
A DNA methylation haplotype block landscape in human tissues and preimplantation embryos reveals regulatory elements defined by comethylation patterns
- Yan Feng1,4,
- Zhiqiang Zhang2,3,4,
- Yuyang Hong1,
- Yi Ding1,
- Leiqin Liu1,
- Siqi Gao1,
- Hai Fang2 and
- Jiantao Shi1
- 1Key Laboratory of RNA Science and Engineering, Shanghai Institute of Biochemistry and Cell Biology, Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences, Shanghai 200031, China;
- 2Shanghai Institute of Hematology, State Key Laboratory of Medical Genomics, National Research Center for Translational Medicine at Shanghai, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China;
- 3School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200030, China
-
↵4 These authors contributed equally to this work.
Abstract
DNA methylation and associated regulatory elements play a crucial role in gene expression regulation. Previous studies have focused primarily on the distribution of mean methylation levels. Advances in whole-genome bisulfite sequencing (WGBS) have enabled the characterization of DNA methylation haplotypes (MHAPs), representing CpG sites from the same read fragment on a single chromosome, and the subsequent identification of methylation haplotype blocks (MHBs), in which adjacent CpGs on the same fragment are comethylated. Using our expert-curated WGBS data sets, we report comprehensive landscapes of MHBs in 17 representative normal somatic human tissues and during early human embryonic development. Integrative analysis reveals MHBs as a distinctive type of regulatory element characterized by comethylation patterns rather than mean methylation levels. We show the enrichment of MHBs in open chromatin regions, tissue-specific histone marks, and enhancers, including super-enhancers. Moreover, we find that MHBs tend to localize near tissue-specific genes and show an association with differential gene expression that is independent of mean methylation. Similar findings are observed in the context of human embryonic development, highlighting the dynamic nature of MHBs during early development. Collectively, our comprehensive MHB landscapes provide valuable insights into the tissue specificity and developmental dynamics of DNA methylation.
DNA methylation, a crucial epigenetic modification, plays an important role in maintaining cell identity across diverse tissue types (Robertson 2005; Ziller et al. 2013). In normal cells, DNA methylation is dependent on local CpG density. The human genome can be segmented into distinct functional elements based on mean methylation levels: unmethylated regions with high CpG density (UMRs; such as CpG islands), low-methylated regions (LMRs), partially methylated domains (PMDs), and high-methylated regions (HMRs) (Burger et al. 2013). UMRs, which are often proximate to core promoters, play a pivotal role in transcriptional regulation. Recently, a study of 205 healthy tissues identified unique methylation markers predominantly in UMRs (Loyfer et al. 2023). Long UMRs, spanning >3.5 kb, are also known as DNA methylation canyons (DMCs) and are intricately connected with highly conserved and developmentally important genes (Jeong et al. 2014; Wiehle et al. 2016). LMRs show an average methylation of 30% (Stadler et al. 2011) and are often located distally to promoters and enriched in binding sites for cell type–specific transcription factors.
Recent advances in sequencing-based techniques, such as whole-genome bisulfite sequencing (WGBS), have allowed DNA methylation to be measured at the level of individual reads, yielding single-nucleotide resolution (Bock 2012; Smith et al. 2017). Unlike the conventional approach of quantifying mean methylation, which aggregates the detected signal, WGBS has the potential to characterize DNA methylation haplotypes (MHAPs) (Zhang et al. 2021b). These MHAPs are defined as CpGs on the same read fragment originating from a single chromosome. Several read-level metrics have been proposed to quantify the characteristics of MHAPs in a heterogeneous population of cells. For instance, the proportion of discordant reads (PDR) quantifies methylation heterogeneity, explaining more expression variation than mean methylation, as exemplified in chronic lymphocytic leukemia (Landau et al. 2014).
In addition to the locally disordered methylation described above, adjacent CpGs on the same DNA molecules can be highly correlated, forming methylation haplotype blocks (MHBs) characterized by comethylation at the fragment level (Guo et al. 2017). The comethylation pattern can be determined using linkage disequilibrium (LD) analysis of epialleles, with LD R2 calculated based on phased DNA methylation data (Shoemaker et al. 2010). MHBs are characterized by a predominance of fully methylated or unmethylated MHAPs in the sequencing reads. MHBs have shown promise as plasma DNA biomarkers for noninvasive early cancer detection (Wu et al. 2022). Although MHBs are associated with enhancers (Guo et al. 2017), their functional roles remain largely unexplored, especially in the regulation of cell type–specific gene expression in normal tissues and during embryonic development.
Here, we identified MHBs from 17 representative normal tissues and human preimplantation embryos using well-curated WGBS data sets. We aim to characterize the tissue-specific regulatory potential of MHBs through large-scale data integration. We examined MHB enrichment in open chromatin regions, association with gene expression, and tissue-specific genes in comparison to known DNA methylation–associated regulatory regions such as UMRs and LMRs.
Results
A landscape of MHBs established for 17 representative human normal tissues
Local CpG sites can show comethylation, quantified by LD of epialleles in DNA MHAPs, with LD R2 calculated from phased DNA methylation statuses (Supplemental Fig. S1A). For example, if two CpG sites covered by 18 sequencing reads are either both methylated (n = 11) or both unmethylated (n = 7), they are considered significantly comethylated (R2 = 1, P = 0.0008) (Fig. 1A, left panel). These comethylated CpG regions are called DNA methylation haplotype blocks (MHBs) (Guo et al. 2017), characterizing CpG interdependence within a heterogeneous population (Fig. 1A, right panel; Supplemental Fig. S1B,C). To systematically investigate MHB profiles across human tissues, we characterized read-level methylation patterns across 17 representative normal tissue types using 72 expert-curated WGBS samples (Fig. 1B; Supplemental Tables S1, S2). We identified a total of 109,978 MHBs with a minimum of five CpGs required per block (Fig. 1C; Supplemental Table S3), mostly with low (<0.2) or intermediate (0.2∼0.8) methylation levels across all tissue types (Fig. 1D). More MHBs were discovered in the colon and placenta than in other tissues, and this observation was independent of sequencing depth (Supplemental Fig. S2A). The majority of these MHBs were <100 bp, with a median length of 50–70 bp (Supplemental Fig. S2B). Approximately 25% of tissue MHBs were located in promoters, indicating their potential regulatory functions, which are further supported by their prevalence in distal regions where enhancers are located (Fig. 1E). To validate the robustness of the MHBs identified in each tissue, we curated independent WGBS data sets for adipose, heart, liver, and lung tissues (Supplemental Table S1). Using region-set enrichment analysis via locus overlap analysis (LOLA) (Sheffield and Bock 2016), we showed that MHBs identified from these four tissues were specifically enriched with the highest significance in matched tissues (Fig. 1F; Supplemental Fig. S2C,D). Given the potential sharing of MHBs across different tissues, we combined them into 58,385 nonoverlapping MHBs, which were subsequently categorized into 23 clusters comprising 17 tissue type-specific clusters (n = 42,093) and six common clusters shared by multiple tissue types (n = 16,292) (Fig. 1G). In addition, tissue-specific MHBs showed a tendency toward hypomethylation in their respective tissues of origin (Supplemental Fig. S2E). DNA methylation–mediated genomic imprinting is known to generate parent-of-origin-specific methylation regions, which are frequently identified as MHBs. We compared the MHB clusters to known imprinted loci and found that all clusters contain a very low fraction of imprinted genes, except for cluster 23, which consists of MHBs present in 15 or more tissue types (Fig. 1H; Supplemental Fig. S2F).
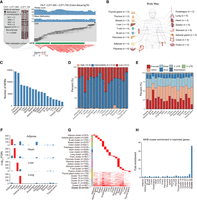
MHB landscapes identified in 17 representative human normal tissues. (A) Schematic of MHB patterns. The left panel shows an example of linkage disequilibrium (LD) between two CpG sites. A region (Chr 7: 2,571,065–2,571,120) with four CpG sites is depicted with a lollipop plot of 18 sequencing reads, in which black circles represent methylated cytosines and white circles represent unmethylated cytosines. The second and third CpG sites are comethylated, as indicated by the signed LD R2. Statistical significance was assessed using a binomial test. N11 indicates the number of MHAPs that are methylated at both CpG sites; the numbers of MHAPs of the other three types are also shown. The right panel shows an example of an MHB in colon tissue (Chr 7: 2,571,000−2,571,700). The top part shows the coverage and mean CpG methylation of each CpG site, and the middle part displays the DNA methylation status of the individual fragment, in which black and white represent methylated and unmethylated CpG sites, respectively. The bottom part shows the heatmap of the signed LD R2 score between pairs of covered CpG sites. (B) Body map showing analyzed normal tissue types and sample sizes. Panel created with BioRender (https://www.biorender.com). (C) Number of MHBs identified per tissue type. (D) Bar plots illustrating the proportion of MHBs in low (<0.2), intermediate (0.2–0.8), and high (>0.8) methylation groups. (E) Bar plots illustrating the proportions of MHBs annotated to different genomic features. (F) Validation of MHBs in four tissue types using independent data sets. Enrichment was determined by the R package LOLA, using the union of MHBs from all tissue types as the background. To preclude computing the logarithm of zero, FDR values of zero were converted to 10−300 before logarithmic transformation. (G) Categorization of MHBs into 23 nonoverlapping clusters. The number of regions in each cluster is shown. (H) A bar plot illustrating the enrichment of MHBs in the loci of imprinted genes, with fold enrichment evaluated using rGREAT.
Tissue MHBs represent distinct regulatory elements
Motivated by the enrichment of MHBs within regulatory regions such as promoters and enhancers (Supplemental Fig. S3A), we compared MHBs to open chromatin regions, which are known to be accessible to regulatory proteins (Buenrostro et al. 2013). In 15 out of 17 tissue types, >60% of MHBs overlapped with chromatin regions defined as accessible by ATAC-seq in their respective tissue types (Supplemental Fig. S3B; Supplemental Table S2). This observation was further supported by our analysis of nucleosome-free regions (NFRs) (Tarbell and Liu 2019), which covered >48% of MHBs in 14 out of 17 tissue types (Supplemental Fig. S3C).
Segmented DNA methylation states, such as UMRs and LMRs, are known to be enriched in active regulatory elements (Lister et al. 2009). We examined the overlap of tissue MHBs and DNA methylation region annotations, including UMRs, LMRs, PMDs, and HMRs (Supplemental Table S2). DNA methylation states were defined purely by mean methylation, and most of the genome was annotated as PMDs (Supplemental Fig. S3D; Zhou et al. 2018). Our results show that MHBs are mainly located in UMRs, LMRs, and PMDs, which collectively accounted for ∼75% of MHBs in each tissue type (Supplemental Fig. S3E). Next, we compared the enrichment of open chromatin in MHBs and DNA methylation state regions while controlling for confounding factors such as region size and methylation level. MHBs showed greater enrichment in open chromatin than any other DNA methylation–associated regions, including UMRs and LMRs (Fig. 2A). For instance, at a mean methylation level of 0.25, 81.5% of CpG sites in MHBs were covered by ATAC-seq peaks, whereas the proportions in UMRs and LMRs were 53% and 60.5%, respectively. This pattern was highly robust across all individual tissue types (Supplemental Fig. S4), suggesting that MHBs represent regulatory elements defined by DNA methylation patterns rather than by average methylation levels. When open chromatin regions were categorized based on their overlap with UMRs/LMRs, MHBs, or neither, those coexisting with MHBs displayed greater enrichment for H3K4me3 and H3K27ac histone modifications (Fig. 2B; Supplemental Fig. S5A).
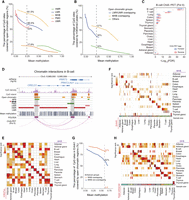
Tissue MHBs are enriched in regulatory elements. (A) Enrichment of open chromatin regions in MHBs as a function of mean methylation. The enrichment score was calculated as the percentage of CpG sites covered by open chromatin regions. (PMD) Partially methylated domain, (LMR) low-methylated region, (UMR) unmethylated region, and (HMR) high-methylated region. (B) Enrichment of H3K4me3 modification in open chromatin regions that overlap MHBs. Open chromatin regions were categorized based on overlap with UMRs/LMRs, MHBs, or neither. The enrichment score was calculated as the percentage of CpG sites covered by H3K4me3 ChIP-seq peaks. (C) Enrichment of B cell ChIA-PET (Pol II) tags in tissue MHBs. The enrichment was tested using the R package LOLA, with the union of MHBs from all tissues as the background. (D) An example genomic track showing that chromatin interactions tend to occur in MHB regions. Tracks of CpG density, mean methylation, MHBs, methylation-associated regions (PMD, LMR, UMR, and HMR), NFR, open chromatin regions, and chromatin interaction in this region (Chr 3: 9,969,000–9,995,000) for B cells are shown. (NFR) Nucleosome-free region. (E,F) Tissue-specific enrichment of MHBs in H3K27ac peaks and EnhA1 regions in matched tissue types. (G) Greater enrichment of open chromatin regions in enhancers with MHBs. The enhancers represented by EnhA1 regions were divided into two groups based on whether they overlap with MHBs. Compared with EnhA1 regions without MHBs, enhancers overlapping MHB regions are more enriched in open chromatin regions, regardless of the distribution of mean methylation. The enrichment score is calculated as the percentage of CpG sites covered by regions of open chromatin. (H) Tissue-specific enrichment of MHBs in super-enhancer regions in matched tissue types. Enrichment tests in E, F, and H were performed using the R package LOLA, with the union of MHBs from all tissues as the test background. The resulting adjusted P-values for significant enrichment (FDR < 5%) were ranked across all tissue types. The black rectangle highlights the top one tissue in E, the top five in F, and the top three in H. Nonsignificant (FDR > 5%) results are shown in white. Enrichment tests in A, B, and G were performed with the computeCpgCov function in mHapSuite.
To explore whether MHBs mediate long-range chromatin contacts, we compared MHBs to publicly available data sets generated by chromatin interaction analysis by paired-end tag sequencing (ChIA-PET). Chromatin contacts in B cells (ENCFF507KYL) were most significantly enriched in B cell MHBs, indicating a tissue-specific regulatory role of MHBs (Fig. 2C). One case in point is the MHB downstream from CRELD1 (Chr 3: 9,969,000–9,995,000), which ChIA-PET indicated contacts the promoter region through long-range interactions. This MHB showed partial methylation and was flanked by HMRs (Fig. 2D).
To further investigate the regulatory roles of MHBs, we examined the overlap of tissue MHBs with annotations for seven different histone modifications (Supplemental Table S1), including H3K4me3, H3K27ac, H3K4me1, H3K36me3, H3K27me3, H3K9me3, and H3K9ac. Cumulatively, >70% of MHBs in 15 out of 17 tissues overlapped H3K4me3, H3K27ac, or H3K4me1 peaks, suggesting their roles in promoters and enhancers (Supplemental Fig. S5B). LOLA revealed that MHBs showed significant enrichment in H3K27ac peaks from matched tissues in 12 out of 17 tissue types (Fig. 2E). As a control, we observed a similar pattern for LMRs but not for other DNA methylation–associated elements, including UMRs, PMDs, and HMRs (Supplemental Fig. S5C).
We also compared the patterns of MHBs and chromatin annotated as active enhancers (EnhA1) by The ENCODE Project (The ENCODE Project Consortium 2012; Boix et al. 2021). Tissue-specific enrichment was observed in 15 out of 17 tissues (Fig. 2F; Supplemental Fig. S6). To explore the functional changes in enhancers associated with the presence of MHBs, we categorized enhancers based on MHB overlap. MHB-overlapping enhancers displayed greater enrichment in open chromatin regions. For example, at a mean methylation of 0.6, 82% of CpG sites in MHB-overlapping enhancers were covered by ATAC-seq peaks compared with 62% for nonoverlapping enhancers (Fig. 2G). Furthermore, we examined the tissue specificity of MHBs through comparison to cis-regulatory elements (CREs) previously defined using single-cell ATAC-seq (Zhang et al. 2021a). These CREs, selected from 65 cell clusters spanning 15 tissue types, were subjected to LOLA, which revealed that MHB enrichment in matched tissues ranked among the most significant for 13 out of 15 tissue types (Supplemental Fig. S7). Finally, we compared MHBs to super-enhancers, a unique enhancer type with unusually high levels of mediator binding, as measured by ChIP-seq (Pott and Lieb 2015). Tissue-specific enrichment was observed in all 15 tissue types, further supporting the notion that MHBs represent regulatory elements associated with DNA methylation patterns (Fig. 2H; Supplemental Fig. S8).
DNA motifs identified in tissue MHBs
Given the enrichment of regulatory elements in MHBs, we next sought to identify MHB-associated transcription factors through DNA motif analysis. Using the union of all tissue MHBs as background, 58 significant motifs were identified, of which 53 were enriched exclusively in one specific tissue (Fig. 3A; Supplemental Table S4). Our predictions were supported by data from previous studies (Kohyama et al. 2009; Grainger et al. 2010; El-Khairi et al. 2012; Haldar et al. 2014; Shan et al. 2017; Elmen et al. 2020; Hunter et al. 2022), showing the tissue-specific relevance of the predicted factors. For example, the motif for CDX2, an intestine-specific transcription factor critical for intestinal epithelium development and differentiation, particularly in the colon (Grainger et al. 2010), was significantly enriched in colon MHBs (P = 1 × 10−4). The motif for CNOT4, which regulates cardiac gene expression and protein levels (Elmen et al. 2020), was significantly enriched in heart MHBs (P = 1 × 10−4). The motif for LIN28A, an RNA-binding protein that is highly expressed in germ cells during early human ovary development (El-Khairi et al. 2012), was significantly enriched in ovarian MHBs (P = 1 × 10−3). The motif for SPIC, a SPI1 (also known as PU.1)-related transcription factor that controls the development of white blood cells such as macrophages (Kohyama et al. 2009; Haldar et al. 2014), was significantly enriched in spleen MHBs (P = 1 × 10−7). The motif for RUNX3, a key factor for the activation of the cytotoxic program in T cells (Shan et al. 2017), was significantly enriched in the MHBs of T cells (P = 1 × 10−3). Finally, motifs for both isoforms of HNF4, a master regulator of liver development encoded by the genes HNF4A and HNF4G (Hunter et al. 2022), were significantly enriched in liver MHBs (P = 1 × 10−7).
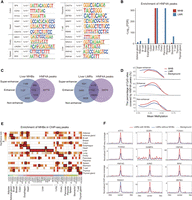
DNA motifs identified in tissue MHBs. (A) Representative DNA motifs enriched in tissue MHBs. HOMER was used to identify significantly enriched motifs in tissue-specific MHBs, with the union of MHBs as the test background. The top 10 motifs with P-values < 0.05 were retained for each tissue. (B) Enrichment of liver HNF4A ChIP-seq peaks in MHBs and LMRs. Enrichment tests were performed using the R package LOLA, with the union of MHBs and LMRs as the test background. To preclude computing the logarithm of zero, FDR values of zero were converted to 10−300 before logarithmic transformation. (C) Venn diagram showing the overlap between MHBs or LMRs and HNF4A ChIP-seq peaks in the liver. Liver MHBs and LMRs were categorized into three groups based on overlap with super-enhancers and enhancers (EnhA1). (D) Enrichment of HNF4A ChIP-seq peaks in liver MHBs is independent of the presence of known enhancers. Enrichment scores were defined as the percentage of CpG sites covered by open chromatin regions. Enrichment analysis was performed with the computeCpgCov function in mHapSuite. (E) Enrichment of MHBs with ENCODE ChIP-seq peaks. Enrichment tests were performed using the R package LOLA, with the union of MHBs as the test background. The resulting P-values for significant enrichment were ranked across all tissue types (FDR < 5%), and the top three are indicated by black rectangles. (F) Normalized transcription factor ChIP-seq signal around the centers of LMRs in liver tissue. ChIP-seq signals of transcription factors in liver LMRs with or without MHBs are shown in red and blue, respectively. To control for confounding factors, only regions with similar methylation levels (0.3–0.45) were considered.
To validate the regulatory motifs identified in tissue MHBs, we compared them to transcription factor binding regions defined by ChIP-seq assays. Specifically, we analyzed HNF4A binding sites that were previously profiled in liver tissue using ChIP-seq (obtained from the NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] under accession number GSE96260). We found that these regions (83,721 peaks) were specifically and significantly enriched within liver MHBs with a high level of significance (P < 1 × 10−322). This tissue specificity was not observed for LMRs, as HNF4A binding sites were similarly enriched in liver and pancreas LMRs (Fig. 3B). In addition, when liver MHBs were categorized into three groups based on their overlap with super-enhancers and conventional enhancers (Fig. 3C), we observed significant enrichment of HNF4A peaks in all three groups of liver MHBs (Fig. 3D). As a control, LMRs showed lower levels of enrichment in two out of three groups compared with MHBs when the same analytical procedure was applied. The enrichment of HNF4A peaks within nonenhancer groups may be attributable to the presence of MHBs in promoter regions and incomplete coverage of enhancers in current databases.
We additionally validated another predicted transcription factor, HNF4G, in liver MHBs using public ChIP-seq data (GEO; GSE105440) (Supplemental Fig. S9A–C). Furthermore, we focused on RUNX3, whose motif was enriched in T cell MHBs (P < 1 × 10−107). As only public B cell RUNX3 ChIP-seq data (GEO; GSM1010893) were available (Pope et al. 2014), we used these data for comparison. We found that RUNX3 peaks were enriched with the highest level of significance in B cell MHBs, followed by T cells (P < 1 × 10−287) (Supplemental Fig. S9D–F). As a complementary approach to validate the above findings, we compared MHBs to peaks of chromatin-associated proteins, such as transcription factors and their coactivators, using ChIP-seq data from The ENCODE Project (Supplemental Table S1). We found that MHBs tended to be enriched in ChIP-seq peaks from the matched tissues with the highest significance (Fig. 3E; Supplemental Fig. S10). For example, of 14 transcription factor-binding sites profiled in liver tissue, 13 showed peak enrichment specifically in liver MHBs and LMRs. Moreover, when stratified by MHB overlap, LMRs overlapping MHBs showed stronger ChIP-seq signals for all factors except RAD21 (Supplemental Fig. S11A). As a control, when we selected regions with similar mean methylation (0.3∼0.45) (Supplemental Fig. S11B), similar results were observed (Fig. 3F).
Tissue MHBs informative of gene expression
It is well established that promoter DNA methylation is negatively correlated with gene expression. Therefore, we investigated whether MHBs are associated with gene expression. For each tissue, promoters were categorized into three groups by mean methylation: low methylation (<0.2), intermediate methylation (0.2–0.8), and high methylation (>0.8). Each group was further divided based on the presence of MHBs in promoters. For each group with similar DNA methylation levels, the presence of an MHB correlated with higher gene expression across nearly all tissue types examined (Fig. 4A; Supplemental Fig. S12; Supplemental Table S2). The most significant distinctions in expression were observed in the high-methylation group (P = 1.8 × 10−9), followed by the intermediate-methylation (P = 1 × 10−7) and low-methylation groups (P = 0.024) (Fig. 4B). To validate our findings, we used the group assignment from T cell WGBS data to examine the distribution of gene expression in scRNA-seq data (GEO; GSE98638) (Zheng et al. 2017). We found that genes with MHBs showed significantly higher expression levels than did genes with similar mean methylation levels but without MHBs (Fig. 4C; Supplemental Fig. S13A; Supplemental Table S2).
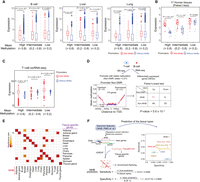
Tissue MHBs informative of gene expression. (A) Distribution of gene expression in promoters with different DNA methylation patterns in B cell, liver, and lung tissues. Promoters were categorized into low- (<0.2), intermediate- (0.2–0.8), and high-methylation (>0.8) groups. Expression was compared between promoters with and without MHBs within each group. Statistical significance was evaluated by the two-sided t-test. (B) The overall association of MHBs with gene expression across 17 tissue types was evaluated with the paired t-test. (C) MHB associations with scRNA-seq gene expression in T cells stratified by mean methylation levels. (D) MHBs are associated with differential gene expression. DMRs in T cells and B cells were identified using metilene (FDR < 5%, Δbeta > 0.1). The bottom left panel shows the average methylation around TSSs of differentially expressed genes with nearly identical DNA methylation in promoter regions (Δbeta < 0.01). The bottom right panel is a 2 × 2 contingency table categorizing genes by differential expression and MHB status in T cells. Statistical significance was evaluated using a Fisher's exact test. (E) Enrichment of MHBs with tissue-specific genes. Enrichment was tested using the R package rGREAT. The resulting adjusted P-values were ranked across all tissue types. Black rectangles indicate the top-ranked tissue with the highest significance. (F) Comparison of tissue type prediction performance for MHBs versus other methylation-associated genomic features. The left panel illustrates the prediction process using different genomic features. The right dot plot shows the prediction sensitivity and specificity.
We further investigated the associations of MHBs with differentially expressed genes (DEGs). We first identified 2009 DEGs (|log2FC| > 1, FDR < 5%) using public B cell (n = 4) and T cell (n = 8) RNA-seq data. Then, comparing DNA methylation between T cells and B cells, we identified 110 DEGs that were not annotated with any differentially methylated regions (DMRs) (FDR > 5%, Δbeta < 0.01) (Fig. 4D; Supplemental Table S5). Among 99 genes without MHBs in T cells, 44 were up-regulated, whereas all 11 genes with MHBs in T cells were significantly up-regulated (P = 5.6 × 10−4, Fisher's exact test) (Fig. 4D; Supplemental Fig. S13B).
To investigate whether MHBs are enriched in the regulatory loci of tissue-specific genes, we used GTEx data (The GTEx Consortium 2020) to define such genes. Of the 17 tissue types with defined MHBs, 14 were represented in the GTEx data set (Supplemental Table S6). We performed gene set enrichment analysis using the rGREAT tool (McLean et al. 2010) and found that MHB enriched tissue-specific genes in 13 out of 14 tissues examined, yielding a prediction sensitivity of 92.86%. Predictions were considered false positives when the highest enrichment was observed for unmatched tissue types. We identified two false positives for MHBs, resulting in a prediction specificity of 98.9% (Fig. 4E). Compared with other DNA methylation–associated regulatory elements, MHBs showed the highest sensitivity and comparable specificity (Fig. 4F; Supplemental Fig. S13C). Recently, a DNA methylation atlas of normal human tissues was used to identify cell type–specific markers for 39 cell types (Loyfer et al. 2023). In terms of prediction sensitivity, MHBs were highly competitive compared with the data from this atlas and outperformed it even when the top 25, 250, or 1000 markers were used (Fig. 4F).
Regulatory importance of MHBs in human embryonic development
To further explore the regulatory roles of MHBs in human tissues, we characterized their dynamics during human embryonic development. Using a publicly available data set, we identified MHBs in early human embryos at different developmental stages (Fig. 5A; Supplemental Fig. S14A; Supplemental Table S2) and validated these results by analyzing single-cell data (FDR < 5%) (Guo et al. 2014; Zong et al. 2022). Among the nine stages examined, five showed robust enrichment in the same cell type with the highest significance (Fig. 5B).
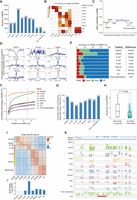
Regulatory importance of MHBs in human embryonic development. (A) Number of MHBs identified from bulk bisulfite sequencing of early human embryos. (B) Validation of MHBs using single-cell bisulfite sequencing data. An enrichment test was performed using the R package LOLA, with the union of single-cell MHBs as the test background. Each set of single-cell MHBs was tested against all sets of bulk MHBs; the cell type with the highest significance is highlighted by a black rectangle. (C) Temporal dynamics of mean methylation in MHB regions during early human embryonic development. (D) Normalized signals representing chromatin accessibility proximal to the center of MHB regions. (E) Annotation of MHBs to DMRs. DMRs were identified by comparing the test and reference groups. (Hyper) Hyper-DMR, (Hypo) hypo-DMR, (NC) no significant change. (F) Percentage of MHBs situated near DMRs. (G) MHBs tend to be located in DMR loci. Bar plots show the percentage of DMR-nonoverlapping MHBs that are located within DMR-defined loci. (H) The presence of MHBs in promoter regions is associated with gene activation. The expression of genes with and without MHBs was compared among genes with high methylation (>0.8) in promoters. Statistical significance was evaluated using the Wilcoxon rank-sum test. (I) A heatmap showing the expression of stage-specific genes. (J) Enrichment of MHBs in stage-specific genes in matched stages. The enrichment test was performed using the R package rGREAT. (K) An example genomic track displaying the known maternal imprinted control region KCNQ1. Mean DNA methylation and MHB tracks are shown for each stage in the region (Chr 11: 2,720,544–2,722,271).
We observed that although the global mean methylation levels were higher in sperm than in oocytes (Supplemental Fig. S14B), an inverse trend was evident in MHB regions (Fig. 5C), suggesting a distinct role for MHBs in early embryonic development. Given the observation that MHBs were enriched in regions of open chromatin in somatic tissues, we tested whether a similar pattern persisted in early human embryos. We analyzed a single-cell data set that quantifies chromatin accessibility through GCH site (GCA/GCT/GCC) methylation (Li et al. 2018). We found that the centers of MHB regions showed peaks of GCH methylation in all cell types after fertilization (Fig. 5D), indicating an enrichment of MHBs in open chromatin. We observed a lower level of GCH methylation in the centers of MHBs in sperm, which could be explained by a higher degree of chromatin packing at the global scale, including within the regions around transcription start sites (TSSs) (Supplemental Fig. 14C).
Next, we investigated the relationship between MHBs and DMRs that were identified by comparing adjacent embryonic stages. Approximately 25% of MHBs overlapped DMRs at each stage, except zygotes, in which 75% overlapped (Fig. 5E; Supplemental Fig. S14D). Furthermore, >40% of MHBs in most stages were localized within 5 kb of DMRs (Fig. 5F). When we focused on MHBs not overlapping with DMRs, >50% were located within the gene loci defined the DMRs (Fig. 5G).
Finally, we investigated the association between gene expression and MHBs in early human embryos. For a group of genes with highly methylated (>0.8) promoter regions, we found that the presence of MHBs was associated with a higher level of gene expression (P = 0.0028) (Fig. 5H; Supplemental Table S7). Moreover, we observed that MHBs at each stage were enriched within the loci of stage-specific genes during embryonic development (Fig. 5I,J; Supplemental Table S8). Imprinted regions, characterized by differential DNA methylation between maternal and paternal alleles, were often identified as MHBs. For instance, multiple MHBs were consistently identified at various stages of human preimplantation embryo development in the locus of KCNQ1, a known imprinted gene (Fig. 5K; Guo et al. 2014). Three MHBs in this locus were identified in sperm, which is haploid, indicating that these three MHBs are maintained through mechanisms other than imprinting.
Discussion
To date, DNA methylation–associated regulatory elements have been identified mainly using the distribution of mean methylation, which has resulted in the identification of UMRs and LMRs located in promoters and enhancers, respectively. In this study, we characterized MHBs with our previously developed toolkit (Ding et al. 2022) in 17 normal somatic tissues and human preimplantation embryos and showed that they represent a distinct type of regulatory element defined by DNA methylation patterns. MHBs are more enriched in open chromatin regions than previously defined DNA methylation–associated regulatory elements, such as UMRs and LMRs, regardless of their mean methylation levels. MHBs show tissue-specific enrichment in enhancers, as represented by H3K27ac ChIP-seq peaks, CREs identified by single-cell ATAC-seq, and chromatin states learned by ChromHMM and annotated as enhancers. This finding was further supported by the observed tissue-specific enrichment in super-enhancers. Furthermore, MHBs are enriched in tissue-specific genes with high sensitivity.
When MHBs were categorized into three groups based on overlap with super-enhancers and conventional enhancers, nonenhancer groups also showed enrichment for transcription factor binding peaks. This could potentially be explained by incomplete coverage of enhancers in current databases, as nonenhancer LMRs displayed a similar enrichment pattern. Additionally, ∼25% of MHBs were located in promoter regions, which also harbor enrichment for transcription factor binding sites.
In individual samples, the presence of MHBs is associated with higher expression levels of genes with similar methylation in their promoters, indicating regulatory effects that are independently explained by MHBs. Globally, promoter methylation was negatively correlated with gene expression, particularly for gene promoters without MHBs. For gene promoters with MHBs, gene expression did not clearly correlate with promoter methylation. A potential explanation is that gene promoters with MHBs tend to overlap with open chromatin regions. This allows the engagement of other factors that influence transcription (de Mendoza et al. 2022), including chromatin remodeling complexes and enhancers (Barral and Déjardin 2023), providing an alternative regulatory pathway. Thus, the conventional inverse relationship between promoter methylation and expression appears to be modulated by the dynamic interactions of chromatin remodelers when MHBs are present. This also suggests that genes with MHBs can be either up-regulated or down-regulated, depending on the function of transcription factors or cofactors binding in the open chromatin.
The exact mechanisms underlying MHB generation and maintenance remain unclear despite the described features. Concurrent DNA methylation and demethylation can yield discordant reads (Shi et al. 2021). Thus, mutual exclusivity between these processes (Ginno et al. 2020) may produce fully unmethylated or methylated reads, forming MHBs. Additionally, neuronal enhancers undergo demethylation and are hotspots for DNA single-strand break repair (Wu et al. 2021), whose features resemble those of MHBs. Recently, mathematical modeling and stochastic simulation suggested that local correlation of CpG methylation maintenance rates results in methylation-correlated blocks (Busto-Moner et al. 2020; Ren et al. 2022), potentially contributing to MHB formation.
This study is subject to limitations arising from the data sets and methodology used. First, the inclusion of curated data sets from various sources introduces inherent heterogeneity. Additionally, the used tool does not differentiate between paternal and maternal fragments, potentially misidentifying imprinted regions as MHBs. Approximately 1% of the identified MHBs are estimated to overlap with known imprinted regions. Second, the identification of matched plus and minus strands poses challenges in bulk WGBS data, potentially leading to the identification of hemi-methylated regions as MHBs. Despite these limitations, the findings of our study strongly support the notion that MHBs represent a distinct category of regulatory elements characterized by comethylation patterns. This assertion is robustly shown across human normal tissues and during early human embryonic development.
Methods
Data sources
All data sets used in this study are publicly available, with details listed in Supplemental Table S1. We collected public WGBS samples from 17 different normal human tissues: adipose (n = 4), adrenal gland (n = 5), breast (n = 3), B cell (n = 5), colon (n = 4), esophagus (n = 4), heart (n = 4), lung (n = 5), liver (n = 5), ovary (n = 4), pancreas (n = 4), placenta (n = 6), spleen (n = 5), stomach (n = 6), T cell (n = 4), thyroid gland (n = 2), and thymus (n = 2). Bulk BS-seq data for human early embryos were downloaded from GEO accession number GSE49828 (Guo et al. 2014). Processed ChIA-PET data for B cells were downloaded from ENCODE (The ENCODE Project Consortium 2012; https://www.encodeproject.org) under accession number ENCFF507KYL. ATAC-seq and ChIP-seq data of histone marks were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/), Roadmap (Roadmap Epigenomics Consortium et al. 2015), ENCODE, and CistromeDB (Mei et al. 2017). CREs in normal tissues were taken from a previous study (Zhang et al. 2021a). Active enhancers (EnhA1) and super-enhancers were downloaded from the EpiMap (Boix et al. 2021; https://compbio.mit.edu/epimap/) and SEdb (Jiang et al. 2019; https://bio.liclab.net/sedb/) databases, respectively. Preprocessed bulk RNA-seq data of normal tissues were downloaded from EpiMap (Boix et al. 2021). Single-cell RNA-seq data of T cells were downloaded from GEO accession number GSE98638 (Zheng et al. 2017). When applicable, the genomic coordinates of the downloaded regions were converted to the corresponding hg19 coordinates by liftOver (Kent et al. 2002).
A full list of samples along with their corresponding codes and all processed data, including MHAP files, can be found in Supplemental Table S2. The mHapSuite software is available in the Supplemental Code S1 and at GitHub (https://github.com/yoyoong/mHapSuite).
Data preprocessing
The public raw SRA files were downloaded from NCBI GEO and converted to FASTQ files using the SRA Toolkit (v2.9.1) followed by quality control using FastQC (v0.11.8). Sequence adapters and low-quality reads were trimmed by Trim Galore! (v0.6.2; https://github.com/FelixKrueger/TrimGalore) in paired-end or single-end mode with default parameters. Trimmed sequences were aligned to hg19 with BSMAP (Xi and Li 2009) with the following options: -q 20 -f 5 -r 0 -v 0.05 -s 16 -S 1. Duplicate reads were marked by Sambamba (v0.7.1) (Tarasov et al. 2015). Mean CpG methylation levels were extracted using MethylDackel (v0.5.0; https://github.com/dpryan79/MethylDackel).
Identification of DMRs
DMRs were identified by using metilene (v0.2-8; -t 10 -c 2 -m 5) (Jühling et al. 2016). Only DMRs with Q-values < 0.05, Δbeta > 0.1, and five or more CpG sites were considered for further analysis. The genome of each tissue type was segmented into PMDs, LMRs, and UMRs by using the MethylSeekR tool (version 1.32.0) with default parameters (Burger et al. 2013). Excluding UCSC-annotated genomic gaps, the remaining regions were defined as HMRs.
Identification of DNA MHBs
BAM files were converted to MHAP files with mHapTools (v1.0) (Zhang et al. 2021b). MHBs were identified using the “MHBDiscovery” module of mHapSuite (v2.0; https://github.com/yoyoong/mHapSuite), a Java implementation of mHapTk (Ding et al. 2022), with the default parameters (‐‐window 5 ‐‐r_square 0.5 ‐‐p_value 0.05). Genome-wide tracks were built with the “genomeWide” module of mHapSuite with default parameters (‐‐minK 1 ‐‐maxK 10 ‐‐K 4).
Motif enrichment
HOMER (Heinz et al. 2010) software was used to identify enriched motifs in tissue-specific MHBs using the union of MHBs from all tissues as the background. For each tissue type, the top 10 most significant motifs were selected for further analysis (P < 0.05).
Region set enrichment analysis
Statistical analysis was conducted using R statistical software version 4.1.0, together with Bioconductor library version 3.14 (R Core Team 2021). P-values were adjusted using the Benjamini–Hochberg procedure, resulting in Q-values or FDR (Benjamini and Hochberg 1995). The R package LOLA (Sheffield and Bock 2016) (v1.22.0) was used to test the enrichment of a query region set in a database of reference region sets. Q-values <5% were considered to indicate statistical significance. To show tissue specificity, Q-values were ranked, and the top k tissues with the highest significance were highlighted, where k was dependent on the database size, typically representing ∼5% of the total number of tissues analyzed. In case of tied ranks, more than k tissues may be highlighted. The specific k used in each data set is indicated in the figure legends.
The computeCpgCov function in mHapSuite was used to evaluate enrichment of a query region set in open chromatin regions while controlling for region size and mean methylation levels. First, query region CpG sites were binned by mean methylation levels. Then, in each quantile, the fraction of CpG sites covered by open chromatin regions was calculated. The resulting curve indicated the fraction of CpGs covered by open chromatin across stratified mean methylation levels.
For the enrichment of genomic features in tissue MHBs, a permutation-based method was used to evaluate the enrichment of a reference set within a query set. Briefly, the “shuffle” function in BEDTools (version 2.25.0) (Quinlan and Hall 2010) was used to generate 1000 random sets matching the query set size. The expected overlap was the average overlap between the reference and random sets. The enrichment score was defined as the ratio of observed and expected numbers of overlapping regions. The permutation test P-value was defined as the fraction of random sets that overlap more regions with the reference set than the query set.
Prediction of tissue specificity with methylation-associated genomic features
Tissue-specific genes were identified by applying the R package TissueEnrich (Jain and Tuteja 2019) to the GTEx data set (The GTEx Consortium 2020). Each gene was assigned a basal regulatory domain of 5 kb upstream of and 1 kb downstream from the TSS. This regulatory domain was extended in both directions to the nearest gene's basal domain, up to a maximum of 1 Mb (McLean et al. 2010). The rGREAT R package (Gu and Hubschmann 2023) with default parameters was used to assess the enrichment of tissue-specific genes in defined sets of regions, including MHBs, UMRs, LMRs, PMDs, HMRs, and Atlas (Loyfer et al. 2023). We calculated the enrichment rank for each tissue with tissue-specific genes, and the top-ranked tissue type was considered the predicted tissue. The sensitivity and specificity were determined to evaluate the tissue specificity prediction performance of different methylation-associated genomic features.
Genome build statement
For convenient integrative analysis with existing data, this study used hg19 as the reference genome. As benchmarked with a subset of samples, using a newer genome assembly such as hg38 does not change the overall results.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This study is supported by the National Natural Science Foundation of China (grant IDs: 32270691 and 32170663) and Innovative Research Team of High-Level Local Universities in Shanghai.
Author contributions: H.F. and J.S. conceived and designed the study. Y.F. and Z.Z. performed the data analysis. Y.H. developed mHapSuite, which was extensively used in this study. Y.D., L.L., and S.G. contributed to data curation and preprocessing. All authors discussed the results and contributed to the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278146.123.
- Received June 1, 2023.
- Accepted November 3, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








