
Localizing unmapped sequences with families to validate the Telomere-to-Telomere assembly and identify new hotspots for genetic diversity
- Brianna Chrisman1,2,
- Chloe He3,
- Jae-Yoon Jung4,
- Nate Stockham5,
- Kelley Paskov3,
- Peter Washington1,
- Juli Petereit2 and
- Dennis P. Wall3,4
- 1Department of Bioengineering, Stanford University, Stanford, California 94305, USA;
- 2Nevada Bioinformatics Center, University of Nevada, Reno, Nevada 89557, USA;
- 3Department of Biomedical Data Science, Stanford University, Stanford, California 94305, USA;
- 4Department of Pediatrics (Systems Medicine), Stanford University, Stanford, California 94305, USA;
- 5Department of Neuroscience, Stanford University, Stanford, California 94305, USA
Abstract
Although it is ubiquitous in genomics, the current human reference genome (GRCh38) is incomplete: It is missing large sections of heterochromatic sequence, and as a singular, linear reference genome, it does not represent the full spectrum of human genetic diversity. To characterize gaps in GRCh38 and human genetic diversity, we developed an algorithm for sequence location approximation using nuclear families (ASLAN) to identify the region of origin of reads that do not align to GRCh38. Using unmapped reads and variant calls from whole-genome sequences (WGSs), ASLAN uses a maximum likelihood model to identify the most likely region of the genome that a subsequence belongs to given the distribution of the subsequence in the unmapped reads and phasings of families. Validating ASLAN on synthetic data and on reads from the alternative haplotypes in the decoy genome, ASLAN localizes >90% of 100-bp sequences with >92% accuracy and ∼1 Mb of resolution. We then ran ASLAN on 100-mers from unmapped reads from WGS from more than 700 families, and compared ASLAN localizations to alignment of the 100-mers to the recently released T2T-CHM13 assembly. We found that many unmapped reads in GRCh38 originate from telomeres and centromeres that are gaps in GRCh38. ASLAN localizations are in high concordance with T2T-CHM13 alignments, except in the centromeres of the acrocentric chromosomes. Comparing ASLAN localizations and T2T-CHM13 alignments, we identified sequences missing from T2T-CHM13 or sequences with high divergence from their aligned region in T2T-CHM13, highlighting new hotspots for genetic diversity.
The human reference genome has been one of the major successes of modern genomics and has been heavily relied on to study how genetic variation contributes to disease (Lander et al. 2001; International Human Genome Sequencing Consortium 2004; Venter et al. 2015; Schneider et al. 2017). However, despite its achievements, the current release of the human reference genome (GRCh38) is incomplete in two major ways: It is still missing >200 Mb of heterochromatic sequence (Altemose et al. 2014), and it does not represent the full spectrum of genetic diversity that exists in the human species. Because of this incompleteness, reads originating from heterochromatic regions of the genome or from alternative haplotypes not well represented on the reference are not analyzed in most genomic studies. On a population scale, we therefore understand much less about the role these regions play in health and disease, as well as the amount of genetic diversity present in these regions (Miga 2019; Ebert et al. 2021; Miller et al. 2021). Reads originating from these regions, as well as from viruses and bacteria in hosts or massively parallel sequencing reagents (Chrisman et al. 2022a,b), collectively make up the unmapped read space.
We sought to use unmapped reads from whole-genome sequences (WGSs) to identify human genomic sequences that do not align to the current human reference genome (GRCh38) and to identify which region of the genome they belonged to, the process by which we refer to as localization. Originally, we aimed to develop an algorithm to localize unmapped reads to coarse regions of the genome, with the hope that these regions could function as “bins” and that from these bins, we could ultimately assemble longer contigs that represented alternative haplotypes or sections of the genome missing from GRCh38. We presented a proof-of-concept version of such a localization algorithm for nonrepetitive sequences in autosomes, with discussion about what a final de novo step would look like (Chrisman et al. 2021).
While we were developing our algorithm, the Telomere-to-Telomere (T2T) consortia was finalizing the release of T2T-CHM13, the first gapless assembly of a human genome (Miga et al. 2020; Nurk et al. 2022). Rather than attempting to de novo assemble low-reliability contigs from scratch, we used our results to validate and analyze T2T-CHM13, which is likely slated to become the next official human reference genome. The T2T-CHM13 assembly relies on high-fidelity long-read sequencing (HiFi sequencing) and on novel graph-based algorithms for stitching reads together (Nurk et al. 2022). Our localizations rely on short-read sequencing and a novel maximum likelihood algorithm that uses sequences from families. Thus, our algorithm for localizing sequences is an orthogonal approach to the T2T's assembly strategy and can help validate the T2T's work. We also use our results to understand genetic diversity in relation to the T2T-CHM13 reference genome, especially in regions that were previously gaps in GRCh38 and have thus been understudied. In this paper, we present a modified version of our proof-of-concept algorithm, show accuracy and precision using a validation data set, localize subsequences from unmapped reads, and compare the localization results with the results of T2T-CHM13 alignment.
Specifically, we extended our original algorithm to be able to perform on tandem repeats, which are present in telomeres and centromeres and are highly variable across individuals (Lu et al. 2021), and to handle sequences originating from the X and Y Chromosomes. We present an algorithm for sequence location approximation using nuclear families (ASLAN). ASLAN uses the phasing information of siblings and the distribution of a given k-mer across the reads of family members in order to build a maximum likelihood model and identify the most probable region of the genome a k-mer originates from. In this work, we (1) show ASLAN's precision and accuracy on two validation data sets, (2) use ASLAN to localize sequences from WGS that did not map to GRCh38, and (3) compare ASLAN's localizations with alignments to the newly released T2T-CHM13 assembly, in order to better understand regions of high genetic diversity.
Results
ASLAN localizes subsequences using a maximum likelihood model of k-mer distributions and family phasings
We propose an algorithm for sequence location approximation using nuclear families (ASLAN). ASLAN requires a large WGS data set of nuclear families, ideally with multiple children. We use ASLAN with the iHART data set (Ruzzo et al. 2019), a large WGS data set from families with autistic children (4501 individuals from 1010 families), that our group curated originally to study the genetic components of autism. To our knowledge, this is one of the largest familial WGS data sets in the world and offers a unique opportunity for family-based analysis such as ASLAN and others (Chrisman et al. 2018; Paskov et al. 2021; Chrisman et al. 2022a,b). The iHART collection includes the raw WGS from the individuals, as well as the aligned and variant-called data (VCF format) in reference to GRCh38. A detailed description of the iHART data preprocessing pipeline is given in the Methods. ASLAN builds a maximum likelihood model that uses family phasing information to identify the genomic region of origin of subsequences extracted from WGS reads from this large data set of nuclear families (Fig. 1A).
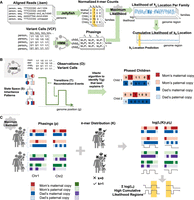
Pipeline for ASLAN and its components. (A) Overall pipeline for extracting k-mers, phasing families, and localizing k-mers based on phasings and k-mer distributions. (B) Simplified schematic of the hidden Markov model used for the phasing algorithm, in which the goal is to identify the inheritance patterns and recombination points that best explain the variant calls in a family. (C) Simplified schematic of the maximum likelihood model to identify the most likely region of a genome that a k-mer originates from, given the distribution of the k-mer and phasing patterns within and across families.
We first phase the children—identify which copy of their mother's and father's chromosomes a child inherited at a given region—using a hidden Markov model (HMM) (Fig. 1C). In this HMM, the state space is the parental copies of a chromosome a child has at any given region, transitions are recombination points, and observations are the variant calls. A Viterbi algorithm (Forney 1973) is used to walk through the HMM to identify the parental copy each child inherited at a given genomic region that best explains the variant calls in the family. Next, for a given 100-bp subsequence (a length that balances uniqueness with likelihood of appearing within a read) (see Supplemental Methods), we extract the number of times it occurred in each individual's WGS reads. A detailed description of the phasing pipeline is given in the Methods and in Supplemental Methods S1.
We then build a maximum likelihood model that identifies the most likely genomic origin of the 100-mer, given the distribution of the 100-mer and the phasings within and across families, assuming Mendelian inheritance patterns (Fig. 1B). We validate the accuracy of ASLAN using a synthetic data set, as well as a data set of real reads from already known locations, and then apply ASLAN to 100-mers extracted from the unmapped read space of the iHART data set. We illustrate the logic behind ASLAN in Figure 1 and a full mathematical description in the Methods and Supplemental Methods S3.
ASLAN accurately localizes validation sequences with high precision
We validated ASLAN on two data sets: (1) a synthetically generated data set and (2) a data set using k-mers extracted from ALT sequences in the decoy genome. The decoy genome is a published list of sequences included in the latest human reference genome release, containing contigs that were not able to be included in the reference genome during assembly but are common enough to warrant including as extra contigs in the decoy to speed up alignment. One set of sequences, the ALT sequences, consists of sequences from known locations in the genome where there are high amounts of genetic diversity that cannot be captured in a linear reference genome (Zheng-Bradley et al. 2017). It is the reference genome's first foray into pangenomes, although the ALT sequences capture only a very small amount of genetic diversity from a handful of regions.
Each data set contained k-mer counts and a location label for each k-mer. The goals of analyzing ASLAN's performance on these validation data sets were to understand ASLAN's performance and to tune ASLAN's hyperparameters to optimize the balance between precision and accuracy. In this case, precision refers to the size of the region to which ASLAN localizes a 100-mer. For the most part, ASLAN uses biologically computed hyperparameters. The expected number of occurrences of a k-mer is computed using the length of the human genome and a sample's read depth, and inheritance probabilities are derived from simple Mendelian inheritance rules. As described in our related paper (Paskov et al. 2023), the phasing algorithm uses parameters measured or derived by the literature: the maternal and paternal recombination rates (Hussin et al. 2011), and the variant-calling error rate of our data set (Paskov et al. 2021). The only hyperparameter we use is in the final step of the maximum likelihood model: After we build a cumulative likelihood distribution for the likelihood that a k-mer originates from each region of the genome (about 225,000 possible regions based on the recombination points found within the families), rather than select a single genomic region, we select a range of neighboring regions. The size of this range is governed by the distribution of cumulative likelihoods and a hyperparameter λ (see Methods) (see Fig. 1). During validation, we also sought to identify which value of λ gave ASLAN the best balance of precision and accuracy.
We first validated our method on a synthetically generated data set, which simulated theoretical observed counts of k-mers given various genomic regions of origin, number of tandem repeats, and prevalences (see Methods). We found that across all tandem repeat numbers and maximum likelihood hyperparameter values, our algorithm's performance began to degrade after the prevalence of a k-mer became >80%. ASLAN also had poor performance for k-mers with prevalences <1% (Supplemental Fig. S2A,C). Smaller values of λ resulted in faster degradation of performance in terms of localization ability and accuracy, although smaller values of λ also tightened the region length to which k-mers were localized. For the most part, ASLAN did not show a bias toward any particular chromosome, except for Chromosome 6 (Supplemental Fig. S2D). This was expected as Chromosome 6 contains the most recombination points and thus had more possible regions available for the algorithm to incorrectly localize a k-mer. As shown in Supplemental Figure S2, a value of λ = 0.1 gives a good balance of localization ability, accuracy, and precision: Our algorithm localizes >90% of our k-mers, is 92% accurate, and has a median localized region length of <1 Mb.
It is important to note that ASLAN's localization consists of a start and end coordinate for a given k-mer. Although two k-mers might be residing right next to each other, ASLAN is not guaranteed to give the same start and end coordinate for each k-mer, because of the Poisson noise in the k-mer counts and because of ASLAN using a maximum likelihood interval cutoff (Hall and Scala 1990).
We then validated our data set using k-mers extracted from alternative sequences in the decoy genome, contigs that are not on the primary reference genome but have been seen in enough individuals to warrant inclusion as alternative sequences in GRCh38 (Zheng-Bradley et al. 2017). ASLAN performed similarly on this decoy data set. Changing the λ-values altered performance similarly to the synthetic data set. Again, a value of λ = 0.1 provided good balance of localization ability, accuracy, and precision. Using λ = 0.1, ASLAN localized 94% of k-mers with 92% accuracy, as well as a median region length of 1.3 Mb. ASLAN again showed a slight preference toward localizing to Chromosome 6, which has a disproportionate number of recombination points. There are some specific hotspots for incorrect localizations, as shown by the spikes in Chromosomes 6 and 8 in Supplemental Figure S3I, which correspond to k-mers from a handful of alternative haplotypes being consistently localized to those regions.
Only 24 out of 198 alternative haplotypes had their k-mers localized with <90% accuracy. These alternative haplotypes include chr8_KI270813v1_alt, chr12_GL877875v1_alt, chr2_KI270894v1_alt, chr17_KI270907v1_alt, and chr22_KI270878v1_alt. For k-mers from many of these “poorly” localized haplotypes, our algorithm consistently localized the k-mers to another region of the genome. For example, k-mers from chr12_GL877875v1_alt consistently were localized to the beginning of Chromosome 6, and k-mers from chr8_KI270813v1_alt were consistently localized to a region 2 Mb downstream from their annotated location. Given these patterns, we wonder if perhaps there are alternative haplotypes located elsewhere in the genome that are homologous to those in the decoy sequence and are detected by our algorithm.
Many unmapped reads localize to gaps in GRCh38
After validating our algorithm using the synthetic and decoy data sets, we ran ASLAN on 100-mers extracted from the unmapped and poorly aligned WGS reads of 727 nuclear families (we excluded families there were with half siblings or twins or were missing a parent). We outline the criteria for what was considered unmapped or poorly aligned in the Methods. We sampled approximately 100 million 100-mers that were found in at least two samples, and localized them with ASLAN.
Tuned to the λ-parameter selected based on the validation data, ASLAN was able to localize 79% of 100-mers with iHART population prevalences between 20% and 80% (Fig. 2B–D). For all k-mers, which included very low or high prevalence k-mers, ASLAN could localize <20%. This was expected given that very low or very high prevalences of k-mers will not produce enough siblings discordant for a given k-mer, which ASLAN depends on for the likelihood model. Many k-mers from our model had very low prevalences (see Fig. 2A). We suspect such k-mers originate from private familial sequences, sequencing artifacts, and contaminants. Similar to the validation data results, ASLAN localized the unmapped 100-mers to a median region length of 762,052 bases (Fig. 2E). Because validation showed more reliable performance on k-mers between 20% and 80% prevalence and because ASLAN struggled to localize very low and very high prevalence k-mers from the unmapped reads, we focus on these medium prevalence k-mers. The rest of our findings only use k-mers between 20% and 80% prevalence.
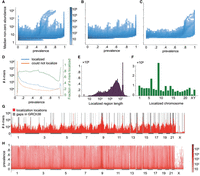
ASLAN performance on unmapped reads. (A) Distribution of prevalence and abundance (median of nonzero counts) for all 100-mers extracted from unmapped reads. (B) Distribution of prevalence and abundance for 100-mers that localized to autosomes. (C) Distribution of male prevalence and abundance for 100-mers that localized the Y Chromosome. (D) Number and fraction of 100-mers that ASLAN could and could not localize, given their prevalences across the iHART population. (E) Distribution of localized region length. (F) Number of k-mers localized to each chromosome. (G) Distribution of localization location in reference to GRCh38, with gaps annotated. (H) Distribution of k-mer localization location and prevalence.
We found that many unmapped 100-mers localized to regions that included gaps in GRCh38 (Fig. 2G,H), more than expected by chance, as shown using a Monte Carlo simulation shuffling the gap locations to generate a null distribution. We found that 56% of localization regions contained at least one gap present in GRCh38 (P < 0.05), and 49% of localizations had a center point that fell within a GRCh38 gap (P < 0.0001). In particular, we see many k-mers localizing to the large gaps representing the centromeres of Chromosomes 1 and 9 (Fig. 2F). We also see many sequences localizing to gaps that represent the short arms of the acrocentric chromosomes (Chromosomes 13, 14, 15, 21, and 22). Even with the highly repetitive nature of heterochromatin, ASLAN is able to identify many subsequences coming from the heterochromatic gaps in GRCh38. These successfully localized subsequences probably include tandem repeat sequences that are unique to a single region of heterochromatin, unique sequences in flanking regions of repetitive sequences, or repeats in heterochromatin that have developed a variant, giving some frequency of the human population a variant on a repeat.
There are several hotspots where sequences localize that are not gaps in GRCh38, namely, Chr2:89 Mb, Chr2:175 Mb, Chr3:167 Mb, Chr3:175 Mb, Chr6:33 Mb, Chr8:13 Mb, Chr8:18 Mb, Chr8:136 Mb, Chr11:1 Mb, Chr11:127 Mb, Chr12:11 Mb, Chr15:20 Mb, Chr19:55 Mb, and Chr20:29 Mb. We describe the functional relevance of these regions in the Discussion. Note that ASLAN's localization consists of a start and end coordinate for a given k-mer. Although two k-mers might be residing right next to each other, ASLAN is not guaranteed to give the same start and end coordinate for each k-mer, because of the Poisson noise in the k-mer counts and because of ASLAN using a maximum likelihood interval cutoff. k-mer 1 might be localized to Chromosome 1 799 Mb–801 Mb, and k-mer 2 might be localized to Chromosome 1 798 Mb–800 Mb, even though both actually come from right around 800 Mb. Therefore, it is difficult to come up with a start and end loci for a “hotspot,” so from here on, we report the nearest 1 Mb or so that resides within a hotspot.
Metacentric chromosomes show high concordance between ASLAN localizations and T2T-CHM13 alignment
During the course of our study, the T2T consortia released the sequence of the T2T-CHM13 genome, the first fully assembled human genome. Well aware of the relevance of T2T-CHM13 to the goals of ASLAN, we decided to align the sequences of the ASLAN-localized 100-mers to the T2T-CHM13 assembly in order to identify regions where ASLAN and the T2T-CHM13 alignment were in high or low concordance and to identify potential sources of genetic diversity, especially in the gapped regions of GRCh38.
We first analyzed ASLAN and T2T-CHM13 concordance. Our thinking was that for k-mers for which ASLAN localizations and T2T-CHM13 mappings agree, we can be more certain that these k-mers truly originate from the specified region. If the T2T-CHM13 alignment score is low, this probably indicates some genetic variants between the set of iHART samples and the T2T-CHM13 genome. If ASLAN localizations and T2T-CHM13 mappings disagree, it might indicate homology between a T2T-CHM13 region and a different region in a subset of the iHART samples. T2T-CHM13 represents only a single genome, and it is important to understand the limits of the single genome, especially in the previously gapped regions where no large-scale analyses of structural variation exist.
We found that ASLAN and T2T-CHM13 showed a high concordance, particularly on the metacentric and submetacentric chromosomes (Chromosomes 1–12, 16–20, and X). Across the entire genome, 69% of sequences mapped to the same chromosome, using both ASLAN localization and T2T-CHM13 alignment (Fig. 3B). For close to 80% of sequences localized to each of the metacentric and submetacentric chromosomes, ASLAN and T2T-CHM13 mappings were concordant: The ASLAN-localized region contained the T2T-CHM13 alignment location (Fig. 3D). Mismatches were most common between the sequences that aligned to the T2T-CHM13 centromeres of Chromosomes 1 and 9 and the short arms of Chromosomes 13–15 and 21–22 (the acrocentric chromosomes, which we discuss below). ASLAN localizations of these mismatches were fairly uniformly distributed across the genome (Fig. 3A,B) with no glaring preferences toward certain chromosomes or regions.
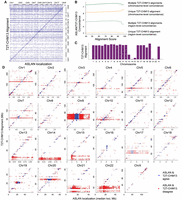
Comparison between ASLAN localizations and T2T-CHM13 alignments. (A) Confusion matrix comparing ASLAN localizations, lifted over to T2T-CHM13 coordinates to T2T-CHM13 alignments of 100-mers extracted from the unmapped reads, binned into 1000 equally sized bins across the genome. (B) Concordance rate between ASLAN localization and T2T-CHM13 alignment versus alignment score, colored by whether or not alignment to T2T-CHM13 was a unique mapping or not. (C) Concordance rate between ASLAN localization and T2T-CHM13 alignment, versus the chromosome to which ASLAN localized. Acrocentric Chromosomes 13–15 and 21–22 show a significantly lower concordance. (D) T2T-CHM13 alignment versus center point of ASLAN localization region, separated by chromosome and colored by whether or not T2T-CHM13 alignment and ASLAN localization were in concordance.
Disagreement between ASLAN localization and T2T-CHM13 alignment may be caused by one of several reasons. First of all, the ASLAN localization may have limited accuracy in certain regions. ASLAN showed 92% accuracy on fairly clean data: synthetic data and data from the primarily euchromatic alternate haplotypes in the decoy genome. On the validation data, ASLAN has an 8% error rate, and it is possible that ASLAN has slightly degraded performance on the k-mers extracted from the unmapped reads, the majority of which seem to originate from heterochromatin. Second, it is possible that a k-mer originating from a haplotype and appearing in a fraction of iHART genomes is not well represented on T2T-CHM13 but does appear in a fraction of the iHART genomes. In this case, the T2T-CHM13 alignment may be incorrect, and the ASLAN localization correct. If the k-mer is highly divergent from any region in T2T-CHM13, we may see a very low alignment score when mapping to T2T-CHM13. However, if the true region of origin has homology with a different region in T2T-CHM13, it might be impossible to determine whether the specific ASLAN localization or T2T-CHM13 is correct. It seems possible that some of the k-mers in our data set are being mismapped to regions in T2T-CHM13. Figure 3C shows that ASLAN-T2T-CHM13 concordance goes up slightly as the alignment score increases. Furthermore, ASLAN versus T2T-CHM13 concordance is much higher for k-mers that uniquely aligned to a single region on T2T-CHM13 compared with those that had multiple similarly scored alignments. We also see lower alignment scores when ASLAN localizations and T2T-CHM13 alignments disagreed (Fig. 4D,E).
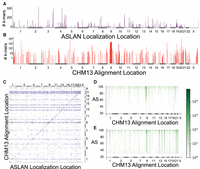
Characterizing nonconcordance between ASLAN localizations and CHM13 alignments and potential hotspots of genetic diversity. (A) Distribution of reads that failed to align to the T2T-CHM13 assembly but that were successfully localized via ASLAN. (B) Distribution of reads for which the localization region predicted by ASLAN contained the location the read aligned to on T2T-CHM13 but for which the T2T-CHM13 alignment score was less than 90. These regions may indicate new hotspots for genetic diversity. (C) Joint-plot of regions where ASLAN localization and T2T-CHM13 alignments were in disagreement with one another and where the T2T-CHM13 alignment score was less than 90. These may indicate sequences that are not represented in the T2T-CHM13 but that are somewhat homologous to a different region in T2T-CHM13 and may be mismapped. (D,E) Loci and alignment score distribution between the T2T-CHM13 alignments for k-mers with ASLAN localizations in agreement with each other (D) and in disagreement with each other (E). We see that k-mers in disagreement have significantly lower alignment scores, suggesting that imperfect alignments to T2T-CHM13 may actually be originating from a human genome sequence not well represented on T2T-CHM13.
Concordance between ASLAN localizations and T2T-CHM13 alignment showed a bimodal distribution across chromosomes (Fig. 3D). Although the metacentric and submetacentric chromosomes showed close to 80% concordance with the T2T-CHM13 alignments, the acrocentric chromosomes (13–15, 21–22) showed <20% concordance. We also saw a much lower concordance in the centromeric and short arm regions of the metacentric and submetacentric chromosomes (Fig. 3D). These two phenomena are likely related and may be caused by a few reasons, assuming the T2T-CHM13 assembly is correct, as it was validated using several different sequencing modalities. We discuss possible reasons for disconcordance and the limitations of both ASLAN and T2T-CHM13 further in the Discussion.
Comparison of ASLAN localizations and T2T-CHM13 alignments highlights new hotspots for genetic diversity
Although T2T-CHM13 is the first assembly of a full human genome, it only represents the sequence of a single genome. We next compared ASLAN's localization results to the T2T-CHM13 alignment in order to analyze genetic variation, particularly in the regions of the genome that have been understudied owing to their absence in the reference genome. We looked at three types of genetic variation: (1) 100-mers that failed to align to anywhere in the T2T-CHM13 assembly but were successfully localized with ASLAN, indicating regions of the human genome where T2T-CHM13 and the genome of other samples are extremely divergent; (2) 100-mers for which T2T-CHM13 alignment and ASLAN localization agree, but that have a low T2T-CHM13 alignment score, indicating regions where large amounts of SNPs or simple genetic diversity exists across samples; and (3) regions where ASLAN localizations and T2T-CHM13 alignments disagree and had low T2T-CHM13 alignment scores, suggesting regions where T2T-CHM13 alignment may not be reliable owing to homologous sequences in regions of the human pangenome not captured on T2T-CHM13.
First of all, we found that a small (1.5%) percentage of our unmapped k-mers that localized successfully with ASLAN failed to align to T2T-CHM13 (Fig. 4A). Of the total 20%–80% prevalence k-mers that did not align to T2T-CHM13, ASLAN was able to localize 88% of them. According to the ASLAN localizations, these k-mers seemed to originate from the Y Chromosome (37%) and the autosomes (63%). It makes sense that many of these k-mers originate from the Y Chromosome, as the T2T-CHM13 is from a hydatidiform mole, and so the assembly does not contain a Y Chromosome. The T2T-CHM13-unaligned k-mers that localized to autosomes with ASLAN seem to originate from specific regions of the genome.
These regions may indicate hotspots for genetic diversity, where a sequence exists in an iHART haplotype that is not well represented on the singular T2T-CHM13 genome. These hotspots include regions around Chr2:176 Mb, Chr3:171 Mb, Chr3:177 Mb, Chr4:117 Mb, and Chr11:7 Mb, as well as overall Chromosome 19 (in GRCh38 coordinates). Unlike the hotspots for localized reads in general, these hotspots for localized but unaligned reads do not seem to have any relationship with known genetic diversity, as cataloged by dbVar and the alternative sequences in the decoy. Notably, within the hotspot around Chr3:177 Mb is the repeat-containing gene TBL1XR1. Mutations in TBL1XR1 have been previously associated with autism spectrum disorders (O'Roak et al. 2012). As the iHART data set is from multiplex autism families, it is possible that ASLAN is localizing a structural variant in TBL1XR1 that is not represented on T2T-CHM13 but may be more frequent in families with autism.
We also found many regions where ASLAN localization and T2T-CHM13 alignment seemed to be in agreement but where the T2T-CHM13 alignment had a relatively low alignment score (less than 90) (Fig. 4B). With short query sequences, it can be difficult to tell the reason for a low alignment score. It may indicate that a query sequence does in fact originate from the region of the genome it aligned to but has variation compared with the reference. Or, a low alignment score may indicate that a query originates from another region from the genome entirely, possibly from a genomic region not well represented on the reference genome used in alignment. Given that ASLAN localizations and T2T-CHM13 alignment were the same in these cases, we assumed that these k-mers were the former and truly did belong to the region that they aligned to in T2T-CHM13. The low alignment score would then be caused by genetic variation relative to the T2T-CHM13 reference sequence. (Note that the reason we use alignment score instead of mapping quality is because a low mapping quality [MAPQ] often means that a read may have aligned perfectly to the human reference genome but aligned to many locations perfectly. We are not interested in these reads but rather are interested in reads that failed to align, or had very little overlap, to the human reference genome.)
Again, we saw hotspots for this type of genetic variation. The number of low alignment score k-mers with ASLAN and T2T-CHM13 concordance differed by over five orders of magnitude across different regions of the genome (Fig. 4B). Specifically, we see hotspots within the centromeres of Chromosomes 1, 5, 7, 9, 10, 17, and 20 and near loci Chr8:137 Mb and Chr13:96.5 Mb. The alignment near Chr8:137 Mb is within the long noncoding RNA sequence, LINC02055, and the alignment near Chr13:96.5 Mb is within the gene HS6ST3.
Finally, we analyzed k-mers for which ASLAN localizations and T2T-CHM13 alignment did not agree and T2T-CHM13 AS was low. We discussed these types of sequences in the previous section but revisit them briefly. The disagreement between ASLAN and T2T-CHM13 could result from several reasons: On one hand, they could be a sample of the k-mers that ASLAN localized incorrectly (as seen in the validation data set, ASLAN with the chosen hyperparameter does localize ∼8% of k-mers incorrectly). On the other hand, these k-mers could be originating from a genomic sequence not well captured in T2T-CHM13, with the low alignment score suggesting that the aligned location is not the true origin of the k-mer. As we discussed previously, the lower alignment scores for k-mers that were discordant between T2T-CHM13 alignment and ASLAN localization compared indicate that T2T-CHM13 alignment may be incorrect for at least some of these mismatched mappings. Particular hotspots for discordance and low alignment scores are the k-mers that aligned to the T2T-CHM13 centromeres in Chromosomes 1, 5, 9, 10, and 17 and small regions within the short arms of the acrocentric chromosomes (Fig. 4C).
Aligning a sample's reads to a singular reference genome, even one as complete as T2T-CHM13, does not necessarily give us a full understanding of sample's variants: It may be difficult to distinguish between misalignment and divergence from the reference sequence, and some sequences within a sample may be poorly represented on the reference genome. In the genetically diverse hotspots we highlighted, particular care should be taken when performing alignment to T2T-CHM13, and alternate sequences may be needed to ensure adequate representation of genetically diverse regions in the human genome.
Discussion
Taking advantage of the unique family structure of the iHART data set, we built an algorithm that uses phasing patterns across families to identify probable regions of origin of unmapped sequences. ASLAN shows high performance on validation data sets and localized many unmapped reads that seemingly originate from gaps in GRCh38. Comparing ASLAN localizations with T2T-CHM13 alignments, we identified several regions of interest that may warrant further studies, including high fidelity human genome assemblies: regions where ASLAN localizations and T2T and T2T-CHM13 alignments were in agreement but where the low T2T-CHM13 alignment scores may indicate hotspots for genetic diversity. Additionally, regions where ASLAN localizations and T2T-CHM13 alignments disagree may indicate sequences that are homologous to a T2T-CHM13 region, but they themselves not well represented on the T2T-CHM13. As the field of pangenomics hones in on developing a human reference genome that encompasses genetic diversity, these genetically diverse regions we mention in our results may be regions of the genome to prioritize.
Localization hotspots
As described in the Results, ASLAN localizes reads to many localization hotspots. Localization locations are enriched for gaps in GRCh38, and nongapped locations may be genetic diversity hotspots where the single reference genome fails to capture all sequences.
Some of these hotspots (Chr2:89 Mb, Chr8:13 Mb, Chr15:20 Mb) contain a fix patch, a known incorrectly assembled region of the genome that will be updated in the next release of the reference genome. The subsequences localized to this region probably went originally unmapped owing to the incorrect assembly of GRCh38. Note that we did not use the fix patches in our original alignment, as this is the standard in alignment pipelines. Often fix patches indicate a more complicated change than a simple inserted or alternate sequence, such as a change in coordinate system.
Additionally, several of these regions are well-cataloged hotspots for genetic diversity. They either have alternate haplotypes cataloged in the reference genome (Chr6:33 Mb, Chr19:54 Mb, and Chr12:11 Mb), contain high-frequency structural variations cataloged by dbVar (Chr8:136 Mb, Chr11:1 Mb, Chr11:127 Mb) (Lappalainen et al. 2012), or are likely home to additional uncataloged complex variation.
The localization hotspot on Chromosome 6 (around 33 Mb) is a known hotspot for genetic diversity: It lies within the HLA gene set, the most diverse region of the genome across individuals. The decoy genome lists six specific alternative haplotypes in this region, but it is estimated there are many orders of magnitudes more across the human population (Gourraud et al. 2014). These sequences were extracted from reads that did not align well to the primary reference genome or the decoy contigs; therefore, ASLAN is localizing HLA subsequences that are not well represented anywhere on GRCh38. Similarly, Chromosome 19 (around 54 Mb) contains more than 30 alternative haplotypes. This region is home to the killer immunoglobulin receptor (KIR) gene family, which is also involved in immune function and known to be extremely diverse across the human population (Uhrberg 2005). Finally, the hotspot on Chromosome 12 (around 11 Mb) has two additional alternate haplotypes with likely more alternative haplotypes not cataloged in the decoy genome.
The hotspot on Chromosome 8 around 136 Mb is a known hotspot for structural variation, especially within non-European populations, with numerous insertions and deletions with high allele frequency (>0.2) cataloged on dbVar in the long noncoding RNAs (LINC2005 and RP11-149P24.1) The hotspots on the start (1 Mb) and end (127 Mb) of Chromosome 11 also show evidence of widespread genetic diversity. Within the first 1 Mb of Chromosome 11, there are three alternative haplotypes in the decoy genome and likely even more variation not cataloged in the decoy. At the end of Chromosome 11 (127 Mb), there is widespread high-frequency structural variation cataloged in dbVar, particularly in the KIRREL3 gene set and the lnRNA LOC101929473.
While we were conducting this research, the Human Pangenome Research Consortia (HPRC; a group closely related to the T2T) began assembling the first pangenome, using sequencing technology and pipelines similar to the T2T-CHM13 to assemble a draft pangenome of 47 fully phased diploid genomes (Ebert et al. 2021; Liao et al. 2023; Porubsky et al. 2023). Their draft assembly has not only hotspots for genetic diversity and structural variation that agree with many of ASLAN's genetic diversity hotspots but also newly discovered abnormalities in chromosome structure that may explain some of the disagreement between ASLAN localizations and the T2T-CHM13 alignment.
Many of ASLAN localization hotspots corresponded to regions that were discovered to contain high frequencies of structural variants or to correspond to the end of haplotypes as computed by the HPRC (Porubsky et al. 2023). These included (in GRCh38 coordinates) Chr4:50 Mb, Chr7:78 Mb, Chr8:13 Mb, Chr9:4–8 Mb, Chr10:80 Mb, Chr12:11 Mb, Chr12:133 Mb, Chr15:20 Mb, Chr17:38 Mb, Chr19:52, Chr19:58 Mb, and Chr22:24 Mb. Notably, the hotspots on Chromosome 8 and Chromosome 17 fall within the 8p23.1 band and the TBC1D3 gene, respectively. 8p23.1 is a biomedically relevant region that is home to a common deleterious deletion causing congenital defects, and TBC1D3 (Pei et al. 2002) has been implicated in prostate cancer (so strongly that its alias is the prostate cancer protein).
Additionally, many of our localization and genetic diversity hotspots fell within regions that had required numbers of alternative haplotypes to represent their pangenomic form, as computed using the HPRC's assembly (Lee et al. 2023). These include Chr1:150 Mb and 245 Mb, Chr8:88 Mb, Chr4:50 Mb, Chr7:78 Mb, Chr8:12 Mb, Chr11:90 Mb, Chr12:133 Mb, and Chr12:70 Mb.
ASLAN and the upcoming pangenome give strong orthogonal support for various genetically diverse regions that should be prioritized in population studies.
Discordance between ASLAN and T2T-CHM13
Although reads localized to the metacentric and submetacentric chromosomes have high degrees of concordance with T2T-CHM13, the acrocentric chromosomes do not perform as well. There are a number of possible explanations for this. First of all, ASLAN was designed and works well for tandem duplications, repetitive motifs that occur in only a single region of the genome. It is limited in its ability to detect segmental duplications, repetitive motifs that occur in multiple regions of the genome, especially if there are recombination points between repeats. This may be the case for SINEs, LINEs, LTRs, and other nontandem repetitive elements. Second, our phasing algorithm relies on variant calls in relation to the reference genome. We cannot, therefore, precisely identify recombination events that happen in gaps in the reference genome. If a recombination event happened within a gap in GRCh38, our algorithm would at best identify that recombination point to occur right after or right before the gap. If two recombination events occurred within a gap, our phasing algorithm would miss the recombination events entirely. This may be the case for the discordance for the sequences aligned to Chromosome 9 using the T2T-CHM13 reference and localized across the genome using ASLAN. Chromosome 9 has the longest contiguous satellite array (short, tandem repeats), which may have made it hard to phase families in this ∼30-Mb region (Altemose et al. 2022). Additionally, PCR amplification bias of certain low GC-content regions in the centromeres (Aird et al. 2011) may invalidate the assumption that the observed subsequence count follows a Poisson distribution uniformly across the genome.
Most importantly and unbeknownst to us while developing ASLAN, acrocentric chromosomes have high degrees of heterologous recombination across chromosomes. A recent study related to the HPRC assembly showed that the short arms of acrocentric swap chromatin with each other at rates comparable to intrachromosomal recombination rates (Guarracino et al. 2023). This could explain the large degradation in ASLAN/T2T-CHM13 agreement in the acrocentric chromosomes compared with the metacentric and submetacentric chromosomes. A sequence from a short arm of specific acrocentric chromosome may well belong to a different chromosome in the CHM13 cell line than in the majority of individuals in the iHART data set. Furthermore, ASLAN only accounted for intrachromosomal recombination, and this newfound recombination across chromosomes invalidates many assumptions about inheritance patterns (and most phasing algorithms) used by ASLAN in these regions.
Additionally, the HPRC identified regions of common segmental duplications, large high-identity sequences duplicated in nonneighboring regions of the genome (Vollger et al. 2022). Notably, many of these segmental duplication hotspots coincide with regions of the genome where ASLAN and T2T-CHM13 disagreed. In additional to the acrocentric chromosome recombination hotspots, which can effectively be thought of as segmental duplications, the HPRC found high frequencies of segmental duplications near the same spots where ASLAN struggled to localize (Fig. 3D): around Chr1:130 Mb, Chr3:100 Mb, Chr5:70 Mb, Chr7:35 Mb–80 Mb, Chr9:45 Mb–80 Mb, Chr10:50 Mb, Chr17:25 Mb–40 Mb, and Chr20:31 Mb.
Finally, like many sets of reference genomes, T2T-CHM13 and GRCh38 have differences in their coordinate systems: A contiguous segment of genome in GRCh38 may not translate to a contiguous segment of genome in T2T-CHM13. T2T-CHM13 has deletions, insertions, and translocations relative to GRCh38. Thus, a region predicted by ASLAN, a contiguous section of GRCh38, may include several noncontiguous regions of T2T-CHM13, some of which may be on different chromosomes. We used the software package liftOver (Hinrichs et al. 2006) and the chain files provided by the T2T to translate our GRCh38 localizations into T2T-CHM13 coordinates to compare to T2T-CHM13 alignments. Lifting over each base contained in a large region usually results in an output that is split across multiple regions and, often, even multiple chromosomes of a genome. To avoid confusion with this, we lifted over only the ends of our region, which can be specified in the liftOver arguments, which was developed to lift over large regions such as BACs. Therefore, our contiguous regions of GRCh38 were lifted over to contiguous regions of T2T-CHM13, even though it was possible they contained sequences of T2T-CHM13 outside the output regions. If a 100-mer originated from one of these sequences, there may be a mismatch between its T2T-CHM13 alignment and the ASLAN localization when translated to T2T-CHM13 coordinates, even if ASLAN actually performed correctly.
Prioritizing studies of global genetic variation
Similar to several other studies (Miga 2019; Grigorev et al. 2021), our results showed that the telomeres and centromeres in the human genome are major sources of genetic variation. The combination of long-read technology and the T2T-CHM13 reference genome will open up new doors for population-scale studies identifying the roles of these satellite sequences in human health and disease (Miga and Wang 2021). Some of the other highly diverse regions we found fell into regions where others have observed high levels of sequence divergence or structural variation as cataloged in dbVar and the alternative haplotypes in the decoy genome. We also identified additional candidate hotspots for genetic variation that, to our knowledge, have not been previously characterized as major contributors to human genetic diversity, including regions within the TBL1XR1 and HS6ST3 genes and the long noncoding RNA LINC02055.
Although some of the goals of this study were to characterize genetically diverse regions of the human genome, the iHART data set falls prey to many of the diversity and representation issues that modern genomics has been grappling with. The iHART data set, which encompasses a subset of samples from the Autism Genetic Resource Exchange (AGRE) biobank (Lajonchere 2010), primarily contains individuals of European descent: 83.5% of our participants self-identified as white, 2.7% as Asian, 2.9% as Black, <1% each Alaskan native/American Indian and Native Hawaiian/Pacific Islander, 5% mixed race, and 4% unknown. Autism research poses a particularly challenging set of circumstances in data ascertainment, in which socioeconomic and demographic factors play a major factor in the availability of resources for diagnosis, treatment, and connections to research studies (Albert et al. 2017). Nevertheless, not only is the iHART data set limited when it comes to understanding autism genetics of diverse populations, but also using ASLAN on iHART only gives us insight into genetic diversity of a small subset of the global population. Although there are several more representative WGS data sets currently in the making, ASLAN's requirements are fairly niche in that it requires large numbers of multichildren families. To our knowledge, iHART is the one of the only data sets of this kind.
Regions of the genome that are genetically diverse within populations also tend to be genetically diverse across populations, as these hotspots may be in regions that are subject to higher de novo mutation rates or reside in genetic elements that can handle higher mutational loads (Myles et al. 2008; Oleksyk et al. 2008; Nesta et al. 2021). Therefore, although there may be additional hotspots for genetic diversity in other populations, our newly highlighted regions of genetic diversity (Fig. 4A,B) within the relatively ethnically homogeneous iHART population likely generalize to other populations as well. These hotspots of genetic diversity are thus important areas to focus on as long-read technology makes it possible to quickly assemble partial genomes. If these regions are candidates for disease-association studies, the studies may require larger sample sizes or need to include in-depth analysis of structural variation.
Methods
Data preprocessing
iHART data set, GRCh38 alignment, variant calling
We used the iHART WGS collection (Ruzzo et al. 2019), a data set of multiplex autism families containing 1006 families and 4610 individuals. Individuals were sequenced at 30× coverage using Illumina's TruSeq Nano library kits; reads were aligned to build GRCh38 of the reference genome (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/GRCh38_full_analysis_set_plus_decoy_hla.fa, accession GCA_000001405.15) and decoy contigs using BWA-MEM (Li 2013); and variants were called using GATKv3.4. Only biallelic variants that passed GATK's variant quality score recalibration (VQSR) (De Summa et al. 2017) were included in the analysis.
Extracting and realigning unmapped and poorly mapped reads
We excluded secondary alignments, supplemental alignments, and PCR duplicates from downstream analyses. We extracted reads from the iHART genomes that were unmapped or mapped with low confidence. Low-confidence reads were defined as reads marked as improperly paired and with an alignment score below 100. We used alignment score rather than MAPQ in order to select reads not likely to be true alignments to the human reference genome, rather than reads that had ambiguous alignments to GRCh38. Note that a low MAPQ often means that a read may have aligned perfectly to the human reference genome but aligned to many locations perfectly. We are not interested in these reads but, rather, are interested in reads that failed (or had very little overlap) to align to the human reference genome at all. Our pipeline is computationally heavy, and running the entire pipeline using different AS cutoffs would be unnecessarily costly.
We used Kraken2 (Wood et al. 2019) to align the unmapped and poorly aligned reads to the Kraken default (RefSeq) databases of archaeal, bacterial, human (GRCh38.p13), and viral sequences (O'Leary et al. 2016). These reference databases were accessed on February 16, 2021. Kraken2 was run on the unmapped and poorly mapped reads from each sample, using the default parameters. We then removed the reads that Kraken was able to classify as archaeal, bacterial, viral, or human reference sequences to leave us with the ultimately unmapped reads. This corresponded to a median of 4.4 million reads per sample, or 0.6% of the total number of reads.
Localization algorithm
Phasing
Phasing refers to the use of an individual's genetic data to determine which sequences or variants in their genome were inherited from their mother and which from their father. During meiosis, each parent’s two copies of each chromosome are combined in large blocks to form a new chromosomal copy, which is then inherited by the child. In phasing, the goal is to identify which parental copy was inherited by each child at every position of the genome. We used an HMM developed in our related work to phase the families in the iHART data set. The phasing algorithm ultimately outputs for each region of the genome, whether a child inherited copy 1 or copy 2 of their mother's genome in the form of (m1|2, p1|2). We describe the mechanics of the phasing algorithm fully in our other work (Paskov et al. 2023) and briefly in Supplemental Methods S1.
Extracting k-mer counts
Although the read lengths in the iHART data set were 150 bp, attempting to localize an entire sequence of a read runs the risk that a sample's genome contains a specific 150-bp sequence but that no reads from that sample originate from that exact region. Therefore, we instead extracted subsequences from the raw reads. We aimed to extract a subsequence that was short enough to nearly guarantee that if a sample's genome contained the subsequence, the sample's reads would as well, but not so short that a subsequence might occur many times across various regions of a genome. To accomplish this, we chose a k-mer length of 100 bp. The full derivation is shown in Supplemental Methods S2 and Supplemental Figure S1.
From the reads of interest (either reads mapping to the decoy genome in the validation pipeline or reads that are poorly aligned and unmapped), we converted these alignments back to raw reads using bam2fastq and then the fast multithreaded k-mer counter jellyfish (Marçais and Kingsford 2011) to extract and count subsequences of 100 bases from the reads. To reduce the amount of low abundance contaminants and reads originating from sequencing errors (Schirmer et al. 2016; Pfeiffer et al. 2018; Stoler and Nekrutenko 2021), for each sample, we extracted and counted only nonsingleton k-mers. Again, using jellyfish, we cataloged and counted the 100-mers that appeared at least twice in at least two samples in the iHART data set.
Maximum likelihood model
We previously developed and validated a proof-of-concept algorithm to localize 100-bp k-mers extracted from 150-bp reads (Chrisman et al. 2021). We reviewed the mathematics of this maximum likelihood model and discuss the extensions that we added in order to allow localization of tandem repeats and for sequence originating from the sex chromosomes.
The goal of the maximum likelihood model is as follows: For each k-mer, we wish to find its corresponding location (region r) in the genome that best explains the distribution of the k-mer counts in all of the families. We define the distribution of a given k-mer in all samples as K and the distribution of a given k-mer in family f as Kf. Therefore, we want to find the region r that maximizes the likelihood of observing k. We wish to compute the contiguous genome region r, where (K|r) falls above a certain threshold. Assuming Mendelian inheritance and a Poisson distribution of coverage for each family,
this probability can be written as
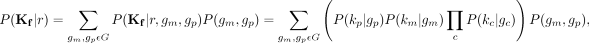 (1)
where km, kp, and kc are the counts of a k-mer from a mother, father, and child, respectively; gm, gp, and gc are the genotypes of the mother, father, and child, respectively. We can use the phasings to compute the probability of a
child's genotype given their parental genotypes.
(1)
where km, kp, and kc are the counts of a k-mer from a mother, father, and child, respectively; gm, gp, and gc are the genotypes of the mother, father, and child, respectively. We can use the phasings to compute the probability of a
child's genotype given their parental genotypes.
We give a full derivation and explanation of assumptions of the maximum likelihood model in Supplemental Methods S3. The code for our localization algorithm can be accessed at GitHub (https://github.com/briannachrisman/alt_haplotypes).
Validation
We validated our algorithm by using two data sets of k-mer distributions with known locations: (1) a synthetically generated distribution of k-mers and (2) k-mer counts extracted from alternative sequences in the decoy reference genome.
We describe how these validation data sets were generated in Supplemental Methods S4. Additionally, our maximum likelihood model is entirely based on biological constants except for one hyperparameter, λ, which determines the length of the region that ASLAN ultimately localizes a subsequence to. We evaluated and optimized our algorithm using different values of λ on both validation data sets and found that λ = 0.1 provided a good balance of accuracy (>90%) and precision (∼1 Mb of resolution). We give a full description of this hyperparameter tuning in Supplemental Methods S4.3.
Unmapped read localization and analysis
Extracting k-mers from unmapped reads
We used the previously described pipeline involving jellyfish to extract 100-mers from the unmapped and poorly aligned reads from iHART. To improve computation time, we sampled about 100 million out of 200 million k-mers (exact number, 104,623,400 out of 234,132,233) under the assumption that many k-mers would overlap and localize to the same region. We assumed that ∼50% sampling would provide sufficient understanding of genetic diversity and missing haplotypes in the T2T assembly while adhering to computational limitations.
Comparing to gaps in GRCh38
We retrieved the current gaps in GRCh38 from the GRCh38 AGP file provided by the UCSC Genome Browser track on February 15, 2022. To statistically test if our reads localized to gaps more often than by chance, we build a null distribution of the fraction of k-mers that localized to gaps in GRCh38 and used it to compute a P-value for the fraction of our k-mers that aligned to gaps. To generate the null distribution, for each cataloged gap in the AGP file (Karolchik et al. 2003), we randomly generated a new gap on the same chromosome and with the same length as the original gap but at a different location. We did this for every gap, and then we computed the percentage of unmapped reads that localized to a region containing a gap in GRCh38. We performed this 10,000 times to generate a null distribution for P-value computation.
Comparing to T2T alignment
We converted the 100-mers from the unmapped reads to FASTA format and aligned them to the T2T's v1.1 release of T2T-CHM13 (chm13.draft_v1.1.FASTA, accessed on February 15, 2022) using BWA-MEM with the default parameters. To compare the results of ASLAN with those of alignment to the T2T-CHM13 assembly, we translated the starts and ends of our localization coordinates to the T2T-CHM13 coordinate system using liftOver (Hinrichs et al. 2006) and the GRCh38-to-T2T-CHM13 chain file (GRCh38.t2t-chm13-v1.1.over.chain.gz, accessed on February 15, 2022). To prevent split liftOver conversions, we set the parameter -ends = 100,000 for lifting over long sequences. We considered T2T-CHM13 alignment and ASLAN localization to be in concordance if the liftOver ASLAN localization and the T2T-CHM13 alignment contained an overlapping region.
Software availability
The code for our localization algorithm, analysis scripts, and figures can be accessed at GitHub (https://github.com/briannachrisman/alt_haplotypes) and as Supplemental Code.
Data access
All raw and processed sequencing data used by from the iHART samples can be found on AnVIL, maintained by NHGRI at https://AnVILproject.org/data/studies/phs001766. Data set access is controlled in adherence to NIH policy and in line with the standards set forth in the individual consents involved in each cohort. The ASLAN localizations, T2T-CHM13 mappings, and the 100-bp sequences have been submitted to Stanford's public research data library and can be accessed at https://purl.stanford.edu/sx779pk7425:
-
ASLAN_localizations.bed—a BED file containing the chromosome, start coordinate, end coordinate, and k-mer index (starting from 0) of the region ASLAN localized each k-mer to, and coordinates follow the GRCh38 system;
-
T2T_mappings.bed—a BED file containing the chromosome, start coordinate, end coordinate, and k-mer index (starting from 0) of each k-mer's primary alignment to the T2T-CHM13 reference, and coordinates have been converted to the GRCh38 system; and
-
kmers.txt.zip—a compressed list of k-mers extracted from the unmapped reads, and the order of these k-mers corresponds to the k-mer index in the previously described BED files.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank The Hartwell Foundation for supporting the creation of the iHART database and the Simons Foundation for additional support for genome sequencing. We thank the New York Genome Center for conducting sequencing and initial quality control of the iHART data set. We thank Amazon Web Services for their grant support for the computational infrastructure and storage for the iHART database. This work has been supported by grants from The Hartwell Foundation and the National Institutes of Health (NIH) (U24 MH081810, R01MH064547, NS101158, NS070911, NS101665, NS095824, S10OD011939, P30AG10161, R01AG17917, and U01AG61356), from the Stanford Precision Health and Integrated Diagnostics Center, and from the Stanford Bio-X Center. This publication was additionally partially supported by grants from the National Institute of General Medical Sciences (GM103440 and GM104944) from the NIH.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277175.122.
-
Freely available online through the Genome Research Open Access option.
- Received August 2, 2022.
- Accepted May 25, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








