpopulations (Fig. 6A), suggesting that the allelic diversity at this subset of STRs might have been subject to either genetic drift, population bottlenecks, or selective pressures. From these, we observed that the vast majority of loci showed divergent alleles in individuals with African ancestry (n = 743, 99.1%), which is consistent with the greater genetic diversity observed in this population (The 1000 Genomes Project Consortium 2015).
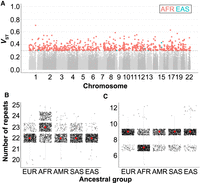
Population stratification of fm-mSTRs. (A) Manhattan plot showing the highest VST index for 126,676 polymorphic STRs across the five tested superpopulations. For each STR and ancestry, VST index was calculated using the individual allele size. VST values are color-coded according to ancestry when VST > 0.3 (gray dashed line). (B,C) The allele size distribution for di- and pentanucleotide STRs located at the promoter and 5′ UTR regions of the CLEC17A (B; Chr 19: 14,582,958–14,582,997) and CRYBB2P1 (C; Chr 22: 25,448,062–25,448,099) genes, respectively. For each ancestral group, median of allele size is depicted by red dot. (EUR) European, (AFR) African, (AMR) American, (SAS) South Asian, (EAS) East Asian.
We observed a 2.73-fold enrichment (Fisher's exact test P = 0.0073) (Supplemental Table S7) for STRs with high population divergence in our set of fm-mSTRs compared with the whole set of polymorphic STRs. For instance, we observed a TG repeat (Chr 5: 131,204,743–131,204,761) located within the promoter region of CLEC17A [MIM: 616838], where the largest alleles are exclusively present in individuals with African ancestry (Fig. 6B). Similarly, we observed a GCGTG repeat within the 5′ UTR of CRYBB2P1 [MIM: 123620], which is involved in multiple types of cataracts, where the smallest alleles are present in individual alleles of African ancestry (Fig. 6C).
Fine-mapped mSTRs contribute to human phenotypic variation
To provide insights into the molecular mechanisms underlying phenotypic variation, we next investigated the potential involvement of fm-mSTRs in human traits. Using SNV genotypes from the regions flanking fm-mSTRs (±250 kb), we computed the local LD structure of each locus to identify tag SNVs that strongly correlated with mSTR genotypes (r2 ≥ 0.8) and then compared these SNVs against published GWAS signals. Of the 585 fm-mSTRs defined by CAVIAR, 187 (31.9%) (Supplemental Fig. S12A) were well tagged by at least one nearby SNV. As expected, we observed both (1) an inverse trend between the number of different alleles of fm-mSTRs and their ability to be tagged by nearby SNVs and (2) that tagging SNVs tended to lie in close proximity to the fm-mSTRs (Supplemental Fig. S12B).
From the GWAS catalog, we focused on 139,337 autosomal variants with P < 5 × 10−8 derived from 3377 published GWAS studies. Direct comparison between these GWAS signals and the set of SNVs that tag fm-mSTRs revealed 48 overlaps, representing a set of fm-mSTRs that may contribute to 109 different traits (Supplemental Table S8). These include a wide range of health-related traits and multiple human diseases. For instance, we observed that the previously described AG repeat (Chr 6: 42,959,499–42,959,540) located in the promoter region of GNMT [MIM: 606628] (Fig. 5) associated with Apolipoprotein B levels and low-density lipoprotein (LDL) cholesterol levels. In line with this association, murine studies have shown that deficiency in GNMT results in hyperlipidemia, with Gnmt−/− showing higher LDL levels compared with WT mice (Liao et al. 2012). We also observed fm-mSTRs associated with behavioral, neurodegenerative, and autoimmune diseases. For instance, a GT repeat located (Chr 15: 58,750,646–58,750,679) in the promoter region of ADAM10 [MIM: 602192] was linked to Alzheimer's disease (AD) and schizophrenia. ADAM10 encodes for a metalloprotease that is involved in the cleavage of the amyloid-β (Aβ) protein precursor. Transgenic mice have shown that overexpression of human ADAM10 leads to an excess of Aβ extracellular deposition (Yuan et al. 2017), which is the primary cause of AD according to the amyloid hypothesis. We also observed an association between a CCG repeat (Chr 7: 99,144,016–99,144,048) in the promoter of SMURF1 [MIM: 605568] and inflammatory bowel disease (IBD). SMURF1, which encodes for an E3 ubiquitin ligase and acts as a mediator of autophagy (Lassen and Xavier 2017), has been identified as a risk locus for IBD (Jostins et al. 2012; Lassen and Xavier 2017).
Discussion
In this study, we performed genome-wide QTL mapping between the length of polymorphic STRs and local DNA methylation levels in a discovery cohort, followed by replication analysis in a second independent cohort. After application of stringent statistical thresholds, we identified a total of 19,129 cis mQTLs that comprise pairwise associations between 11,870 mSTRs and 10,714 CpGs, 90% of which were replicated in a second cohort. We then used conditional analysis and a statistical fine-mapping approach to investigate the causality of our candidate mSTRs, which determined that the length variation of 585 (4.9%) of these candidate loci represents the most likely genetic driver of methylation variation at the tested CpGs. Finally, we observed that our set of fm-mSTRs are enriched for loci showing evidence of population differentiation among major ancestral groups, and identified 48 that lie in strong LD with nearby SNVs that have been associated with human traits by prior GWAS, suggesting these STRs may potentially be the true causal variants at these loci.
Several possible mechanisms by which variation of an STR might regulate local DNA methylation can be proposed. A logical mechanism implies the alteration of CpG density in a given locus by the insertion of CpG-containing repeats. This is the proposed mechanism for some repeat expansion disorders such as fragile X syndrome [MIM: 300624], where expansion of the CGG repeat in the 5′ UTR of FMR1 [MIM: 309550] becomes methylated and results in transcriptional silencing (Sutcliffe et al. 1992). A second possible mechanism could be that STR variation modulates the binding of transcription factors (TFs) to specific DNA motifs by creating or disrupting them, modifying the affinity of the TF for the DNA in a copy-dependent manner, or altering the spacing between flanking regulatory regions (Bagshaw 2017). Although this mechanism seems to be the most obvious by which STRs can impact gene expression, it can also be applied to DNA methylation as we and others have recently shown that the binding of TFs can impact local DNA methylation profiles (Onuchic et al. 2018; Martin-Trujillo et al. 2020). Changes at the sequence level can also influence the regional structure of the DNA by inducing noncanonical DNA formations such as cruciforms, hairpins, or G-quadruplexes (Wells 2007), which can have functional impacts on the genome (Mao et al. 2018; Georgakopoulos-Soares et al. 2022a,b). Furthermore, variation of the length of STRs can result in changes at both DNA and chromatin level. For instance, GAA expansions that underlie Friedreich's ataxia (FRDA) [MIM: 229300] (Hannan 2018) are associated with a local depletion in nucleosomes, increased DNA methylation, and a repressive chromatin structure (Zhao et al. 2015). Understanding the molecular mechanisms through which STR variation regulates local epigenetics can expand our knowledge of the molecular basis underlying the control of DNA methylation, which remains elusive.
The LD structure of the human genome results in the presence of large haplotype blocks that can make the determination of causal variants challenging. For example, variation in DNA methylation that is associated with polymorphic STRs could potentially be explained by additional nearby variants present in the locus such as SNVs. Under this scenario, in association analysis such as QTL mapping, genetic variation corresponding to the causal variant can be captured by tagging SNVs. Applying both conditional analysis and a Bayesian fine-mapping method (CAVIAR), we were able to discriminate a set of 585 mSTRs from our candidate list (4.9%) as the likely main genetic drivers of DNA methylation variation at the tested loci. Although this represents a small fraction of the total mQTLs we identified, these results are in line with those described by Fotsing et al. (2019), in which ∼10% of the STRs identified as transcriptional regulators of nearby genes showed the highest causality score compared with other variants within a given locus. However, we cannot rule out the possibility that more of our mSTR candidates represent the actual causal variant as (1) some STRs might be refractory to CAVIAR analysis owing to their effect size and/or the sample size of our cohort, (2) we applied stringent criteria to consider an mSTR as causal that can yield false-negative results, and, finally, (3) a fraction of our candidate mSTRs can regulate DNA methylation coordinately together with other variants, as was recently shown for SNV eQTL (Abell et al. 2022). Although conditional analysis has been widely used in fine-mapping applications to identify variants that act as independent regulators in a given locus, this approach presents some limitations compared with statistical fine-mapping methods, including the following. First, results obtained from conditional analysis are likely to be influenced by the MAF of the lead SNV. For instance, conditional analysis can lack the statistical power to detect independent signals owing to the reduced sample size upon conditioning for a lead SNV. This is particularly relevant in cases in which the allele frequency of the lead SNV is common across the population. Conversely, where the lead SNV is rare, conditioning tends to lack sensitivity to detect secondary signals. Collectively, this can lead to an inaccurate identification of variants that can act as independent regulators. Second, conditional analysis does not consider local LD structure, and thus, lead SNVs can be mistakenly selected. Third, variants need to be interrogated individually, which is computationally intensive. In contrast, statistical fine-mapping methods such as CAVIAR allow the integration of LD structure and association statistics in the model, allowing the evaluation of multiple variants simultaneously and providing an estimate of causality for each. In summary, our results highlight the importance of integrating fine-mapping analysis in association analysis in order to discriminate the genuine causal variants from the whole bulk of signals and prioritize them for further analysis or functional studies.
Using tagging SNVs (r2≥ 0.8) as a proxy for our fm-mSTRs, we identified 48 fm-mSTRs that are potentially linked to a variety of human traits and diseases. It should be noted that in this study, we could only evaluate STRs that were well tagged by flanking SNVs, suggesting that additional STRs that are not well tagged by nearby SNVs could potentially underlie human phenotypes. Importantly, it has been postulated that missing heritability can be partially attributed to variants in the human genome that are poorly assayed by standard genotyping methods (Hannan 2010). Therefore, the addition of STR variation to existing genetic models has the potential to explain a fraction of the missing heritability evident in SNV-based studies (Manolio et al. 2009). Currently, with the availability of large biobanks that include sequencing data from thousands of individuals together with extensively annotated phenotype data, it is now possible to perform analysis of well-powered phenome-wide association studies to comprehensively evaluate the contribution of STR variation to human phenotypes, which will likely lead to the identification of novel disease-relevant STRs. Annotation of trait/disease modifying STRs with data obtained from QTL studies, similar to the work presented here, will provide insights into the molecular mechanism underlying a particular human trait. However, it should be noted that association does not equate with causality; therefore, additional functional validation will be required to definitively prove causality between STR-directed DNA methylation and the associated traits.
Despite the robustness of our findings, there are some limitations in our study. One is the lack of allelic DNA methylation information as a result of the use of array data, which provides an aggregate measure from both alleles. Because of this limitation, despite HipSTR providing individual genotypes for each allele, we used averaged TR allele sizes as input for association analysis. Another limitation arising from the use of DNA methylation arrays is their limited genome coverage, as the Illumina 850K array only samples ∼3% of the CpGs present in the human genome, which are heavily biased toward those mapping within regulatory regions such as gene promoters or enhancers. An alternative to arrays would be the use of whole-genome bisulfite sequencing or long-read sequencing data (Loman et al. 2015). Although these approaches would allow the assessment of allelic DNA methylation across the whole genome, available sample sizes for these data sets are currently limited. In addition to these technical limitations, our association test assumed linearity between the two studied variables, namely, that changes in DNA methylation are proportional to STR length. Therefore, we had limited power to capture more complex nonlinear effects of STR alleles on DNA methylation, such as those previously described in yeast (Vinces et al. 2009). Another limitation in our study is the lack of statistical power to capture associations between rare STR alleles and flanking SNVs. However, the presence of rare alleles across our set of polymorphic STRs might also be limited as unusually long alleles are unlikely to be captured by the HipSTR algorithm, which is only able to provide genotypes for STR-spanning reads, and therefore, large and rare expanded STR alleles will be refractory to genotyping. Furthermore, we calculated the LD structure between STR and SNV genotypes using as an input the average of allelic copy number for STRs, which may lead to increased error in LD estimates at some STR loci. For example, at an STR that has alleles of eight, 10, and 12 copies, an individual who is homozygous for the 10-copy STR allele will be considered the same as an individual who is heterozygous for eight- and 12-copy STR alleles, as in both cases, the average STR allele size is 10. Finally, throughout this study, we used both genotype and methylation data derived from whole-blood samples, which likely give inherently limited insight into epigenetic regulation given that DNA methylation profiles can be highly cell-, tissue-, and developmental-stage specific. However, to date, blood-based studies represent the most common QTL mapping analysis owing to the availability of larger sample sizes and, consequently, greater statistical power.
In summary, our findings expand our previous work on promoter STRs and show that a fraction of STRs act as regulators of the genome function by modulating DNA methylation levels in cis, which ultimately can represent the molecular mechanisms underlying phenotypic variation and disease.
Methods
Description of the cohorts
For this work, we used DNA methylation and PCR-free WGS data sets from individuals collected by the PCGC (discovery cohort) and PPMI (replication cohort). All individuals included in this study provided proper informed consent for research use at the time of sample collection. Ethical approval for collecting blood samples was granted by the ethics committees on human studies of the institutions involved. This study was approved by the Institutional Review Board (IRB) of the Icahn School of Medicine at Mount Sinai under HS 20-00153.
PCGC cohort
WGS and DNA methylation data for 249 individuals were selected from the cohort collected by the PCGC. An extensive description of PCGC samples as well as further details about sample collection can be found in a summary publications released by the PCGC (Pediatric Cardiac Genomics Consortium et al. 2013; Hoang et al. 2018). Briefly, the PCGC cohort comprises individuals aged from newborn to 47 yr (mean, 8.2 yr) diagnosed with a range of congenital heart defects; conotruncal and left-sided obstructive lesions were the two most common diagnoses. Illumina 150-bp paired-end WGS data generated via PCR-free libraries from peripheral blood DNA (average of 36× genome coverage, range 25×–39×) were downloaded from the NCBI database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/ phs001138.v1.p2). Peripheral blood methylomes generated with the Illumina 850K array were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/; accession number GSE159930), normalized and processed as described previously (Martin-Trujillo et al. 2020). DNA methylation measurements were represented by β-values ranging from zero (unmethylated) to one (fully methylated). To ensure the accuracy of our DNA methylation measurements, we excluded any probe that mapped to multiple genomic locations according to BSMAP (Xi and Li 2009). We also used the genotypes obtained from GATK analysis of the WGS data in each sample and excluded β-values for any CpG that contained an SNV within either the probe-binding site or the interrogated CpG. After these filters, β-values for a total of 821,035 autosomal CpGs were used in downstream analyses.
We performed quality control (QC) of DNA methylation data and excluded four samples that showed divergence between self-reported and array-inferred gender using DNA methylation data from the sex chromosomes. No other samples were considered as outliers based on principal component analysis (PCA), density plots using DNA methylation of autosomal CpGs as an input (Supplemental Fig. S13A,B), or minimum call rate (>90%). A total of 245 samples were retained for downstream analysis. All PCGC data were aligned to hg19.
PPMI cohort
Data used in the preparation of this article were obtained from the PPMI database (https://www.ppmi-info.org/access-data-specimens/data), corresponding to 489 individuals for whom Illumina WGS and DNA methylation data generated with the Illumina 850K array were available (Marek et al. 2018). Applying the aforementioned QC protocols for DNA methylation data, we excluded one sample, retaining 488 samples for downstream analyses (Supplemental Fig. S14A,B). For 458 of these individuals, whole-blood expression profiling consisting of DESeq2 (Love et al. 2014) normalized read counts (RNA sequencing [RNA-seq]) for 54,576 autosomal transcripts were also available. From these data, expression data for 18,861 expressed transcripts were selected (median normalized read counts ≥ 10 across all samples) for downstream analysis. Similar to DNA methylation data, we performed a QC of RNA-seq data and excluded a total of 53 samples based on PCA, density plots, and expression profiles using normalized read count of the selected transcripts, retaining expression data for 405 PPMI samples (Supplemental Fig. S15).
Whereas Illumina WGS and RNA-seq data were aligned to hg38, DNA methylation data were aligned to hg19, respectively. Before our analysis, data aligned to hg19 were converted into hg38 coordinates using the liftOver tool provided by UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgLiftOver).
Genotyping and QC of STR genotypes in PCGC and PPMI cohorts
We used HipSTR (v0.4) to generate diploid STR genotypes (allele sizes) for each sample in the PCGC cohort at 1,490,769 autosomal STRs with motif size ranging between 1 and 6 bp, which are included in the catalog provided by HipSTR algorithm (Willems et al. 2017). Briefly, using aligned WGS reads as input, HipSTR generates a locus-specific stutter model to reduce potential noise derived from amplification and uses it together with haplotype-based alignments to generate diploid STR repeat length estimates for each individual and STR. Unlike algorithms that are able to genotype STRs with allele size greater than the read length (150 bp) (Tang et al. 2017; Dashnow et al. 2018; Mousavi et al. 2019), HipSTR is limited to genotype STR alleles smaller than the read length as it only uses reads that completely enclose the repeat and have sufficient anchoring sequences at both flanks (Willems et al. 2017).
HipSTR genotyping was performed simultaneously across all 249 samples in separate batches of 800 STRs. The resulting genotypes were concatenated using the concat function of BCFtools (v1.9) (Danecek et al. 2021) and subjected to QC per sample and STR. Only estimates that fulfilled default HipSTR quality parameters were retained (https://hipstr-tool.github.io/HipSTR/#default-filtering; ‐‐min-call-qual 0.9 ‐‐max-call-flank-indel 0.15 ‐‐max-call-flank-stutter 0.15 ‐‐max-call-allele-bias -2 ‐‐min-call-strand-bias -2). After applying these filters, we focused on those STRs with (1) a minimum genotyping rate of ≥50%, (2) motif length of ≥2 bp, and (3) a minimum variation within the cohort, defined as three or more observed alleles and a nonmodal allele frequency (NMAF) ≥ 0.1, that is, frequency of the major allele <0.9. After this selection, a total of 132,092 polymorphic STRs were retained. Within each sample, we used the average length of the two alleles at each locus for each STR for subsequent analysis, including QC and association analyses.
To identify outlier samples, we performed QC analysis of the STR genotypes using density plots and PCA based on STR length per sample and locus as an input, as well as genotyping rate per sample (Supplemental Fig. S13C–E).
For the 489 samples in the PPMI cohort for which PCR-free WGS and DNA methylation data were available, allele size estimates for the set of polymorphic STRs were generated, filtered, and processed using the same parameters as described above for the PCGC cohort. Five samples were removed as these either were outliers on the density profile of STR lengths or had a low genotyping rate (<90%). After these filters, a total of 484 samples were retained for further analyses (Supplemental Fig. S14C,D).
WGS-derived SNV genotypes
For PCGC samples, SNVs were called using GATK (v2.7) from BWA-MEM-aligned WGS reads following best practices (Li and Durbin 2009; McKenna et al. 2010; DePristo et al. 2011; Van der Auwera et al. 2013). For any analysis involving SNV genotypes derived from PCGC samples, we focused on biallelic SNVs that passed GATK filters with minor allele frequency (MAF) ≥ 0.05, quality score (Q) ≥ 30, read depth ≥10, alternate allele fraction of 0.2–0.8 for heterozygous samples, and Hardy–Weinberg equilibrium P ≥ 0.001.
For PPMI samples, genotypes for SNVs were extracted from the VCF file directly downloaded from https://www.ppmi-info.org/.
Before association analysis with either DNA methylation levels or STR length, SNV genotypes were converted into dosage of the alternate allele (homozygous for major allele = 0, heterozygous = 1, and homozygous for minor allele = 2).
Mapping cis-methylation STRs in the PCGC cohort
After excluding samples that did not fulfill our QC criteria for either STR genotypes or DNA methylation data, 245 samples from the PCGC cohort were used for association analysis between STR length and DNA methylation levels. For this analysis, we selected variable CpG sites (defined as those with standard deviation of β-values >0.02) that were located within ±50 kb from the set of 132,092 polymorphic STRs. Focusing on those STR:CpG pairs for which both DNA methylation and STR genotypes were available for at least 50% of the samples, we performed association testing for a total of 1,950,668 STR:CpG pairs, involving 387,641 different CpG sites within the flanks of 131,635 polymorphic STRs (average of 14.8 CpGs per STR).
Given that DNA methylation levels can be confounded by multiple factors such as cell type, age, gender, and ancestral background, we adjusted the methylation β-values for age, gender, the top two ancestry-related principal components derived from PCA of SNVs (Pedersen and Quinlan 2017), and blood cell fractions estimated directly from the methylation data using the Houseman method (Supplemental Fig. S13F–H; Houseman et al. 2012). The resulting residuals were used to test the association between DNA methylation and STR length (based on the average of allele size) using the lm function in R. Multiple testing correction for the number of STR:CpG pairs evaluated was applied using the Bonferroni method.
Finally, STRs were annotated with closest gene and enhancers using the NCBI RefSeq and GeneHancer tracks obtained from UCSC Genome Browser. All annotations were performed using BEDTools closest function (v2.29.0) (Quinlan and Hall 2010) and custom R scripts.
Replication of mSTRs in the PPMI cohort
To replicate our findings, we used available WGS data and whole-blood DNA methylation profiles generated with the Illumina 850K array in 484 samples selected from the PPMI cohort, as described previously for the PCGC cohort. DNA methylation levels were adjusted for confounding factors, including age, gender, ancestry determined by the top three PCs derived from high-confidence SNVs, and estimated blood cell fractions inferred from DNA methylation as described previously (Supplemental Fig. S14F,G; Houseman et al. 2012). The resulting residuals were used to test for association with STR length using the lm function in R.
Evaluation of the accuracy of HipSTR genotypes for validated mSTRs
We tested the accuracy of HipSTR genotypes in 14 samples for which both Illumina WGS and PacBio HiFi long-read sequencing data generated by the Human Genome Structural Variation Consortium (HGSVC; https://www.internationalgenome.org/human-genome-structural-variation-consortium/) were available. Here, we performed analysis of Illumina WGS data using HipSTR and compared these against genotypes derived from PacBio HiFi long reads using the Tandem Repeat Genotyper algorithm (TRGT; https://github.com/PacificBiosciences/trgt) generated from the same 14 individuals. Before profiling STRs with TRGT, raw reads were first merged using pbmerge (pbbam; https://github.com/pacificbiosciences/pbbam/), converted into FASTQ format using bam2fastq (https://github.com/pacificbiosciences/bam2fastx/), and, finally, aligned using minimap2 v2.17 (Li 2018). Aligned reads were then used as input into TRGT, which provides allelic genotypes for a predefined set of TRs from long-read sequencing data. Using the catalog of STRs provided by TRGT, we were able to genotype a total of 4839 mSTRs. To ensure robustness in these STR genotypes, we removed any that were supported by fewer than five overlapping reads. We also genotyped this set of 4839 mSTRs using HipSTR to analyze Illumina WGS data generated for these same 14 individuals. We then compared the repeat length, namely, average STR copy number, across 4167 validated mSTRs for which both HipSTR and TRGT genotypes were available for 10 or more individuals. Concordance between STR genotypes obtained from these two sequencing technologies was calculated using Spearman's rank correlation test.
Statistical fine-mapping of causal variants
We next sought to determine whether the identified mSTRs represented the causal genetic variants responsible for the variation of DNA methylation. To test this, we first performed a conditional analysis and, second, calculated the causal probability for each mSTR and additional genetic variants that have the potential to act as a functional element in the region (associated CpG ± 250 kb) using CAVIAR (Hormozdiari et al. 2014).
Conditional analysis
Using the same set of PCGC samples used in our initial association test, at each mSTR locus, we performed a conditional analysis using an identical statistical model as used in our initial STR:CpG association analysis after stratifying samples based on their genotype at the SNV that showed the strongest association with DNA methylation levels of the mSTR-associated CpG. Briefly, to identify the lead SNV for each mSTR-associated CpG, we first selected and extracted genotypes derived from WGS for all SNVs located within ±250 kb of the tested CpG. Then, after converting these genotypes into dosages based on the alternate allele count (zero, homozygous for alternate allele; one, heterozygous for alternate allele; two, homozygous for alternate allele), we performed pairwise association between each SNV genotype and the residuals obtained after adjusting β-values for confounding factors (see Mapping cis-Methylation STRs in the PCGC Cohort) using lm R function. Subsequently, once the lead SNV was identified, we repeated the STR:CpG association analysis, retaining only those samples that were homozygous for the major allele of the lead SNV (defined as the SNV with the most significant P-value). mSTRs were considered as independent regulators of DNA methylation levels when the resulting association upon conditioning had nominal P < 0.05 and the same directionality as in the unconditioned analysis.
CAVIAR
To further fine-map mSTRs within each mQTL locus (associated CpG ± 250 kb), we applied CAVIAR (Hormozdiari et al. 2014) to calculate the causal probability for our candidate mSTR and the top 300 most strongly associated SNVs with the β-values of the targeted CpGs. Briefly, CAVIAR is able to infer the causal probability for each of these 301 variants using the following as input: (1) the magnitude and direction of the association between them and the tested phenotype and (2) the LD structure across the locus. For each mQTL locus, the top 300 SNVs were selected on the basis of the resulting P-value obtained from pairwise SNV:CpG associations (as described above). Before use of CAVIAR, biallelic genotypes of the selected 300 SNVs were converted into dosages according to their alternate allele content (zero, one, and two) as described earlier. Then, local LD structure was computed and defined as the square of the Pearson correlation coefficient (r2) obtained from pairwise associations between genotypes among the selected genetic variants. Whereas dosage for the alternate allele was used for SNV:SNV pairwise associations, STR length (average genotype of allele size) against SNV dosage was used for STR:SNV association tests.
We considered the mSTR as a causal variant when the tested STR showed the highest probability across all tested candidate variants and had probability of >0.3. We referred to these mSTRs as fm-mSTRs.
Cross-platform DNA methylation validation of fm-mSTRs
To test the suitability of the DNA methylation measurements obtained from the Illumina 850K array in our QTL mapping, we performed a replication analysis of the set of fm-mSTRs using genome-wide methylation data extracted from long-read ONT sequencing data generated by the HGSVC from 36 samples for which Illumina sequencing were also available.
After downloading the files containing likelihood ratios (log_lik) calculated using the call-methylation script (https://nanopolish.readthedocs.io/en/latest/quickstart_call_methylation.html) on ONT reads, we calculated the methylation frequency using the calculate_methylation_frequency.py script provided by Nanopolish (Simpson et al. 2017). To ensure robust DNA methylation estimates, we focused on methylation values that had a minimum coverage of 10 reads with alignments on both forward and reverse strands and that had likelihood ratios for methylation (log_lik_ratios; more than zero methylated and fewer than zero unmethylated) that were less than −2 or greater than +2. Using these criteria, we calculated methylation values as the fraction of reads supporting methylation in relation to the total number of reads overlapping a given site for 581 CpGs associated with 516 unique fm-mSTRs.
For the set of 36 individuals with DNA methylation data derived from ONT sequencing reads, genotypes for the same set of 516 fm-STRs were generated from Illumina WGS data using HipSTR. After performing QC of the STR genotypes, we calculated pairwise correlation between STR genotypes and the DNA methylation values extracted from ONT reads using the lm function in R for each of the 600 STR:CpG pairs for which both DNA methylation values and HipSTR genotypes were available in at least 10 individuals (median of 34 individuals per STR:CpG pair).
Population stratification of functional STRs
We used available WGS data from 2000 individuals that were sequenced to high coverage using PCR-free protocols as a part of the 1KGP (The 1000 Genomes Project Consortium 2010; https://www.internationalgenome.org/). For each of these samples, we generated allele size estimates for our set of polymorphic STRs that were informative for DNA methylation (n = 131,635) using HipSTR (Willems et al. 2017). After applying the same QC steps as described previously (Supplemental Fig. S16), we retained genotypes for 126,171 STRs with at least 100 genotyped individuals for each major ancestral group. QC-filtered genotypes were then used to compute the VST index for the selected STRs as previously described using the diploid allele size per sample (Redon et al. 2006). Briefly, the VST measures the proportion of variance attributable to variation population. VST values range from zero to one, with higher values indicating allelic differentiation across the tested populations. P-values were calculated by permutation testing (n = 1000 permutations) where samples were randomly considered either as tested or as background population. STR alleles were considered as population specific when VST > 0.3 and P < 0.01.
Mapping cis-expression STRs in the PPMI cohort
After excluding samples that did not fulfill our QC criteria for either STR genotypes, DNA methylation, or RNA-seq data, 405 samples from the PPMI cohort were used for association analysis between STR length and expression levels of transcripts located within ±250 kb from the set of 131,635 polymorphic STRs. Focusing on those STR:transcript pairs for which both expression and STR genotypes were available for at least 50% of the samples, we tested for association a total of 425,385 STR:transcript pairs, involving 18,749 different transcripts within the flanks of 85,417 polymorphic STRs (median of four transcripts per STR).
Similar to our QTL mapping using DNA methylation data, we adjusted normalized RNA-seq read counts for age, gender, the top three ancestry-related principal components derived from PCA of SNVs (Pedersen and Quinlan 2017), the first PC derived from read normalized counts, and the first inferred probabilistic estimation of expression residuals (PEER) (Stegle et al. 2012) factor (Supplemental Fig. S16D). The resulting residuals were used to test the association between expression and STR length (average of allele size) using the lm function in R. Multiple testing correction for the number of STR:transcript pairs tested was applied using the Bonferroni method.
Identification of trait-associated mSTRs
To evaluate the impact of fm-mSTRs on phenotypic variation and human traits, we first identified local SNVs (fm-mSTR ±250 kb) that are in strong LD with our fm-mSTRs (r2 ≥ 0.8), and then, using these SNVs as surrogates for our fm-mSTRs, we determined their overlap with reported GWAS signals.
Identification of tagging SNVs
In brief, the genotypes corresponding to biallelic SNVs that are separated <250 kb of the tested fm-mSTRs were obtained from WGS data, filtered, and converted into dosages according to the alternate allele content (zero, one, or two). The LD between resulting SNV dosages and STR length (averaged allele size) was computed using Pearson's correlation. SNVs that were strongly associated with STR (r2 ≥ 0.8, henceforth termed tagging SNVs) were retained and considered as surrogates for their corresponding fm-mSTRs.
Colocalization of GWAS signals with tagging SNVs (GWAS-tagging SNVs)
SNVs associated with human traits (P < 5 × 10−8) were obtained from the GWAS catalog (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/gwasCatalog.txt.gz) and overlapped with our collection of tagging SNVs using BEDTools (v2.29.0) (Quinlan and Hall 2010). We considered an fm-mSTR as the most likely causal variant responsible for the phenotypic variation when an SNV identified by GWAS and the STR-tagging SNV were the same. All statistical analyses were performed using R statistical software (v3.5.3) (R Core Team 2019).
Data access
The DNA methylation, gene expression, and WGS data used in this study are available using the links provided above (see Methods). We are working with the relevant data owners to make the STR genotype data available, and the current study can be reproduced using the available data and codes provided. The scripts for quality control of data, QTL mapping, and fine-mapping can be found at GitHub (https://github.com/amartint/Methylation-associated-STRs) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
First and foremost, we thank all individuals who donated biospecimens and participated in this work for their willingness to contribute to scientific research. We thank all the investigators and consortiums who facilitated access to the data sets deposited in the dbGaP repository (http://www.ncbi.nlm.nih.gov/gap). The Pediatric Cardiac Genomics Consortium (PCGC) program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health (NHLBI, NIH), U.S. Department of Health and Human Services through grants UM1HL128711, UM1HL098162, UM1HL098147, UM1HL098123, UM1HL128761, and U01HL131003. This manuscript was not prepared in collaboration with investigators of the PCGC, has not been reviewed and/or approved by the PCGC, and does not necessarily reflect the opinions of the PCGC investigators or the NHLBI. In addition to data sets generated from PCGC, data used in the preparation of this article were obtained from the Parkinson's Progression Markers Initiative (PPMI) database (https://www.ppmi-info.org/access-data-specimens/data). For up-to-date information on the study, visit https://www.ppmi-info.org/about-ppmi. PPMI, a public-private partnership, is funded by the Michael J. Fox Foundation for Parkinson's Research and funding partners, a full list of which can be found at https://www.ppmi-info.org/about-ppmi/who-we-are/study-sponsors/. This work was supported in part by NIH grant R01NS105781 to A.J.S. and post-doctoral and early-career fellowships to A.M.T. from the American Heart Association (18POST34080396) and the NHLBI Biodata Catalyst (5120339), respectively. Research reported in this paper was supported by the Office of Research Infrastructure of the National Institutes of Health under award number S10OD018522. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions: A.M.T., P.G., N.P., B.J., and A.J.S. conceived the project and performed the bioinformatic analysis included in this study. A.M.T. and A.J.S. wrote and revised the manuscript. All authors read and approved the final draft of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277057.122.
- Received June 23, 2022.
- Accepted December 19, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








