
Unsupervised cell functional annotation for single-cell RNA-seq
- 1Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA;
- 2Meakins-Christie Laboratories, Department of Medicine, McGill University Health Centre, Montreal, Quebec, H4A 3J1, Canada;
- 3Machine Learning Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA
Abstract
One of the first steps in the analysis of single-cell RNA sequencing (scRNA-seq) data is the assignment of cell types. Although a number of supervised methods have been developed for this, in most cases such assignment is performed by first clustering cells in low-dimensional space and then assigning cell types to different clusters. To overcome noise and to improve cell type assignments, we developed UNIFAN, a neural network method that simultaneously clusters and annotates cells using known gene sets. UNIFAN combines both low-dimensional representation for all genes and cell-specific gene set activity scores to determine the clustering. We applied UNIFAN to human and mouse scRNA-seq data sets from several different organs. We show, by using knowledge about gene sets, that UNIFAN greatly outperforms prior methods developed for clustering scRNA-seq data. The gene sets assigned by UNIFAN to different clusters provide strong evidence for the cell type that is represented by this cluster, making annotations easier.
The large increase in studies profiling RNA sequencing data in single cells (Tanay and Regev 2017) raises several computational challenges. One of the first, and most important, steps in the analysis of such studies is cell type assignment (Clarke et al. 2021). Several methods have been developed for such assignment, including (semi-)supervised and unsupervised methods. (Semi-)supervised methods mainly use previously annotated data sets to annotate new data sets (Abdelaal et al. 2019). This is performed by directly classifying each cell (Alavi et al. 2018; Pliner et al. 2019), by learning an alignment between the data sets (Kiselev et al. 2018), or by joint training using multiple data sets to classify groups of cells in a new study (Brbić et al. 2020; Hu et al. 2020).
Although supervised methods are useful in some cases, they cannot be applied to all cases because reference data sets are not available for most organs, tissues, and conditions. Another challenge with supervised methods is their inability to identify new cell types, which is often one of the major goals of the study (Abdelaal et al. 2019). Thus, the most popular way to annotate single-cell data is by using unsupervised methods. These are often based on clustering cells in a low-dimensional space and manually annotating each cluster using known marker genes or cluster specific differentially expressed genes. Several methods for clustering single-cell data have been developed and used. These include SIMLR (Wang et al. 2017), which clusters cells by using multiple kernel functions to construct a similarity matrix between cells; Leiden clustering (Traag et al. 2019) and Seurat v3 (Stuart et al. 2019), which use k-nearest neighbor (k-nn)–based graph partitions to group cells; SCCAF (Miao et al. 2020), which refines initial clusters using a self-projection-based approach; and methods based on deep neural networks, such as DESC (Li et al. 2020), which uses autoencoders to reduce the dimensions of the data and then clusters cells in the reduced dimension space.
Although several clustering methods have been developed and used for single-cell RNA sequencing (scRNA-seq) data, to date these methods have only relied on the observed expression data. However, there are several additional complementary data sets that can be used to improve clustering and reduce noise-related grouping. Specifically, gene sets (Subramanian et al. 2005) have been compiled to characterize many processes, pathways, and conditions. Although the exact processes or functions that are activated in specific cells or clusters are unknown, we can use these sets to guide the grouping of cells by placing more emphasis on coexpression of genes in known sets when clustering single-cell data. Because cells of the same type likely share many of the biological processes, such design can both improve the identification of good clusters and help in annotating them based on the function of the sets associated with each cluster.
Here we introduce Unsupervised Single-cell Functional Annotation (UNIFAN) to simultaneously cluster and annotate cells with known biological processes (including pathways). For each cell, we first infer its gene set activity scores based on the coexpression of genes in known gene sets. We also use an autoencoder that outputs a low-dimensional representation learned from the expression of all genes. We combine both the low dimension representation and the gene set activity scores to determine the cluster for each cell. The process is iterative, and we define a target function and show how to learn model parameters to optimize it. In addition to the cell clusters, the method also outputs the gene sets associated with each cluster, and these can be used to annotate and assign cell types to different clusters.
Results
We developed UNIFAN to simultaneously cluster and annotate cells (and cell clusters) with known biological processes or pathways. By integrating prior information about gene sets with observed expression data, UNIFAN can improve clustering results while simultaneously making the clusters more interpretable. Figure 1 presents an overview of UNIFAN. The method starts with inferring gene set activity scores for each single cell, based on the expression levels of genes in known gene sets. Next, UNIFAN clusters cells by using the learned gene set activity scores and a low-dimensional representation of the expression of all genes in the cell. This is performed using an autoencoder-based neural network. The “annotator” part of this network uses the gene set activity scores to guide the clustering such that cells sharing similar biological processes are more likely to be grouped together. For details on the architecture of UNIFAN and on how parameters are learned for UNIFAN, see Methods.
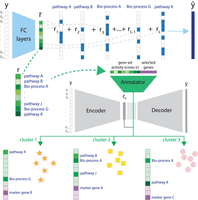
Overview of UNIFAN. (Top) Using the expression levels for genes in a cell y, UNIFAN first infers gene set activity scores (r), using an autoencoder. The decoder is composed of binary vectors with values indicating if a gene belongs to a known gene set or not. (Middle) Next, UNIFAN clusters cells by using the learned gene set activity scores and a low-dimensional representation of the expression of all genes in the cell (ze). For this, it uses an autoencoder-based neural network, which contains two parts: the cluster assignment part (gray) and the “annotator” (green). The cluster assignment part assigns a cell to clusters based on the low-dimensional representation (ze), whereas the “annotator” refines clustering and annotates clusters with biological processes and highly variable genes. (Bottom) Cells assigned to different clusters characterized by selected gene sets and genes. (FC layers) Fully connected layers; (Bio-process) biological process.
UNIFAN correctly clusters cells and identifies relevant biological processes
We first evaluated if UNIFAN can accurately cluster cells and reveal key pathways and cellular functions activated in cells assigned to different clusters. For this, we used the “pbmc28k” scRNA-seq data set (Methods). UNIFAN clusters successfully captured different cell types when compared with manual annotations (adjusted Rand index [ARI] 0.81, normalized mutual information [NMI] 0.77). Figure 2, A through C, presents UMAP (Becht et al. 2019) visualizations of ze output from UNIFAN for each cell. As can be seen, clusters are mostly composed of cells from the same type, which is a large improvement over other methods, including Leiden clustering (shown in Fig. 2C), as we discuss below. By relying on known gene sets, UNIFAN is robust to noise and mainly focuses on relevant coexpressed sets of genes, leading to much more coherent clusters. We observed similar results for the other data sets we tested as can be seen in Supplemental Figures S6 through S8.
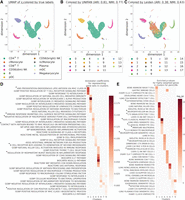
UNIFAN accurately clusters cells and correctly identifies biological processes/pathways. Results presented for the “pbmc28k” data set. (A–C) UMAP visualization of the low-dimensional representation ze of cells output from UNIFAN: (A) colored by true cell type labels, (B) colored by the clusters found by UNIFAN, and (C) colored by Leiden clustering. (D) Coefficients learned by the annotator for highly ranked gene sets for some of the clusters. (E) Enrichment P-values of cell type marker sets in the highly weighted genes learned by the annotator. Here we show the result from the best run for both UNIFAN and Leiden. Because of space limits, some gene set names in D and E are truncated (marked with *). For the full names, see Supplemental Tables S2 and S3.
To annotate cell clusters, we examined the coefficients assigned by the “annotator” to different gene sets for each cluster. Figure 2D presents some of the top-ranked sets for the different clusters. We observe that for cluster 0, the set “GOBP POSITIVE REGULATION OF T CELL RECEPTOR SIGNALING PATHWAY” is assigned a large weight, and this cluster is annotated as CD4+ T cells in the original paper. For cluster 5 (which mainly contains CD8+ T cells), one of the top-scoring sets is “REACTOME NEF MEDIATED CD8 DOWN REGULATION.” Cluster 1 cells were labeled as CD56 (dim) natural killer (NK) cells in the original paper. UNIFAN correctly assigns “GOBP REGULATION OF NK CELL MEDIATED IMMUNITY” and “KEGG NK CELL MEDIATED CYTOTOXICITY” as two of the top gene sets for this cluster. Cluster 3 and 6 correspond to classical monocyte (cMonocyte) and nonclassical monocyte (ncMonocyte), respectively. Although UNIFAN assigns biological processes related to “antigen presentation” and “inflammation” to both clusters, the biological process related to wound healing, “GOBP REGULATION OF INFLAMMATORY RESPONSE TO WOUNDING,” only appears in cluster 6. One of the main differences between ncMonocyte and cMonocyte is their role in wound healing (Schmidl et al. 2014), and so, such assignment can make it much easier to correctly annotate this cluster of cells. In addition to the gene sets, we also evaluated genes highly weighted by the annotator by comparing them to known cell type marker sets. As shown in Figure 2E, the most enriched cell type marker sets for each cluster correspond very well to the true cell labels, indicating that UNIFAN can indeed identify the marker genes for each cell type (cluster).
We observed similar performance in terms of cluster annotations for other data sets we tested. For example, for the “Atlas lung” data set, UNIFAN successfully separated macrophage (cluster 2) and alveolar macrophage (cluster 5) as shown in Supplemental Figure S6A. The annotator selected “GOBP MACROPHAGE FUSION,” “WP MACROPHAGE MARKERS,” and “GOBP NEGATIVE REGULATION OF RESPONSE TO INTERFERON GAMMA” for both clusters. It also selected “GOBP REGULATION OF COLLAGEN FIBRIL ORGANIZATION” for cluster 8, which agrees well with the labels of cells in that cluster (fibroblasts). It selected “GOBP CILIUM MOVEMENT” for cluster 10, again in agreement with the type of cells in this cluster (ciliated). Similarly, the most enriched cell type marker sets for each cluster, learned from the highly weighted genes, corresponded very well to the true cell type labels (Supplemental Fig. S6E).
UNIFAN improves upon prior methods
We compared UNIFAN's clustering performance on all data sets with several prior methods proposed for clustering scRNA-seq data. The number of cells in the data sets we used to compare the methods ranges from 366 (aorta in Tabula Muris) to 96,282 (“Atlas lung” data set), and so, they can provide a good representation of the scRNA-seq data sets being analyzed by researchers. The methods we compared included two graph-based methods, Leiden clustering (Traag et al. 2019) and Seurat v3 (Stuart et al. 2019); a kernel-based method, SIMLR (Wang et al. 2017); a self-projection-based method, SCCAF (Miao et al. 2020); and a deep-learning-based method, DESC (Li et al. 2020). In addition to these unsupervised methods, we also compared UNIFAN to two (semi-)supervised deep-learning methods: MARS (Brbić et al. 2020) and ItClust (Hu et al. 2020). We allow these two (semi-)supervised methods to use the true labels for 5% of the cells in a data set for model learning. We also compared CellAssign (Zhang et al. 2019), which uses known cell type markers for cell type assignment. For each data set, we ran each method 10 times using different initializations. Results are presented in Figure 3 and Supplemental Figure S9. As can be seen, for all data sets, UNIFAN outperforms all other unsupervised methods regardless of the evaluation metric being used (e.g., average ARI/NMI of UNIFAN and the best-performing unsupervised prior method on “pbmc28k”: UNIFAN 0.72/0.74, Leiden 0.37/0.62; on “HuBMAP spleen”: UNIFAN 0.75/0.71, DESC 0.31/0.64; on “Tabula Muris”: UNIFAN 0.70/0.75, SIMLR 0.53/0.65). The large improvement may result from the ability of UNIFAN's to focus on the more relevant sets of coexpressed genes rather than on coexpression that may result from noise or the large number of genes being profiled.
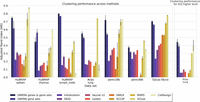
UNIFAN significantly outperforms other methods. “UNIFAN genes & gene sets” is the default UNIFAN version using both gene set activity scores and a subset of genes as features for the annotator. “UNIFAN gene sets” uses only the gene set activity scores. “Initialization” is the initialization clustering results. The others are the prior methods we used for comparison. For the Tabula Muris data, we take the average over all tissues. For tissue-specific results, see Supplemental Figures S11 and S12. The “Atlas lung” data provide two levels of cell type annotations, and so, we show results for both (less detailed annotation comparison shown on the right). SIMLR was unable to cluster the “pbmc68k” and “Atlas lung” data because it ran out of memory. CellAssign does not have an average over all tissues for “Tabula Muris” because some of the tissues in that data set do not have matched cell type marker genes. For details, see Supplemental Methods.
As for the (semi-)supervised methods, UNIFAN outperforms MARS in all data sets and improves on ItClust for most of them as well (e.g., average ARI/NMI of UNIFAN and the best-performing [semi-]supervised prior method on “HuBMAP thymus”: UNIFAN 0.45/0.55, ItClust 0.37/0.46; on “HuBMAP lymph_node”: UNIFAN 0.80/0.71, ItClust 0.44/0.48; on “pbmc28k”: UNIFAN 0.72/0.74, ItClust 0.70/0.70). UNIFAN is worse than ItClust for “pbmc68k” (average ARI/NMI: UNIFAN 0.38/0.54, ItClust 0.45/0.57) and “Atlas lung” (average ARI/NMI: UNIFAN 0.55/0.69, ItClust 0.74/0.78). However, even for “Atlas lung,” UNIFAN improves on ItClust when using another, likely more robust, level of cell annotations (six general cell types vs. 38 used in the initial comparison) as shown on the right of Figure 3 and Supplemental Figure S9. For two other data sets, the performance is comparable (average ARI/NMI on “HuBMAP spleen”: UNIFAN 0.75/0.71, ItClust 0.73/0.75; on “Tabula Muris”: UNIFAN 0.70/0.75, ItClust 0.78/0.74).
For the CellAssign comparison, UNIFAN outperforms CellAssign on most data sets (average ARI/NMI on “HuBMAP lymph_node”: UNIFAN 0.80/0.71, CellAssign 0.05/0.15; on “Atlas lung”: UNIFAN 0.55/0.69, CellAssign 0.12/0.24; on “pbmc68k”: UNIFAN 0.38/0.54, CellAssign 0.35/0.53). CellAssign was unable to annotate some tissues in the Tabula Muris data that do not have matched cell type markers in the database (e.g., adipose tissues). For those having matched markers, UNIFAN performs better than CellAssign in the majority of them, as shown in Supplemental Figures S11 through S14. UNIFAN is worse than CellAssign for “HuBMAP spleen” (average ARI/NMI: UNIFAN 0.75/0.71, CellAssign 0.87/0.72) and “HuBMAP thymus” (average ARI/NMI: UNIFAN 0.45/0.55, CellAssign 0.60/0.56), for both of which, the markers of many cell types are well documented in the marker database. For “pbmc28k,” it is hard to tell which method performs better given the disagreements between ARI and NMI (average ARI/NMI: UNIFAN 0.72/0.74, CellAssign 0.73/0.66).
Overall, these results indicate that UNIFAN provides more accurate cell type identification results especially in cases in which true cell type labels or cell type markers are not available or very few are known.
To further evaluate the different parts of UNIFAN in order to determine which input or processing is contributing the most to its success, we compared different versions of UNIFAN. These included “UNIFAN no annotator,” which is composed of only the clustering part without the annotator; “UNIFAN random,” which uses randomly generated features for the annotator and several other variations differing in the biological features used by the annotator, including “UNIFAN gene sets,” which uses only gene set activity scores; and “UNIFAN genes & gene sets” (the default version), which uses both gene set activity scores and the selected genes.
As shown in Figure 3 and Supplemental Figure S10, the two “UNIFAN” variations using gene sets constantly outperformed the other versions which either did not use an annotator or used randomly generated values as features for the annotator. These results indicate that the use of the annotator to focus on the relevant coexpressed sets of genes is crucial to the performance of UNIFAN.
UNIFAN identifies novel cell subtypes
We tested the ability of UNIFAN to identify novel cell subtypes. For this, we looked at clusters identified by UNIFAN that are combined in the original annotations. Such clusters represent unique cell subtypes according to UNIFAN, whereas in the original analysis all cells in these clusters are assigned to the same cell type (for an example, see Fig. 4). We found a number of such cases and have looked at their biological relevance.
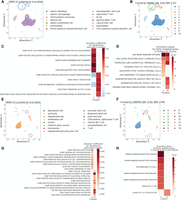
UNIFAN identifies novel cell subtypes for the HuBMAP thymus data and the Brain_Non-Myeloid data in Tabula Muris. (A–D) Results for the HuBMAP thymus data. (E–H) Results for the Brain_Non-Myeloid data in Tabula Muris. (A,B,E,F) UMAP visualization of the low-dimensional representation ze of cells from UNIFAN. Cluster 4 and 10 are circled for the HuBMAP thymus data. Cluster 6 and 12 are circled for the Brain_Non-Myeloid data in Tabula Muris. (A,E) Colored by original cell type labels. (B,F) Colored by the clusters identified by UNIFAN. (C,G) Coefficients learned by the annotator for highly ranked gene sets for the two clusters. (D,H) Enrichment P-values of cell type marker sets in the highly weighted genes learned by the annotator.
The first example is from UNIFAN's results of the HuBMAP thymus data set. Figure 4, A and B, shows a UMAP (Becht et al. 2019) visualization of ze output from UNIFAN for each cell, colored by the true cell types and clusters found by UNIFAN, respectively. UNIFAN clusters 4 and 10 (circled in Fig. 4A,B) were both initially labeled as “splenic fibroblast.” Gene sets and markers selected by UNIFAN for both clusters (Fig. 4C,D) agree with the initial assignment and include “TRAVAGLINI LUNG ADVENTITIAL FIBROBLAST CELL.” They differ, however, in other selected gene sets and markers, indicating that they may represent different subtypes. Cluster 4 cells are enriched for thymus stromal, stellate, and endothelial cell markers, whereas cluster 10 cells are enriched for markers related to mesenchymal stromal cells (MSCs), including “RUBENSTEIN SKELETAL MUSCLE FBN1 FAP CELLS,” and “MURARO PANCREAS MESENCHYMAL STROMAL CELL” (FAP cells, a short form for fibro/adipogenic progenitors, are also a type of MSCs) (Wosczyna et al. 2019).
Examining UNIFAN's results for the Brain_Non-Myeloid data set from Tabula Muris (The Tabula Muris Consortium 2018) highlights another novel subtype. In this data set, we looked at cells labeled as “neuronal stem cell” (shown by the circled clusters in Fig. 4E,F). UNIFAN assigns these cells to two clusters, 6 and 12. Cluster 6 seems to indeed contain mostly neuronal stem cells because gene sets selected for it are related to the differentiation processes of multiple different cell types (e.g., “GOBP NEUROBLAST DIFFERENTIATION” and “GOBP CEREBRAL CORTEX GABAERGIC INTERNEURON DIFFERENTIATION”) (see Fig. 4G), indicating its multipotency. The marker set “FAN EMBRYONIC CTX NSC 2” (NSC is the abbreviation for neuronal stem cells) is also enriched in the selected genes (Fig. 4H). Cluster 12, however, is likely related to oligodendrocyte precursor cells (OPCs) as the selected genes for this cluster are enriched for “ZHONG PFC C4 PTGDS POS OPC” (Fig. 4H). Gene sets related to oligodendrocyte functions, including “GOBP POSITIVE REGULATION OF TRANSMISSION OF NERVE PULSE” (Fields 2008), are also selected by UNIFAN for this cluster (Fig. 4G).
We also conducted simulation experiments to investigate if UNIFAN is robust when the data contain novel cell types using novel pathways that have not been included in the pathway database used by UNIFAN. Results presented in Supplemental Figure S18 show that UNIFAN accurately identifies the correct cell types for such simulated data and is able to identify several of the pathways used by the simulated cell types. For details, see Supplemental Results.
Models are transferable across tissues and species
Because different tissues from the same species or the same tissue across species may share cell types, we next explored if an autoencoder for gene set activity scores that is pretrained on one tissue/species can also be useful for another tissue/species. The importance of such pretraining is that training of the autoencoder for gene set activity scores of UNIFAN is time consuming, and so if this can be performed offline (i.e., using prior data), then the application of the method to a new data set can be much faster.
For this, we pretrain a gene set activity scores model using all available human data sets and apply the learned model to infer the gene set activity scores for Tabula Muris mouse data sets. We then run the clustering and annotation using these inferred scores and compare the results with those inferred from a model that was directly trained on the Tabula Muris data as discussed above. Given we focus on the usefulness of gene set activity scores, we use only these scores as features for the annotator (“UNIFAN gene sets”) for this comparison.
Figure 5 presents the results. As expected, we see an overall decline in the average performance over tissues when comparing the results of pretrained and de novo models. However, for those mouse tissues that are also profiled in the human data sets we used, we observe similar performance when using the pretrained human model. This is most apparent for spleen, lung, and a few adipose tissues, including subcutaneous adipose tissue (SCAT) and gonadal adipose tissue (GAT), as shown in Figure 5 and Supplemental Figures S15 and S16. These adipose tissues contain many immune cell types that are also present in many of the human tissues we used for pretraining (spleen, thymus, lymph nodes, and PBMC). We further tested pretrained models for individual tissues (i.e., training using spleen in human and testing only on spleen in mouse). As shown in Figure 5, for such analysis the performance is even better for the most part compared with using the generally trained model. The only exception is thymus, where the mouse and human annotations differ significantly in the data sets we used. The major cell type in the Tabula Muris thymus data (thymocyte) does not appear in the HuBMAP human thymus data.
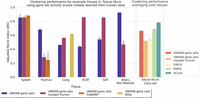
Performance on the Tabula Muris data when using gene set activity scores models pretrained on human tissues. The plot on the left shows the ARI for some example tissues, and the one on the right shows the average ARI across tissues. All versions of UNIFAN methods use models pretrained on human tissues except for “UNIFAN gene sets,” which used models trained on the same data sets as we discussed before. “UNIFAN gene sets merged human” uses the model pretrained on all available human tissues. “UNIFAN gene sets HuBMAP” uses the model pretrained on the corresponding HuBMAP tissue (HuBMAP spleen or thymus). “UNIFAN gene sets Atlas” uses the model pretrained on the “Atlas lung” data set. We included only the best-performing prior methods on the Tabula Muris data (SIMLR, MARS, ItClust) for comparison. We see that the model pretrained on human data is helpful for mouse gene set activity scores inference and for clustering, specifically for tissues having similar cell types between human and mouse such as spleen and lung. For thymus and Brain_Non-Myeloid, whose cell types are not well represented in the pretraining set, the performance drops.
Discussion
Cell type assignment is one of the most important steps in scRNA-seq analysis. In most cases, such assignment is performed by first clustering cells and then assigning each cluster with a cell type based on differentially expressed genes or the expression of known cell type markers.
Here we present UNIFAN, which improves both clustering and cluster annotations by using a large collection of gene sets (Subramanian et al. 2005). UNIFAN infers gene set activity scores and uses them to regularize the clustering of cells. Such design improves the ability to identify biologically meaningful coexpressed genes and to use these to group cells. In addition to leading to improved clustering, UNIFAN also assigns a subset of the gene sets to clusters, which can help characterize their cell types. Finally, by relying on known functional gene sets, UNIFAN can further refine cell subtype assignment, allowing it to obtain a better resolution for cell type assignments. As we have shown for both human and mouse data sets, such analysis can lead to the identification of novel cell subtypes, which may be important as more scRNA-seq data accumulates.
We compared UNIFAN to several popular methods for clustering scRNA-seq data using data sets spanning a large number of organs from both human and mouse. As we show, UNIFAN consistently outperforms other methods across these data sets. We also compared UNIFAN with two (semi-) supervised methods for cell type identification (MARS [Brbić et al. 2020] and ItClust [Hu et al. 2020]) and a method based on known cell type markers (CellAssign) (Zhang et al. 2019). The semi-supervised methods require at least some labeled data, whereas CellAssign uses as input known cell type markers. Still, as we show in Figure 3 and Supplemental Figure S9, UNIFAN outperforms these methods for most data sets. We also observed that, for CellAssign and ItClust, when very high-quality marker sets and labels were available for specific tissues, the performance of these two methods was better than UNIFAN. Thus, we conclude that although UNIFAN is better for general use, in cases in which high-confidence marker lists or labels are available for a certain tissue in a certain species, methods using labels or markers, such as ItClust or CellAssign, may be better. We also analyzed the gene sets selected by UNIFAN for various clusters and showed that they match well with the known cell types (Supplemental Figs. S5–S8, S17).
Analysis of the various parts of UNIFAN identified the annotator and the gene sets and genes it uses as the main sources for the improvement. The fact that adding variable genes as input improves performance is likely the result of the fact that current gene sets, although very useful, are incomplete. It is likely that we are still missing from current collections sets of genes characterizing some less-known biological processes. In such cases, the selected genes capture groupings that are missed by the known gene sets.
UNIFAN can be slow on large data sets (Supplemental Table S4 in Supplemental Results). The main time-consuming part is training the gene set activity score model for the data being clustered. To speed up the analysis, we have applied UNIFAN to a new data set using a gene set activity score model pretrained on another data set. This greatly reduced run time (Supplemental Results) but did lead to drop in performance for tissues whose cell types were not well represented in the pretraining data set. As we obtain more data from tissues and conditions, we expect that we can further improve the ability to use pretraining to improve runtime.
Methods
Data sets and data preprocessing
We used both human and mouse data sets from several tissues to test our method. The human samples include three scRNA-seq data sets from The Human BioMolecular Atlas Program (HuBMAP) consortium (HuBMAP Consortium 2019). These include “HuBMAP spleen,” “HuBMAP thymus,” and “HuBMAP lymph_node.” We use SCANPY (Wolf et al. 2018) for the data preprocessing, leading to 34,515 cells and 26,092 genes for “HuBMAP spleen,” 22,367 cells and 24,396 genes for “HuBMAP thymus,” and 24,311 cells and 20,946 genes for “HuBMAP lymph_node.” The “Atlas lung” uses the healthy control samples from Adams et al. (2020). After filtering, this data set is composed of 96,282 cells and 17,315 genes. The “pbmc28k” data are from Van Der Wijst et al. (2018) and have 25,185 cells and 19,404 genes. The “pbmc68k” data are from Zheng et al. (2017), having 68,551 cells and 17,788 genes. Mouse data sets are from the Tabula Muris paper (The Tabula Muris Consortium 2018). Following Brbić et al. (2020), we end up with 21 data sets each for a single tissue. They all have 22,904 genes, and the number of cells ranges from 366 (aorta) to 4433 (heart). For the preprocessing details, see Supplemental Methods.
In addition to expression data, UNIFAN uses gene sets to guide clustering. For this, we use 7481 gene sets derived from the GO Biological Process ontology (termed c5.go.bp in MSigDB) (Subramanian et al. 2005), 2922 gene sets from pathway databases (c2.cp in MSigDB) (Subramanian et al. 2005), and 335 sets of targets of transcription factors from Ernst et al. (2007). Names for biological process sets start with “GOBP.” Pathway sets use a prefix representing the pathway database they are extracted from (e.g., “KEGG,” “WP,” “REACTOME”). We purposely did not use cell type marker gene sets (c8.all in MSigDB) because we wanted to keep the method unsupervised, and marker lists are often based on DE analysis of labeled cell type data.
Clustering and annotating single cells using gene sets
To enable the use of prior knowledge of gene function and regulation for clustering single cells, we developed a deep-learning model, UNIFAN. For each single cell, UNIFAN first infers gene set activity scores associated with this cell using the input gene sets. Next, UNIFAN clusters cells by using the learned gene set activity scores and a reduced dimension representation of the expression of genes in the cell. The gene set activity scores are used by an “annotator” to guide the clustering such that cells sharing similar biological processes are more likely to be grouped together. Such a design allows the method to focus on the key processes when clustering cells and so can overcome issues related to noise and dropout while simultaneously selecting marker gene sets that can be used to annotate clusters.
Learning gene set activity scores for cells
For each cell, we first infer its gene set activity scores, 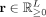 (L: number of gene sets), which represent the activity of known biological processes or pathways in the cell. For this, we design
a special autoencoder whose decoder, instead of being fully connected, is composed of a binary matrix D ∈ ℝG×L, where G is the total number of genes profiled. Each column in D corresponds to a known gene set for a biological process or pathway where the values are indicators for whether a gene belongs
to this set or not. For a cell with expression y, the encoder, which is composed of fully connected layers, outputs a low-dimensional representation, r. r is then multiplied by the binary matrix D, which leads to a reconstructed expression vector,
(L: number of gene sets), which represent the activity of known biological processes or pathways in the cell. For this, we design
a special autoencoder whose decoder, instead of being fully connected, is composed of a binary matrix D ∈ ℝG×L, where G is the total number of genes profiled. Each column in D corresponds to a known gene set for a biological process or pathway where the values are indicators for whether a gene belongs
to this set or not. For a cell with expression y, the encoder, which is composed of fully connected layers, outputs a low-dimensional representation, r. r is then multiplied by the binary matrix D, which leads to a reconstructed expression vector, 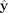 , as shown in Figure 6. Values in r serve as weights/coefficients for known gene sets for this cell. Parameters for the fully connected encoder are optimized
such that the combination of the gene sets, weighted by r, can be used to reconstruct the observed expression y for all genes in the cell. Thus, r can be seen as the activity levels of pathways and processes in the cell.
, as shown in Figure 6. Values in r serve as weights/coefficients for known gene sets for this cell. Parameters for the fully connected encoder are optimized
such that the combination of the gene sets, weighted by r, can be used to reconstruct the observed expression y for all genes in the cell. Thus, r can be seen as the activity levels of pathways and processes in the cell.
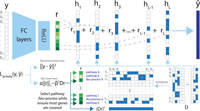
Assigning single-cell gene set activity scores using an autoencoder. The autoencoder is designed such that the decoder is composed of binary vectors with values indicating if a profiled gene belongs to a known gene set or not. The output of the encoder, r, serves as coefficients for the gene set vectors, showing how related a cell is to a known pathway/biological process. r thus can be seen as the gene set activity scores for this cell. The set cover loss is designed to select uncorrelated pathways/processes to better annotate cells. (FC layers) Fully connected layers, (Bio-process) biological process.
To construct the gene set matrix D, which serves as an input to UNIFAN, we collected gene sets representing biological processes (including canonical pathways
and targets of specific regulators) from MSigDB (Subramanian et al. 2005) and Ernst et al. (2007), which resulted in a total of roughly 10,000 sets. We expect that only a small subset of these biological processes is active
for each single cell, and so, we use regularization to select active gene sets for each cell. First, we constrain r to be nonnegative by using ReLU for the output layer, which results in most values in r being assigned to zero. Next, we use a regularizer inspired by the classical set cover algorithm, which aims to find the
least number of sets that cover all elements (in our case, profiled genes of the cell). By using this regularizer, we aim
to find a small subset of nonoverlapping gene sets that can cover as many of genes as possible (Lu et al. 2008). For this, our regularizer optimizes the following function: 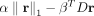 , where α and β are hyperparameters (for selecting values for hyperparameters in our model, see section Training UNIFAN and
Hyperparameter Selection). Using mean-squared error for the reconstruction loss, our overall loss function for a single cell
is
, where α and β are hyperparameters (for selecting values for hyperparameters in our model, see section Training UNIFAN and
Hyperparameter Selection). Using mean-squared error for the reconstruction loss, our overall loss function for a single cell
is
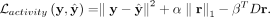
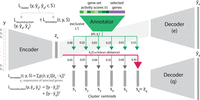
Clustering cells using gene set activity scores
To cluster cells using the inferred gene set activity scores, we use an autoencoder-based method. It is composed of two parts: an expression-based cluster assignment part (Fig. 7, “gray” parts) and the “annotator” part (Fig. 7, “green” parts), which uses the gene set activity scores discussed above as input.
Jointly clustering and annotating cells. The autoencoder contains two parts: the cluster assignment part (gray) uses a low-dimensional representation, ze, to assign a cell to clusters; the “annotator” (green) uses the learned gene set activity scores and selected genes’ expression to refine clustering and annotate clusters. Gene sets and genes selected as predictive by the annotator, in turn, provide useful annotations for each cell cluster. We set the number of clusters M as six in this figure for illustration purposes.
The cluster assignment part only uses the expression profile for each cell. It consists of an encoder and two decoders (Decoder
[e] and [q] in Fig. 7), modified based on Fortuin et al. (2019) and Van den Oord et al. (2017). For a single cell, we first use an encoder on the expression of genes in the cell y, resulting in a low-dimensional representation, ze, as shown in Figure 7. After initialization, we start with a guess of M clusters and cluster centroids. Among M cluster centroids S = {s1, s2, …, sM}, we identify the centroid closest to ze by first calculating the Euclidean distances between ze and all centroids and then transforming the distances using a t-distribution kernel 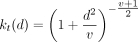 , following Li et al. (2020), Xie et al. (2016), and Van der Maaten and Hinton (2008). d stands for the distance, and v stands for the degrees of freedom, which is fixed at 10 for all experiments. We then take the closest centroid as the discrete
representation zq of the cell and assign the cell to the corresponding cluster. We obtain the reconstructed expression
, following Li et al. (2020), Xie et al. (2016), and Van der Maaten and Hinton (2008). d stands for the distance, and v stands for the degrees of freedom, which is fixed at 10 for all experiments. We then take the closest centroid as the discrete
representation zq of the cell and assign the cell to the corresponding cluster. We obtain the reconstructed expression 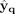 using decoder (q) which only takes zq as input, and so, all cells in the same cluster have the same reconstructed expression. We optimize the reconstruction error
using decoder (q) which only takes zq as input, and so, all cells in the same cluster have the same reconstructed expression. We optimize the reconstruction error
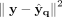 to find the best zq, cluster centroids S, and decoder (q), in a manner similar to finding the best cluster centroids in k-means clustering.
to find the best zq, cluster centroids S, and decoder (q), in a manner similar to finding the best cluster centroids in k-means clustering.
Because we assign cell clusters using k-means (i.e., discrete assignment), the encoder cannot be learned using backpropagation. To enable the iterative refinement
of model parameters using gradients, we follow Fortuin et al. (2019) by adding another decoder, decoder (e). Decoder (e) takes ze as input and outputs another reconstructed expression, 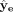 . By optimizing
. By optimizing 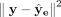 , we can now update ze and the encoder. The overall loss function for a single cell in the cluster assignment part is thus
, we can now update ze and the encoder. The overall loss function for a single cell in the cluster assignment part is thus 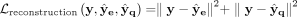 . All neural networks mentioned above are composed of fully connected layers.
. All neural networks mentioned above are composed of fully connected layers.
So far, we only discussed clustering using expression data. We next use the learned gene set activity scores for each cell
to refine cluster assignments as well as to annotate cell clusters. For this, we add an “annotator,” a logistic classifier,
to the network model. For each cell, the annotator uses the gene set activity scores r for that cell as input and outputs p(r), the probability of the cell being assigned to each cluster. We use the annotator's output to refine the cluster assignment
by adding 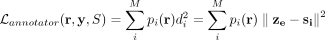 to the existing loss function. Because it uses both, the low-dimensional representation of the cell ze and the cluster centroids S, such loss encourages cells being assigned to clusters based on the probability specified by p(r). In other words, by using
to the existing loss function. Because it uses both, the low-dimensional representation of the cell ze and the cluster centroids S, such loss encourages cells being assigned to clusters based on the probability specified by p(r). In other words, by using 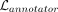 and the annotator, we are using prior knowledge about gene membership in key biological processes to guide the dimension
reduction and cluster assignment. Gene sets selected as predictive by the annotator, in turn, provide useful annotations for
each cell cluster.
and the annotator, we are using prior knowledge about gene membership in key biological processes to guide the dimension
reduction and cluster assignment. Gene sets selected as predictive by the annotator, in turn, provide useful annotations for
each cell cluster.
To allow the selection of marker genes for each cluster, we also tested the use of the UNIFAN with a subset of the most variable
genes selected using Seurat v3 (Stuart et al. 2019). Using such set the annotator loss becomes 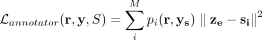 , where ys are the expression of the selected genes. The overall loss function for the cluster assignment part is thus
, where ys are the expression of the selected genes. The overall loss function for the cluster assignment part is thus
 where τ is a weighting hyperparameter.
where τ is a weighting hyperparameter.
The annotator is trained to optimize its own loss function. We use cross-entropy loss to train the annotator: 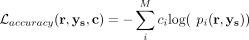 , where ci = 1(cell clustered to i). To select marker gene sets and genes specific to each cluster, we use the exclusive LASSO regularizer (Zhou et al. 2010) for the annotator. The regularizer takes the form of
, where ci = 1(cell clustered to i). To select marker gene sets and genes specific to each cluster, we use the exclusive LASSO regularizer (Zhou et al. 2010) for the annotator. The regularizer takes the form of 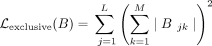 , where B are the parameters of the logistic classifier. Thus, the overall loss function for the annotator is
, where B are the parameters of the logistic classifier. Thus, the overall loss function for the annotator is
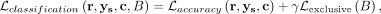 where γ is a weighting hyperparameter.
where γ is a weighting hyperparameter.
Training UNIFAN and hyperparameter selection
The loss functions described in the section above are defined for a single cell. During training, we take the mean of loss over all cells. We first train the autoencoder for the gene set activity scores and obtain the gene set activity scores for all cells. We then pretrain the encoder and the decoder (e) of the autoencoder for clustering on the expression data to obtain an initial low-dimensional representations of the cells. We then run Leiden clustering (Traag et al. 2019) on these representations to obtain a guess of the number of clusters M, the initial cluster assignment, and the cluster centroids S. Both the number of clusters M and cluster centroids S are refined later as part of the training. Specifically, clusters with no cell assigned to them are removed. The annotator is then pretrained using the inferred gene set activity scores and the selected genes, if available. We use the cluster assignment initialized as described above as the true labels to pretrain the annotator.
Finally, we train the annotator together with the cluster assignment part (the encoder and decoder [e] and decoder [q]). In
each epoch, the annotator is trained by using the clustering results as the true label for each cell. The output from the
annotator p(r) is in turn used to evaluate the annotator loss 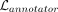 for the cluster assignment part. As described previously, the annotator is optimized using its own loss function, separately
from the cluster assignment part. For details in training, see Supplemental Methods.
for the cluster assignment part. As described previously, the annotator is optimized using its own loss function, separately
from the cluster assignment part. For details in training, see Supplemental Methods.
We use 32 dimensions for the low-dimensional representation ze of a cell. To select the values for hyperparameters, including the neural network configuration and the weighting hyperparameters for the loss functions, we conducted a grid search using the Tabula Muris data set and selected those hyperparameters values leading to the best performance over tissues. Unless specifically mentioned, the same set of values was applied to all data sets in all experiments. For details on how we select the values, see Supplemental Methods. As shown in Supplemental Figures S1 and S2, our method is robust to different choices of hyperparameter values. We also show in Supplemental Figures S3 and S4 that our method is robust to the resolution value used in Leiden clustering initialization and to the number of clustering epochs.
Performance evaluation and comparison to other methods
To evaluate the performance of UNIFAN and to compare it to prior methods, including Leiden clustering (Traag et al. 2019), Seurat v3 (Stuart et al. 2019), SIMLR (Wang et al. 2017), DESC (Li et al. 2020), SCCAF (Miao et al. 2020), MARS (Brbić et al. 2020), ItClust (Hu et al. 2020), and CellAssign (Zhang et al. 2019), we run each method on each data set 10 times with different initializations. For the Tabula Muris data, we run methods on each tissue separately. We use the ARI and NMI implemented in scikit-learn (Pedregosa et al. 2011) to compare clusters with ground truth annotations. Because calculating ARI for large data sets is time consuming, we use stratified random sampling when computing ARI for large data sets (more than 5 × 104 cells).
For the two (semi-) supervised methods MARS and ItClust, for each data set, we use random sampling stratified according to the true cell types to select 5% of the cells for training where the methods take also the true labels as input, following Wei and Zhang (2021). We leave the remaining 95% of the cells for testing and use these to calculate ARI and NMI. We resample the cells for training each time we run the two methods. For CellAssign, which takes known cell type markers as input, we use the marker gene sets from PanglaoDB (Franzén et al. 2019), as described by Zhang et al. (2019). Using all available cell type markers as input for a data set increases run time dramatically, and so, we only use cell type markers for the tissue that best matches the tissue of the data set (e.g., we use marker genes for “Immune system” for the data set “pbmc28k”). For details, see Supplemental Table S1.
For details on how we used prior methods, including hyperparameter settings for these methods and for information on the evaluation strategy, including how we compute enrichment P-values of the cell type marker sets in the highly weighted genes learned by the annotator, see Supplemental Methods. Also, for details on how we conducted the simulation experiments, see Supplemental Methods.
Software availability
UNIFAN is written in Python using PyTorch (Paszke et al. 2017). It is available under a MIT license at GitHub (https://github.com/doraadong/UNIFAN) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was partially supported by National Institutes of Health grants OT2OD026682, 1U54AG075931, and 1U24CA268108 to Z.B.-J., and by a National Science Foundation grant CBET-2134999 to Z.B.-J.
Author contributions: D.L., J.D., and Z.B.-J. developed the method; D.L. implemented the software and performed the analysis; and all authors analyzed the results and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276609.122.
- Received January 17, 2022.
- Accepted June 10, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








