
Coordination of zygotic genome activation entry and exit by H3K4me3 and H3K27me3 in porcine early embryos
- Guowei Bu1,2,6,
- Wei Zhu1,2,6,
- Xin Liu1,2,6,
- Jingjing Zhang1,2,
- Longtao Yu1,2,
- Kai Zhou1,2,
- Shangke Wang1,2,
- Zhekun Li1,2,
- Zhengang Fan1,2,
- Tingting Wang1,2,
- Taotao Hu1,2,
- Ruifeng Hu1,2,
- Zhiting Liu1,2,
- Tao Wang1,2,
- Linhui Wu1,2,
- Xia Zhang1,2,
- Shuhong Zhao2 and
- Yi-Liang Miao1,2,3,4,5
- 1Institute of Stem Cell and Regenerative Biology, College of Animal Science and Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, China;
- 2Key Laboratory of Agricultural Animal Genetics, Breeding and Reproduction (Huazhong Agricultural University), Ministry of Education, Wuhan 430070, China;
- 3Hubei Hongshan Laboratory, Wuhan 430070, China;
- 4Shenzhen Institute of Nutrition and Health, Huazhong Agricultural University, Shenzhen 518120, China;
- 5Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518120, China
-
↵6 These authors contributed equally to this work.
Abstract
Histone modifications are critical epigenetic indicators of chromatin state associated with gene expression. Although the reprogramming patterns of H3K4me3 and H3K27me3 have been elucidated in mouse and human preimplantation embryos, the relationship between these marks and zygotic genome activation (ZGA) remains poorly understood. By ultra-low-input native chromatin immunoprecipitation and sequencing, we profiled global H3K4me3 and H3K27me3 in porcine oocytes and in vitro fertilized (IVF) embryos. We observed sharp H3K4me3 peaks in promoters of ZGA genes in oocytes, and these peaks became broader after fertilization and reshaped into sharp peaks again during ZGA. By simultaneous depletion of H3K4me3 demethylase KDM5B and KDM5C, we determined that broad H3K4me3 domain maintenance impaired ZGA gene expression, suggesting its function to prevent premature ZGA entry. In contrast, broad H3K27me3 domains underwent global removal upon fertilization, followed by a re-establishment for H3K4me3/H3K27me3 bivalency in morulae. We also found that bivalent marks were deposited at promoters of ZGA genes, and inhibiting this deposition was correlated with the activation of ZGA genes. It suggests that promoter bivalency contributes to ZGA exit in porcine embryos. Moreover, we demonstrated that aberrant reprogramming of H3K4me3 and H3K27me3 triggered ZGA dysregulation in somatic cell nuclear transfer (SCNT) embryos, whereas H3K27me3-mediated imprinting did not exist in porcine IVF and SCNT embryos. Our findings highlight two previously unknown epigenetic reprogramming modes coordinated with ZGA in porcine preimplantation embryos. Finally, the similarities observed between porcine and human histone modification dynamics suggest that the porcine embryo may also be a useful model for human embryo research.
Histone modifications are critical epigenetic marks to instruct the chromatin state associated with temporal and spatial gene expression during maternal-to-zygotic transition and cell fate commitments (Dahl et al. 2016; Liu et al. 2016; Zhang et al. 2016, 2018; Zheng et al. 2016; Wang et al. 2018; Xia et al. 2019; Zhu et al. 2019). Compared to DNA methylation, which has similar down-trend dynamics in early embryos from different mammals (Wang et al. 2014; Zhu et al. 2018; Ivanova et al. 2020), histone modification dynamics exhibit great plasticity and species specificity during preimplantation development.
In mouse, paternal H3K4me3 and H3K27me3 are rapidly erased upon fertilization, whereas maternal H3K4me3 and H3K27me3 maintain their broad domains after fertilization, especially those in distal regions with poor DNA methylation, termed partially methylated domains (PMDs) (Dahl et al. 2016; Zhang et al. 2016; Zheng et al. 2016). In contrast, H3K4me3 accumulates as sharp peaks in human oocytes and embryos during zygotic genome activation (ZGA) but accumulates as broad domains before ZGA initiation (Xia et al. 2019). Through either KMT2B (H3K4me3 methyltransferase) depletion or KDM5B (H3K4me3 demethylase) overexpression (Andreu-Vieyra et al. 2010; Zhang et al. 2016), transcriptional quiescence in mouse oocytes can be relieved through H3K4me3 removal, which suggests a repressive feature for broad H3K4me3 modification. Given that broad H3K4me3 reshapes into sharp peaks during mouse and human ZGA (Dahl et al. 2016; Zhang et al. 2016; Xia et al. 2019), we surmise that broad H3K4me3 controls ZGA entry in early embryos. However, there is only a correlation analysis between broad H3K4me3 and ZGA in human embryos (Xia et al. 2019); no direct experimental evidence has been provided to support our hypothesis.
In addition, maternal H3K27me3, which is another repressive mark, is inherited at all preimplantation stages in mouse (Zheng et al. 2016) and controls gene imprinting in a DNA methylation-independent manner (Inoue et al. 2017a; Santini et al. 2021). Unlike that in mouse, H3K27me3 is globally removed during human ZGA (Xia et al. 2019), suggesting a possible influence of H3K27me3 on ZGA initiation. In zebrafish, the parental epigenome of H3K4me3 and H3K27me3 are extensively erased before ZGA, and re-established as the zygotic epigenome at promoters after ZGA (Zhang et al. 2018; Xia et al. 2019; Zhu et al. 2019). These current studies suggest that histone modification dynamics during ZGA are highly divergent among species. Thus, it is necessary to investigate the epigenetic reprogramming specificities that are orchestrated with ZGA in different vertebrates.
Pig (Sus scrofa domesticus) is widely regarded as an important agricultural resource and alternative model for biomedical studies. Compared with rodent models, pig shares more similarities in anatomy, physiology, and genetics with human beings. By combining genome-editing and somatic cell nuclear transfer (SCNT) technologies, an increasing number of pig models for human organ donors and disease research have been constructed (Yan et al. 2018; Yue et al. 2021). More importantly, porcine and human embryos possess similar preimplantation features, such as consistent timings for ZGA (four- to eight-cell stages) and blastocyst formation (6–7 d after fertilization) (Xue et al. 2013; Yan et al. 2013; Kong et al. 2020). In contrast, mouse embryos show different timings for ZGA (two-cell stage) and blastocyst formation (3.5 d after fertilization) (Xue et al. 2013). These observations indicate that pig is a good proxy animal model for human embryology study. Moreover, the establishment of porcine embryonic stem cells (ESCs) and pig cloning also hold great potential in agricultural breeding and regenerative medicine. However, the efficiencies for deriving porcine ESCs from blastocysts (∼30%) (Gao et al. 2019) and producing full-term cloned piglets (∼3%) (Ruan et al. 2018; Yu et al. 2018) are low. This limitation can be attributed to the unknown epigenetic information during porcine embryogenesis, which inspires us to profile histone modification landscapes in porcine early embryos.
Here, by using ultra-low-input native chromatin immunoprecipitation and sequencing (ULI-NChIP-seq) (Brind'Amour et al. 2015), we present high-resolution maps of H3K4me3 and H3K27me3 for porcine oocytes and early embryos. Aiming to understand the relationships between ZGA and histone modifications, we focus on the dynamics of H3K4me3 and H3K27me3 at promoters of ZGA genes in porcine early embryos and reveal the epigenomic features that safeguard ZGA entry and exit. Additionally, by comparative analysis, we further aim to expand the epigenetic reprogramming modes coordinated with ZGA among different species and provide valuable resources for the applications of porcine embryo technologies.
Results
Profiling of H3K4me3 and H3K27me3 in porcine oocytes and early embryos
To illuminate the dynamic changes of H3K4me3 and H3K27me3 during porcine preimplantation development, we performed ULI-NChIP-seq in porcine germinal vesicle (GV) and metaphase II (MII) oocytes, and in vitro fertilized (IVF) embryos at pre-ZGA (zygotes [1C] and two-cell embryos [2C]), peri-ZGA (four-cell [4C] and eight-cell [8C] embryos) (Cao et al. 2014) and post-ZGA stages (morulae [Mo] and blastocysts [Bl]) (Fig. 1A). The strong correlation between replicates indicated the good reproducibility of these data sets (Supplemental Fig. S1A; Supplemental Table S1).
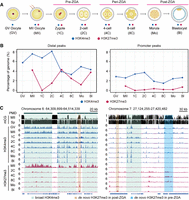
Profiling of H3K4me3 and H3K27me3 in porcine oocytes and early embryos. (A) Experimental design of the developmental stages at which samples were collected for H3K4me3 and H3K27me3 ULI-NChIP-seq analyses. Samples are porcine germinal vesicle (GV) and metaphase II (MII) oocytes, and in vitro fertilized (IVF) embryos at pre-ZGA (zygotes [1C] and two-cell embryos [2C]), peri-ZGA (four-cell [4C] and eight-cell [8C] embryos), and post-ZGA stages (morulae [Mo] and blastocysts [Bl]). (B) Line plots showing the percentage of genomic regions accumulated by H3K4me3 and H3K27me3 in distal and promoter regions. (C) Genome browser snapshots showing the dynamics of H3K4me3 and H3K27me3 in porcine oocytes and early embryos, and DNA methylation levels (5-methylcytosines in CpG [mCG]) in porcine oocyte and spermatozoa. Genomic regions containing oocyte partially methylated domains (PMDs, left panel) and gene promoters (right panel) are shown.
First, immunofluorescence analysis revealed that strong H3K4me3 signal in MII oocytes and pre-ZGA embryos was reduced in peri-ZGA embryos and was recovered in post-ZGA embryos (Supplemental Fig. S2). Comparably, the genomic coverage and width of H3K4me3 peaks reached the lowest level during ZGA (Fig. 1B; Supplemental Fig. S1B). Meanwhile, oocyte H3K4me3 enrichment was decreased upon fertilization but turned to an increase at promoters after ZGA (Supplemental Fig. S1B). More importantly, both broad and sharp forms of H3K4me3 were individually detected at PMDs and promoters in porcine oocytes, and these broad H3K4me3 domains were reshaped into sharp peaks during ZGA (Fig. 1C; Supplemental Fig. S3A,B). A previous study has detected the broad H3K4me3 domains in mouse oocyte PMDs, which are also reshaped during ZGA (Supplemental Fig. S3C,D; Zhang et al. 2016). Therefore, porcine oocytes and embryos possessed H3K4me3 dynamics in oocyte PMDs similar to those in mice.
Next, the dynamic changes of H3K27me3 coverage and peak width indicated that there were two rounds of H3K27me3 removal and establishment in porcine embryos (Fig. 1B; Supplemental Fig. S1B). In line with the immunostaining results (Supplemental Fig. S2), H3K27me3 became nearly absent in porcine zygotes and eight-cell embryos and recovered in two- to four-cell embryos and post-ZGA embryos. In the genome browser view (Fig. 1C), broad H3K27me3 domains were observed in oocyte PMDs and were significantly removed after fertilization. Moreover, H3K27me3 enrichment in pre-ZGA embryos was not colocalized with those peaks established in post-ZGA embryos (Fig. 1C), and we would dissect these two rounds of H3K27me3 reprogramming in the following results. Because H3K27me3 exhibited a low-complexity feature in zygotes (Supplemental Fig. S1C) in accordance with its weak immunostaining signal (Supplemental Fig. S2), we excluded this sample in the following analysis.
Broad H3K4me3 establishment may prevent ZGA initiation in porcine pre-ZGA embryos
In mouse oocytes, broad H3K4me3 domains are already established at gene promoters (Zhang et al. 2016). In contrast, sharp H3K4me3 peaks were observed at promoters in porcine oocytes (Fig. 1C). We thus focused on promoter H3K4me3 enrichment in porcine early embryos. Specifically, 20,566 protein-coding genes were categorized based on their H3K4me3 patterns around transcription start sites (TSS ± 5 kb) in porcine GV oocytes (Fig. 2A; Supplemental Table S2), which were broad H3K4me3 peaks (Cluster 1), sharp H3K4me3 peaks (Cluster 2), and promoters lacking H3K4me3 (Cluster 3). Notably, we found that most H3K4me3 peaks (6936/9320) in Cluster 2 (Cluster 2a) became broader in pre-ZGA embryos (Fig. 2A,E), whereas Cluster 1, Cluster 3, and the rest of Cluster 2 (Cluster 2b) maintained their H3K4me3 patterns after fertilization (Fig. 2A). Moreover, like the broad H3K4me3 domains in oocyte PMDs, broad promoter H3K4me3 peaks in Cluster 2a became sharp during ZGA (Fig. 2A,E).
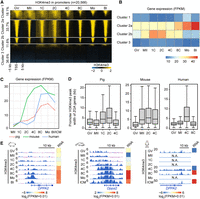
De novo H3K4me3 establishment at ZGA gene promoters upon fertilization. (A) Heat maps showing the H3K4me3 dynamics at all gene promoters (transcriptional start site [TSS] ± 5 kb) in porcine oocytes and early embryos. k-means analysis (k = 3) was performed to classify these promoters into three clusters based on their H3K4me3 intensities in GV oocytes, in which Cluster 2 could be further divided into two subclusters based on their H3K4me3 intensities in zygotes. Gene promoters in each cluster were ordered descending by their H3K4me3 intensities in GV oocytes. (B) Heat map showing the transcriptional levels (average fragments per kb per million reads [FPKM]) of genes in each cluster in porcine oocytes and early embryos. (C) Line plots showing the transcriptional levels (FPKM) of porcine, mouse, and human zygotic genome activation (ZGA) genes in their oocytes and early embryos. (D) Box plots showing the peak width of promoter H3K4me3 for porcine, mouse, and human ZGA genes in oocytes and early embryos. (E) Genome browser snapshots showing the H3K4me3 dynamics and transcriptional levels (FPKM) of orthologous ZGA genes, Dppa2/DPPA2, in porcine, mouse, and human oocytes and early embryos. Broad H3K4me3 was established at ZGA gene promoters in porcine and human pre-ZGA embryos (pig: 1C-2C; human: 4C) and reshaped into sharp peaks in their peri-ZGA embryos (pig: 4C-8C; human: 8C).
A previous study has identified the repressive feature of broad H3K4me3 peaks in mouse oocytes (Zhang et al. 2016). We thus wondered if broad H3K4me3 peaks established in porcine pre-ZGA embryos possesses the same feature. Gene Ontology (GO) analysis indicated that Cluster 1 was enriched for embryonic organ development genes; Cluster 2a was enriched for RNA processing genes; Cluster 2b was not enriched for annotated functions; and Cluster 3 was enriched for immune genes (Supplemental Fig. S3E; Supplemental Table S3). Additionally, the transcriptional patterns of each gene cluster showed that Cluster 2a maintained a steady expression from oocytes to pre-ZGA embryos and was evidently up-regulated during ZGA; Cluster 2b was mainly activated in pre-ZGA embryos; and Cluster 1 and Cluster 3 maintained transcriptionally quiescent states in early embryos (Fig. 2B). These findings suggest that genes in Cluster 2a are associated with ZGA in porcine embryos. We then summarized a list for porcine ZGA genes, as they were highly expressed at the peri-ZGA stage (Fig. 2C; Supplemental Table S4), and found that Cluster 2a contained the highest proportion of ZGA genes when compared to other gene clusters (Supplemental Fig. S3F). We next asked if the establishment of broad H3K4me3 domains was related to the loss of DNA methylation during early development. We found that DNA methylation (Ivanova et al. 2020) maintained comparably low levels at gene promoters in porcine sperm, oocytes, and embryos, whether for all genes or for only genes in Cluster 2a and Cluster 2b (Supplemental Fig. S3G). It suggests that, at least in promoter regions, broad H3K4me3 establishment in Cluster 2a does not accompany DNA methylation loss.
To verify if this broad H3K4me3 at ZGA gene promoters was unique to pig, we also summarized mouse and human ZGA gene lists (Fig. 2C; Supplemental Table S4) and compared the peak width of H3K4me3 at ZGA gene promoters in oocytes and early embryos (Fig. 2D). As exemplified by an orthologous gene, Dppa2/DPPA2 (Fig. 2E), mouse ZGA genes maintained broad H3K4me3 domains at promoters from MII oocytes to two-cell embryos (mouse ZGA stage) (Fig. 2D). In contrast, ZGA gene promoters occupied sharp H3K4me3 peaks in porcine and human oocytes. After fertilization, these peaks became broader in porcine and human (four-cell) pre-ZGA embryos and became sharp again in porcine and human (eight-cell) peri-ZGA embryos (Fig. 2D). These observations imply that the establishment of broad H3K4me3 domains upon fertilization potentially affects ZGA in porcine and human early embryos.
H3K4 demethylase depletion disrupts porcine ZGA and embryonic development
In mice, it has been reported that overexpression of the H3K4 demethylase gene, lysine (K)-specific demethylase 5B (Kdm5b), results in global H3K4me3 removal in oocytes (Zhang et al. 2016), whereas knockdown of Kdm5b leads to the increase of promoter H3K4me3 width in morulae (Dahl et al. 2016; Liu et al. 2016). However, the biological functions of broad H3K4me3 transition during ZGA remain elusive. To figure it out, we first detected the transcriptional patterns of KDM5A, KDM5B, and KDM5C during porcine preimplantation development, as well as their orthologous genes in human and mouse embryos (Supplemental Fig. S4A). We found that KDM5B exhibited conserved activation during ZGA in all three species, wheres Kdm5a and KDM5C were activated during mouse and porcine ZGA, respectively. In contrast, KDM5A is a maternally expressed gene in human and pig. Therefore, we knocked down KDM5B and KDM5C simultaneously (siKDM5B/C) in porcine embryos to examine if broad H3K4me3 transition and ZGA initiation would be affected (Fig. 3A). With 78% and 99% knockdown efficiencies of KDM5B and KDM5C, respectively (Supplemental Fig. S4B), we detected a significant enhancement of H3K4me3 intensity in siKDM5B/C four-cell embryos (Supplemental Fig. S4C,D). Meanwhile, siKDM5B/C embryos could not reach the four-cell, morula, and blastocyst stages efficiently (Fig. 3B,C).
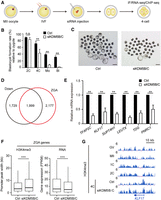
Knockdown of KDM5B and KDM5C disrupts porcine ZGA and embryonic development. (A) Experimental design of KDM5B and KDM5C knockdown in porcine IVF embryos. (B) Bar plot showing the blastocyst formation rates in KDM5B and KDM5C knockdown (siKDM5B/C) and control (Ctrl) groups when compared to the harvested zygotes. Error bars represent the SD. (n = 3, 100 oocytes per repeat.) (**) P < 0.01 by two-tailed t-test, (n.s.) no significance. (C) Microscopy images showing the blastocyst formation in siKDM5B/C and control groups. Scale bar, 200 μm. (D) Venn diagram showing that over half (1999/3728) of down-regulated genes in siKDM5B/C four-cell embryos are overlapped with porcine ZGA genes. (E) Bar plot showing the relative mRNA expression of six porcine ZGA genes in siKDM5B/C and control embryos at the four-cell stage. Error bars represent the SD. (n = 3, 30 embryos per repeat). (**) P < 0.01 by two-tailed t-test. (F) Box plots showing the width of H3K4me3 peaks at ZGA gene promoters and the transcriptional levels (FPKM) of ZGA genes in siKDM5B/C and control groups. (***) P < 0.001 by Wilcoxon test. (G) A genome browser snapshot showing the H3K4me3 enrichment at KLF17 location in siKDM5B/C and control embryos at the four-cell stage.
To determine if this developmental arrest of siKDM5B/C embryos was associated with ZGA failure, we next performed RNA sequencing (RNA-seq) to check the expression status of ZGA genes and identified 3728 down-regulated (siKDM5B/C vs. Ctrl, fold change [FC] > 2, false discovery rate [FDR] < 0.05) differentially expressed genes (DEGs) in siKDM5B/C four-cell embryos (Fig. 3D; Supplemental Table S5). Over half (53.6%) of down-regulated DEGs belong to ZGA genes, and these genes also account for nearly half (47.9%) of all ZGA genes (Fig. 3D), indicating that KDM5B/C depletion showed an extensive influence for ZGA initiation. Furthermore, six down-regulated ZGA genes in RNA-seq data were also confirmed by quantitative PCR (qPCR) (Fig. 3E; Supplemental Fig. S4E,F). Additionally, we overlapped the DEGs between cleavage control embryos with the DEGs between knockdown embryos and found few DEGs derived from the comparison between KDM5B/C knockdown blastocysts versus control blastocysts were overlapped with those from the comparison between cleavage embryos versus control blastocysts (Supplemental Fig. S4G). This analysis ruled out a possible developmental delay resulting from the knockdown experiment. To explore the status of H3K4me3 enrichment after KDM5B/C depletion, we performed H3K4me3 ChIP-seq in siKDM5B/C four-cell embryos and found that broad H3K4me3 domains were maintained in PMDs (Supplemental Fig. S4H,I). More importantly, when compared to the control group, the width of H3K4me3 at ZGA gene promoters was significantly increased, whereas the expression levels of ZGA genes were reduced in siKDM5B/C four-cell embryos (Fig. 3F,G). In sum, our results show that H3K4 demethylase depletion leads to the transition failure from broad H3K4me3 domains into sharp peaks, which partially impairs ZGA gene expression in porcine peri-ZGA embryos. It suggests that broad H3K4me3 controls ZGA entry during porcine preimplantation development.
Distal H3K27me3 deposition in porcine 2- to 4-cell embryos does not correlate with ZGA initiation
In mouse oocytes, distal H3K27me3 peaks were largely deposited in PMDs and maintained at all preimplantation stages (Supplemental Fig. S5A,B; Zheng et al. 2016). In porcine early embryos, distal H3K27me3 exhibited two rounds of removal and establishment (Fig. 1B). We first examined H3K27me3 dynamics in porcine oocyte PMDs and found that it was largely removed upon fertilization, followed by a re-establishment in post-ZGA embryos (Fig. 4A,B). However, this H3K27me3 dynamics could not explain the transient increase of distal H3K27me3 coverage at the four-cell stage (Fig. 1B). We thus identified 73,251 distal H3K27me3 peaks that were de novo established in two- to four-cell embryos and found that they were not observed in either oocytes or embryos of other stages (Fig. 4C,D). These findings indicate that the dynamic of distal H3K27me3 is the superposition of two independent resetting processes: one is H3K27me3 removal upon fertilization, followed by H3K27me3 re-establishment in post-ZGA embryos (Fig. 4A,B); the other is the transient H3K27me3 deposition in two- to four-cell embryos (Fig. 4C,D).
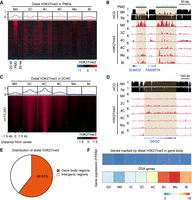
Distal H3K27me3 resetting during porcine preimplantation development. (A) Heat maps and aggregate profile plots showing the dynamics of distal H3K27me3 in porcine oocyte PMDs. PMDs were ordered descending by their H3K27me3 intensities in MII oocytes. (B) A genome browser snapshot showing the removal of distal H3K27me3 in porcine oocyte PMDs upon fertilization, and the H3K27me3 re-establishment in porcine morulae and blastocysts. DNA methylation levels (mCG) in oocyte and spermatozoa are also shown. (C) Heat maps and aggregate profile plots showing the dynamics of distal H3K27me3 established in porcine two- to four-cell embryos. (D) A genome browser snapshot showing the establishment of distal H3K27me3 in porcine two- to four-cell embryos, which was nearly not observed in either oocytes or embryos of other stages. DNA methylation levels (mCG) in oocyte and spermatozoa are also shown. (E) Pie chart showing the genomic distribution of distal H3K27me3 established at the two- to four-cell stages. (F) Heat maps showing the transcriptional levels (FPKM) of porcine ZGA genes and genes covered by H3K27me3 on their gene body regions in porcine oocytes and early embryos.
Given that preventing the H3K4me3 transition influenced porcine ZGA, we thus wondered if transient H3K27me3 deposition could also influence ZGA. We first characterized these distal H3K27me3 peaks through genomic feature annotations and found that about 60% of these peaks were located in gene body regions (Fig. 4E). The transcriptional patterns showed that these H3K27me3-marked genes (Supplemental Table S2) maintained relatively low expression levels from oocytes to blastocysts when compared to ZGA genes (Fig. 4F). GO analysis indicated that these H3K27me3-marked genes were enriched for neuron development (Supplemental Fig. S5C; Supplemental Table S3). To test the biological functions of de novo H3K27me3 deposition in porcine two- to four-cell embryos, we treated pre-ZGA embryos with GSK126, an inhibitor of H3K27me3 writer EZH2 (Supplemental Fig. S5D; Liu et al. 2021). Although GSK126 treatment indeed reduced H3K27me3 intensity in four-cell embryos (Supplemental Fig. S5E,F), we could not find any differentially expressed genes through RNA-seq (Supplemental Fig. S5G). It suggests that, at least, distal H3K27me3 deposition in two- to four-cell embryos is achieved by EZH2, not influencing ZGA gene expression. Additionally, we found that distal H3K27me3 deposition in two- to four-cell embryos was accompanied by another repressive histone mark, H3K9me3 (Supplemental Fig. S5I,J), and the functions of this colocalization still require further investigation.
H3K4me3/H3K27me3 bivalency facilitates ZGA exit in porcine post-ZGA embryos
In mice, although H3K4me3 and H3K27me3 were both present in oocyte PMDs, as few as 2% of PMDs were co-occupied by these two modifications to form bivalency (Fig. 5A; Zheng et al. 2016). In porcine MII oocytes, H3K4me3 and H3K27me3 were also present in oocyte PMDs, and we found that 88% of PMDs were marked as bivalent domains (Fig. 5A). By calculating the Pearson correlation coefficients, the average intensities of H3K4me3 and H3K27me3 were positively correlated in porcine oocyte PMDs but were negatively correlated in mice (Fig. 5B). Genome browser views showed that orthologous genes SLC22A1, SLC22A2, and MAS1 were located in porcine bivalent PMDs, and in mouse PMDs showed no bivalency (Fig. 5C). Moreover, we found that Hox clusters were conservatively enriched in bivalent PMDs in mouse, human, and porcine oocytes (Supplemental Fig. S6). However, these Hox loci underwent total removal of H3K27me3 in porcine two- to eight-cell embryos and in human eight-cell embryos as well. In contrast, H3K27me3 at Hox loci is inherited in mouse early embryos from oocyte genomes.
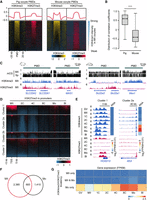
H3K4me3/H3K27me3 bivalency in porcine oocytes and early embryos. (A) Heat maps and aggregate profile plots showing the H3K4me3 and H3K27me3 enrichment in porcine and mouse MII oocytes at their oocyte PMDs. PMDs were ordered descending by their H3K4me3 intensities in MII oocytes. (B) Box plot showing the distribution of Pearson's coefficients between H3K4me3 and H3K27me3 intensities in porcine and mouse MII oocytes at their oocyte PMDs. (***) P < 0.001 by Wilcoxon test. (C) Genome browser snapshots showing the orthologous genomic regions co-occupied H3K4me3 and H3K27me3 in porcine oocyte PMDs, but did not co-occupy these modifications in mouse oocyte PMDs. DNA methylation levels (mCG) in oocyte and spermatozoa are also shown. (D) Heat maps showing the H3K27me3 dynamics at all gene promoters (TSS ± 5 kb) in porcine oocytes and early embryos. Gene clusters were defined in Figure 2A. Gene promoters in each cluster were ordered descending by their H3K27me3 intensities in MII oocytes. (E) Genome browser snapshots showing the H3K4me3 and H3K27me3 dynamics and transcriptional levels (FPKM) of candidate genes for Cluster 1 and Cluster 2a in porcine oocytes and early embryos. (F) Venn diagram showing the oocyte-specific (MII), morula-specific (Mo), and overlapping bivalent gene numbers between porcine MII oocytes and morulae. (G) Heat map showing the transcriptional levels (FPKM) of oocyte-specific (MII only), morula-specific (Mo only), and overlapping (MII & Mo) bivalent genes in porcine oocytes and early embryos.
For promoter H3K27me3, we used the same gene classification in Figure 2A. In porcine MII oocytes, Cluster 1 was partially deposited by broad H3K27me3, whereas the other three clusters showed weak H3K27me3 signal (Fig. 5D,E). After fertilization, promoter H3K27me3 was removed in Cluster 1 and Cluster 2, followed by a re-establishment at the morula stage in Cluster 1 and Cluster 2a but not in Cluster 2b; Cluster 3 partially maintained weak H3K27me3 signal in two- to four-cell embryos and showed nearly no enrichment after the eight-cell stage (Fig. 5D,E). Given that promoter H3K4me3 persisted in its occupation from oocytes to blastocysts in Cluster 1 and Cluster 2 (Fig. 2A), we speculated that H3K4me3/H3K27me3 bivalency was established at promoters of these gene clusters during porcine post-ZGA development, which followed the same preference for accumulating at promoters with higher CpG density as in mouse ICM and ESCs (Supplemental Fig. S7).
To further examine the functions of H3K4me3/H3K27me3 bivalency, we identified 3078 bivalent genes in MII oocytes and 2103 bivalent genes in morulae and found that ∼33.0% (693/2103) of bivalent genes in morulae were shared with oocyte genes (Fig. 5F; Supplemental Table S2). Additionally, morula-specific bivalent genes were activated during ZGA, followed by down-regulation in blastocysts (Fig. 5G). In contrast, oocyte-specific and shared bivalent genes maintained relative transcriptional quiescence in early embryos (Fig. 5G). GO analysis showed that morula-specific bivalent genes were enriched for ribosome biogenesis, whereas both oocyte-specific and shared bivalent genes were enriched for embryonic organ development (Supplemental Fig. S5H; Supplemental Table S3). These results suggest that morula-specific bivalent genes are likely ZGA genes; thus, we hypothesized that promoter H3K27me3 established in post-ZGA embryos may associate with ZGA repression. Indeed, we detected a significant co-occupation of H3K4me3 and H3K27me3 at ZGA gene promoters after porcine ZGA (Supplemental Fig. S8A–C).
In support of our hypothesis, we treated porcine embryos with GSK126 inhibitor to prevent H3K27me3 establishment after ZGA (Supplemental Fig. S9A). GSK126 treatment not only reduced the blastocyst formation rate from 44.0% to 13.7% (Supplemental Fig. S9B,C), the immunostaining signal of H3K27me3 was also reduced in GSK126-treated blastocysts (Supplemental Fig. S9D,E). More importantly, through RNA-seq, we found a significant up-regulation of bivalent genes and ZGA genes in these GSK126-treated blastocysts (Supplemental Fig. S9F,G). For instance, the expression level of ZGA gene AK4 was repressed along withH3K27me3 establishment in post-ZGA embryos (Fig. 5E), whereas GSK126 treatment derepressed its expression (Supplemental Fig. S9H). Furthermore, the transcriptional increment of ZGA genes was also confirmed by qPCR (Supplemental Fig. S9I). These observations support that the H3K4me3/H3K27me3 bivalency established after ZGA is related to ZGA gene repression and, namely, facilitates totipotency exit in post-ZGA embryos.
To verify if this relationship between promoter bivalency and ZGA was unique to pig, we examined the dynamic changes of H3K4me3/H3K27me3 marks at mouse and human ZGA gene promoters. Similar to porcine embryos, promoter H3K4me3 persisted in its accumulation from oocytes to inner cell mass (ICM) cells, the cells derived from mouse and human blastocysts (Supplemental Fig. S8A). Meanwhile, sharp H3K27me3 peaks were established at human ZGA gene promoters in ICM cells, but they were maintained as a weak signal at all pre-implantation stages in mice (Supplemental Fig. S8B). It suggests a common bivalent function to influence ZGA exit in human and pig, as exemplified by the genome browser views for the ZGA genes ZSCAN4, Dppa4, and SPRTN in Supplemental Figure S8C.
H3K4me3 and H3K27me3 are epigenetic barriers to ZGA in porcine SCNT embryos
Recent studies have demonstrated that aberrant epigenetic modifications derived from donor cells hinder SCNT embryonic development (Matoba and Zhang 2018). In this study, we also performed ULI-NChIP-seq using SCNT 4-cell embryos and blastocysts, and compared their H3K4me3 and H3K27me3 landscapes with IVF embryos. For gene promoters, excessive H3K4me3 intensities were found in Cluster 1, whereas H3K4me3 was defectively enriched in Cluster 2a and Cluster 2b in SCNT four-cell embryos (Fig. 6A). Meanwhile, excessive H3K27me3 intensities were found at the promoters of Cluster 1, Cluster 2a, and Cluster 2b in SCNT four-cell embryos, whereas Cluster 3 exhibited weak H3K4me3 and H3K27me3 signal like those in IVF four-cell embryos (Fig. 6A,E). In contrast to promoter H3K4me3, no evident difference of H3K4me3 modification was observed in oocyte PMDs between IVF and SCNT embryos, either in four-cell embryos or blastocysts (Fig. 6B,E). However, SCNT embryos showed abnormally high levels of H3K27me3 intensity in oocyte PMDs during ZGA (Fig. 6B,E). Additionally, aberrant H3K4me3 and H3K27me3 modifications at promoters and oocyte PMDs in SCNT four-cell embryos could be relieved when these embryos reached the blastocyst stage (Fig. 6A,B,E). We thus further compared the transcriptional profiles between IVF and SCNT four-cell embryos and found that there were 78 down-regulated (IVF vs. SCNT, FC > 2, FDR < 0.05) and 569 up-regulated (SCNT vs. IVF, FC > 2, FDR < 0.05) DEGs in SCNT embryos (Supplemental Table S5). Nearly 70% of down-regulated DEGs were ZGA genes (Fig. 6C), and nearly 80% of down-regulated DEGs in PMDs with abnormal H3K27me3 deposition were ZGA genes as well (Fig. 6D). These data indicate that aberrant H3K4me3 and H3K27me3 modifications may impair ZGA initiation in SCNT embryos.

Aberrant H3K4me3 and H3K27me3 modifications in porcine SCNT embryos. (A) Heat maps and aggregate profile plots showing the H3K4me3 and H3K27me3 enrichment at gene promoters in somatic cell nuclear transfer (SCNT) embryos at the four-cell and blastocyst stages, as well as the difference between IVF and SCNT embryos (Δ[SCNT-IVF]: SCNT enrichment values minus IVF enrichment values). Gene clusters were defined in Figure 2A. (B) Heat maps and aggregate profile plots showing the H3K4me3 and H3K27me3 enrichment in oocyte PMDs of SCNT embryos at the four-cell and blastocyst stages, as well as the difference between IVF and SCNT embryos (Δ[SCNT-IVF]). (C) Bar plot showing the fraction of differentially expressed genes (DEGs) between IVF and SCNT four-cell embryos that are porcine ZGA genes. SCNT four-cell embryos contained 78 down-regulated (IVF vs. SCNT, FC > 2, FDR < 0.05) and 569 up-regulated (SCNT vs. IVF, FC > 2, FDR < 0.05). (D) Bar plot showing the fraction of DEGs located in H3K27me3-enhanced PMDs that are porcine ZGA genes. (E) Genome browser snapshots showing the H3K4me3 and H3K27me3 enriched at candidate genes in IVF and SCNT embryos at the four-cell and blastocyst stages. DNA methylation levels (mCG) in oocyte and spermatozoa are also shown.
To assess the potential sources of aberrant histone modifications in SCNT 4-cell embryos, we compared H3K4me3 and H3K27me3 profiles between SCNT four-cell embryos and porcine fetal fibroblasts (PFFs), which are the donor cells for SCNT embryos (Supplemental Fig. S10A,B). We found that PFFs exhibited almost no broad H3K4me3 enrichment in PMDs, but these broad H3K4me3 domains could be successfully established in PMDs in SCNT four-cell embryos (Supplemental Fig. S10B,C, region iii). In contrast, H3K4me3 was excessively enriched at promoters of Cluster 1 after donor cell nuclear transfer, which exhibited sharp peaks on PFF genomes (Supplemental Fig. S10A,C, regions i, ii). Moreover, aberrantly high H3K27me3 enrichment at Cluster 1 promoters and PMDs (Supplemental Fig. S10A–C, region iv) in SCNT four-cell embryos was probably due to the reprogramming failure of H3K27me3 modification on PFF genomes. Nevertheless, the reasons behind this reprogramming failure of SCNT need further investigation.
Furthermore, H3K27me3 modification in oocytes was regarded as DNA methylation-independent imprinting in mice, which controls X Chromosome inactivation (Inoue et al. 2017b) and maternal gene expression (Supplemental Fig. S11A; Inoue et al. 2017a). It was also reported that loss of maternal H3K27me3 imprinting led to developmental defects of mouse SCNT embryos (Matoba et al. 2018), and knocking out H3K27me3-dependent imprinting genes could improve mouse cloning efficiency (Inoue et al. 2010; Wang et al. 2020). In contrast, perhaps due to massive H3K27me3 removal after fertilization (Supplemental Fig. S2), both porcine IVF and SCNT embryos at the same stage showed no enrichment difference of H3K4me3 and H3K27me3 for mouse H3K27me3-dependent imprinting genes, such as Xist, Jade1, Gab1, Sfmbt2, and Smoc1 (Supplemental Fig. S11B). The transcriptional levels of these imprinting genes also showed no significant difference between porcine IVF and SCNT embryos, either in four-cell embryos or blastocysts (Supplemental Fig. S11C; Supplemental Table S5). In contrast, when compared to IVF embryos, mouse SCNT embryos exhibited higher transcriptional levels of H3K27me3-dependent imprinting genes, as well as lower H3K27me3 enrichment on these genes (Inoue et al. 2010, 2017b; Matoba et al. 2018; Wang et al. 2020). Taken together, these observations suggest that H3K27me3-dependent imprinting does not exist in porcine early embryos.
Discussion
Embryogenesis is triggered at fertilization, when a terminally differentiated sperm and an oocyte are fused into a totipotent zygote. A series of important biological events then proceed, like ZGA and cell lineage commitment (Xu et al. 2021). Because histone modifications play a supportive role in gene expression and maintaining cell identity, proper reprogramming of histone modifications from parental genomes to the zygotic genomes is essential for pre-implantation development. Benefiting from the advancement of ChIP methodology made in recent years, histone modification profiling with few cells is feasible for embryo research. In this study, genomic landscapes of H3K4me3 and H3K27me3 in porcine oocytes and early embryos were uncovered by ULI-NChIP-seq, as previously applied in mouse embryos (Liu et al. 2016; Wang et al. 2018). We thus analyzed the reprogramming features of H3K4me3 and H3K27me3 in early porcine development and further elucidated both conserved and divergent concomitant relationships between epigenetic reprogramming and ZGA initiation among mouse, human, and pig (Fig. 7).
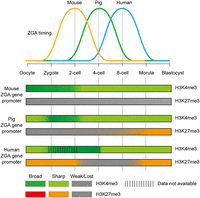
Conserved and divergent coordination between ZGA and histone modifications in porcine, mouse, and human early embryos. Broad H3K4me3 domains are already established at ZGA gene promoters in mouse oocytes, which are inherited upon fertilization and are reshaped into sharp peaks during mouse ZGA (two-cell stage). In contrast, porcine and human oocytes contain sharp H3K4me3 peaks at their ZGA gene promoters. Notably, de novo H3K4me3 establishment is observed at these promoters upon fertilization, resulting in H3K4me3 peaks spreading to form broad domains in pre-ZGA embryos (pig: one- to two-cell stages; human: four-cell stage). Nevertheless, these broad domains are reshaped into sharp form again during porcine and human ZGA (pig: four-cell stage; human: eight-cell stage), and H3K4me3 modification maintains sharp form after ZGA among the three species. For H3K27me3 modification, ZGA gene promoters exhibit weak H3K27me3 signal in mouse oocytes and fertilized embryos at all pre-implantation stages. These gene promoters also exhibit weak H3K27me3 signal in porcine oocytes and fertilized embryos at the two- to eight-cell stages but exhibit strong H3K27me3 signal in human oocytes and pre-ZGA embryos. More importantly, de novo H3K27me3 establishment is observed at ZGA gene promoters after porcine and human ZGA, resulting in promoter bivalency establishment (H3K4me3 and H3K27me3 colocalization) in their post-ZGA embryos.
In mice, broad H3K4me3 domains have been established at both promoters and distal regions in GV and MII oocytes, which maintain in fertilized embryos before ZGA (Dahl et al. 2016; Zhang et al. 2016). However, H3K4me3 occurs as sharp peaks in human GV oocytes, and such broad H3K4me3 peaks only appear in pre-ZGA embryos (Xia et al. 2019). In this study, we determined the existence of both broad and sharp H3K4me3 peaks in porcine GV and MII oocytes. Similar to mouse (Dahl et al. 2016; Zhang et al. 2016), broad H3K4me3 enrichment at PMDs and gene promoters (Cluster 1) was maintained from porcine oocytes to pre-ZGA embryos. For the genes activated immediately after fertilization (Cluster 2b), sharp H3K4me3 peaks maintained at their promoters from oocytes to embryos. Meanwhile, promoter H3K4me3 was reshaped into broad domains before porcine ZGA in Cluster 2a, like the broad H3K4me3 pattern in human pre-ZGA embryos (Xia et al. 2019). Given that sharp H3K4me3 peaks have been reported to be associated with gene activation in mouse embryos (Dahl et al. 2016; Zhang et al. 2016), here, we proposed that genes in Cluster 2b could be regarded as minor ZGA genes in pig, which were immediately activated upon fertilization. In contrast, genes in Cluster 2a were largely major ZGA genes, which were progressively activated in four- to eight-cell embryos in pig along with broad H3K4me3 removal.
More importantly, the H3K27me3 reprogramming pattern in porcine embryonic development shares many similarities to human, but not to mice. First, although broad H3K27me3 was found in porcine oocyte PMDs as previously described in mice (Zheng et al. 2016), its colocalization with broad H3K4me3 appeared to be a unique feature in porcine oocytes, because these two modifications were not colocalized in mouse oocyte PMDs (Zheng et al. 2016). Second, H3K27me3 modification underwent global removal in porcine and human embryos (Xia et al. 2019), rather than being maintained as in mouse embryos until postimplantation (Zheng et al. 2016). Given that maternal H3K27me3 persistence in mouse embryos regulates allelic gene expression (Inoue et al. 2017a), this H3K27me3-mediated imprinting appears to be absent in porcine and human early embryos (Xia et al. 2019). Third, along with the re-establishment of H3K27me3 during lineage commitment, H3K4me3/H3K27me3 bivalent marks not only were deposited at developmental gene promoters but also were deposited at ZGA gene promoters in porcine and human embryos. In contrast, bivalency accumulation at ZGA gene promoters is not observed in mice. Fourth, we checked the transcriptional patterns of H3K27me3 erasers (KDM6A, KDM6B) and writers (EED, EZH2, SUZ12) in mouse, human, and porcine embryos (Supplemental Fig. S12A). Consistent with mouse and human, KDM6A was highly expressed in porcine oocytes, suggesting a conserved role in H3K27me3 demethylation upon mammalian fertilization. Moreover, all of the core H3K27me3 writer components, namely polycomb repressive complex 2 (PRC2) genes, were activated during porcine and human ZGA, possibly resulting in bivalency establishment in porcine and human post-ZGA embryos. In contrast, mouse Eed and Ezh2 were highly expressed in MII oocytes and contributed to H3K27me3 persistence in mouse embryos (Meng et al. 2020). These transcriptional patterns of PRC2 complex genes support the similar H3K27me3 reprogramming during porcine and human embryogenesis. Additionally, we also inspected the PRC1 complex in porcine and mouse embryos (Supplemental Fig. S12B), which has been reported to mediate gene repression in mouse ESCs (Blackledge and Klose 2021). We found that the core components of PRC1 (RNF2 and PCGF family) exhibited steady expression levels at each stage, indicating that PRC1 is not involved in ZGA entry and exit during porcine and mouse pre-implantation development. Taking H3K4me3 dynamics together, these findings indicate that H3K4me3 and H3K27me3 dynamics in porcine embryos show more divergence from mice and more conservation with human. These similarities highlight the potential advantages of using porcine embryos as a substitute animal model for human embryo research.
A recent study (Wang et al. 2022) trained an algorithm to successfully predict histone modifications based on nascent transcription data and found that blocking transcription decreased H3K4me3 and H3K27me3 enrichment from chromatin. These findings suggest that histone modifications serve a supportive, rather than a directly regulatory, role in gene expression. In our study, we delineated the coordinated changes between ZGA initiation and histone modifications and determined that both inhibiting the broad H3K4me3 transition in four-cell embryos and inhibiting H3K27me3 establishment in blastocysts affected ZGA gene expression. However, it is still a limitation to further determine whether histone modifications play a supportive or regulatory role for porcine ZGA. The algorithm provided by Wang et al. (2022) may be suitable only for somatic cells, not for early embryonic development for the following reasons. First, although bivalent genes could be identified based on active transcription alone, this algorithm failed to identify sharp H3K27me3 peaks in stem cells, germ cells, and certain progenitor cells. Second, blocking transcription only decreased H3K27me3 enrichment near EZH2 binding sites but not H3K27me3 accumulation over broad regions in cell line. Third, full-grown oocytes are transcriptionally quiescent (De La Fuente et al. 2004). Thus, which factor maintains broad H3K4me3 and H3K27me3 enrichment when transcription does not extensively initiate during this developmental stage? Fourth, this algorithm cannot explain the repressive function of H3K27me3 to be a DNA methylation-independent imprinting mechanism in mouse embryos. Taken together, we speculate that histone modifications and ZGA initiation are highly interdependent in early embryonic development.
Finally, by comparing porcine IVF and SCNT embryos, we found that anomalous histone modifications were largely deposited in SCNT four-cell embryos, whereas SCNT blastocysts shared similar epigenetic landscapes with IVF embryos. These observations highlight that precise resetting of histone modifications during ZGA is a prerequisite for successful embryonic development. Indeed, H3K27me3 has been confirmed to be a reprogramming barrier in porcine SCNT embryos (Xie et al. 2016). By treating with GSK126, blastocyst formation rates of porcine SCNT embryos are evidently improved in vitro (Xie et al. 2016; Liu et al. 2021). However, it is refractory to balance the hypermethylation and hypomethylation of H3K4me3 in SCNT embryos, which individually occur at developmental and ZGA gene promoters at the same time. Recent advances in SCNT technology have unveiled several aberrant histone modifications on heterochromatin regions and the gene regulatory regions related to ZGA, imprinting, and donor cell identity (Matoba and Zhang 2018). Here, for the first time, we reported that excessive H3K27me3 was deposited in oocyte PMDs during ZGA. In fact, oocyte PMDs belong to distal genomic regions, which may possess inaccessible, gene-poor, and nontranscribed features (Xia and Xie 2020). Thus, PMDs may also contain repressive epigenetic marks like H3K9me3 and DNA methylation to resist SCNT-mediated reprogramming. Efforts are expected to be made to expand the characteristics and divergence of PMDs between maternal genomes and donor genomes. Such studies will provide useful information for improving mammalian cloning efficiency.
In summary, we have generated genome-wide maps of H3K4me3 and H3K27me3 in porcine oocytes and early embryos and uncovered their conservative and distinct reprogramming patterns among mouse, human, and pig. During porcine and human pre-implantation development, our study also determined the coordination of ZGA entry and exit by broad H3K4me3 and H3K4me3/H3K27me3 bivalency, thus providing novel insights into the associations between epigenetic reprogramming and ZGA initiation in mammalian early embryos. The underlying molecular mechanisms of reprogramming diversities among different mammals will require future investigation.
Methods
Animal ethics statements
The experiments were conducted following the guidance of the Animal Care and Use Committee of Huazhong Agriculture University. Porcine ovaries were obtained from prepubertal gilts at a local slaughterhouse and transported to the laboratory within 2 h in 0.9% (w/v) saline at 35°C.
Oocyte collection and in vitro maturation
Follicular fluid with cumulus oocyte complexes (COCs) was aspirated from antral follicles (3–6 mm). COCs with uniform oocyte cytoplasm and several layers of cumulus cells were then picked and rinsed three times in in vitro maturation (IVM) medium (Lai and Prather 2003; Liu et al. 2020). About 30 COCs were transferred into 96-well microplates containing 150 μL IVM medium covered with mineral oil and cultured for 44 h at 38.5°C in a 5% CO2 incubator (Thermo Fisher Scientific). Germinal vesicle oocytes were harvested by removing cumulus cells from fresh COCs in 0.1% (w/v) hyaluronidase for a 5-min vortex. Metaphase II (MII) oocytes with the first polar body extrusion were harvested from matured COCs in the same way.
In vitro fertilization
Fresh semen was washed once with modified Tris-buffered medium (mTBM) by 1900 rpm centrifugation for 4 min. After removing the supernatant, spermatozoa pellets were resuspended in mTBM medium containing 2 mg/mL bovine serum albumin (BSA) and 2 mM caffeine, and diluted to 1 × 106 sperms/mL. A 50-μL sperm suspension and 30 MII oocytes were then added to a 50-μL drop of mTBM medium containing BSA and caffeine. After 6 h postinsemination (hpi) at 38.5°C in a 5% CO2 incubator, 50 putative zygotes were washed and cultured in 500 μL porcine zygote medium-3 (PZM-3) (Lai and Prather 2003; Liu et al. 2020) covered with mineral oil under the same conditions.
Somatic cell nuclear transfer
Somatic cell nuclear transfer was performed as previously described (Lai and Prather 2003; Liu et al. 2020). Briefly, donor cells were porcine fetal fibroblast cells (2–5 passages) cultured in DMEM medium containing 0.5% fetal bovine serum for 72 h. The first polar body and adjacent cytoplasm of MII oocytes were aspirated with a 20-μm glass pipette, and a single donor cell was then injected into the perivitelline space. Reconstructed embryos were transferred into a chamber with electrodes 1 mm apart containing activating medium. Activation was induced with two direct current pulses (1.2 kV/cm, 30 μs) on an Electro-Cell Manipulator ECM2100 (BTX). After activation (0 hpi), 50 embryos were cultured in 500 μL PZM-3 medium as described for IVF.
Embryo collection
Porcine IVF and SCNT embryos at each stage were collected at defined time points: 18 hpi (zygote), 28 hpi (two-cell), 48 hpi (four-cell), 72–76 hpi (eight-cell), 120 hpi (morula), and 150 hpi (blastocyst). For RNA-seq and ULI-NChIP-seq, oocytes or embryos were placed in 0.25% (w/v) pronase in PBS for 30 sec to digest the zona pellucida. Samples were then transferred into 0.2% (w/v) BSA in PBS for 3 min at 38.5°C, and the remaining zona pellucida was removed by pipetting with a 200-μm glass pipette. Zona-free oocytes or embryos were further washed three times in 0.2% (w/v) BSA in PBS to remove pronase before performing downstream experiments.
siRNA microinjection
Three siRNA oligos targeting porcine KDM5B and KDM5C (Supplemental Table S6) were synthesized by GenePharma Co., Ltd. About 10 pL of siRNA mixture (33 μM for each oligo) were injected into zygotes (6 hpi) by FemtoJet 4i (Eppendorf). Development rates of two- to four-cell embryos, morulae, and blastocysts were summarized at 28 hpi, 48 hpi, 120 hpi, and 150 hpi, respectively, and shown as mean ± standard deviation (SD). Four-cell embryos were subjected to immunostaining, RNA-seq, and ULI-NChIP-seq experiments as described below.
GSK126 treatment
To inhibit the establishment of H3K27me3 in pre-ZGA embryos, zygotes were cultured in PZM-3 medium supplemented with GSK126 (Selleck) at a final concentration of 5 μM. Embryos developed to four-cell stages were collected. To inhibit the establishment of H3K27me3 in post-ZGA embryos, four-cell embryos cultured in normal PZM-3 medium were transferred to PZM-3 medium supplemented with GSK126 at a final concentration of 5 μM. Embryos developed to blastocysts were collected. All collected embryos were subjected to immunostaining and RNA-seq experiments as described below.
Immunofluorescence
Oocytes and embryos were fixed with 4% (w/v) paraformaldehyde for 40 min at room temperature, followed by three washes with DPBS containing 0.05% (w/v) polyvinyl alcohol and 0.01% (v/v) Tween-20. Samples were then permeabilized with 0.5% (v/v) Triton X-100 in DPBS for 60 min, blocked with 3% (w/v) BSA in DPBS for 2 h at room temperature, and incubated with primary antibodies for H3K4me3 (Abcam ab8580; 1:500 dilution) or H3K27me3 (Millipore 07-449; 1:500 dilution) overnight at 4°C. After three washes, samples were incubated with Dylight 549 goat anti-rabbit IgG (Abbkine A23320; 1:500 dilution) for 1 h at room temperature. After another three washes, samples were mounted on glass slides with anti-fade medium containing 4,6-diamidino-2-phenylindole (DAPI) and examined with a laser scanning confocal microscope (Zeiss LSM 510). The average fluorescence intensity per unit area within the region of interest was determined by ImageJ software (v1.48). Images were converted into eight-bit format to obtain the mean gray value of region of nuclei in each sample by auto threshold adjustment with default setting.
Quantitative PCR
Total RNA isolation from 30 four-cell embryos or five blastocysts was performed by using a RNAprep Pure Micro kit (TIANGEN DP420). cDNAs were synthesized by a HiScript II Q RT SuperMix kit plus gDNA wiper (Vazyme R223-01), and quantified by ChamQ Universal SYBR qPCR Master Mix (Vazyme Q321-02) on a CFX96 Real-Time PCR Detection System (Bio-Rad). The results from noninjected or nontreated control embryos were set as 1 and were normalized to the internal control gene GAPDH. All primer sequences are listed in Supplemental Table S6. Data are shown as the fold change = 2−ΔΔCt mean ± SD.
ULI-NChIP-seq
ULI-NChIP was performed as previously described (Brind'Amour et al. 2015). For each immunoprecipitation reaction, approximately 200 cells of oocytes or embryos were added into nuclear isolation buffer and MNase master mix to digest chromatin for 7 min at 25°C. Samples were then incubated with antibody-bead complexes in complete immunoprecipitation buffer overnight at 4°C. These complexes were formed by pre-incubating 11 μL Dynabeads Protein G (Invitrogen) with 1 μg antibody of H3K4me3 (Abcam ab8580), H3K27me3 (Millipore 07-449), or H3K9me3 (Abcam ab9989) in complete immunoprecipitation buffer for 6 h at 4°C. After three washes with low-salt wash buffer and high-salt wash buffer, chromatin was dissolved in elution buffer for 2 h at 65°C. DNA was then extracted by phenol-chloroform-isoamyl alcohol (25:24:1) and used for library generation by using a KAPA HyperPrep kit (Roche KK8504) following the manufacturer's instructions. Paired-end 150-bp sequencing was performed on an Illumina HiSeq X-Ten system.
RNA-seq library preparation
Total RNA isolation from 30 IVF embryos was performed by using a PicoPure RNA Isolation kit (Applied Biosystems). cDNA was synthesized and amplified by a SMARTer Pico PCR cDNA Synthesis kit (Clontech) and Advantage 2 PCR kit (Clontech). cDNAs were then fragmented into 300 bp using Covaris M220 sonicator (Covaris) and were end-repaired, adaptor-ligated, and amplified by a VAHTS Universal V6 RNA-seq Library Prep kit (Vazyme NR604-01) according to the manufacturer's instructions. Paired-end 150-bp sequencing was performed on the Illumina HiSeq X-Ten system.
ChIP-seq data processing
After removing sequencing adaptors from ULI-NChIP-seq data by Trimmomtic (v0.39) (Bolger et al. 2014), sequencing data were aligned to porcine genome (SusScr11) with Bowtie 2 (v2.4.1) using parameters “‐‐no-mixed ‐‐no-discordant” (Langmead and Salzberg 2012). All unmapped reads, low mapping quality (MAPQ < 30) alignments, and PCR duplicates were then removed. To make ChIP-seq intensities comparable across different stages with different genomic coverages, we first divided porcine genome into 100-bp bins and normalized the read counts by computing the numbers of fragments per kilobase of bin per million of mapped reads (FPKM). Then, FPKM values across the whole genome were further Z-score-normalized, which was previously performed in ChIP-seq data for mouse (Zhang et al. 2016) and human (Xia et al. 2019). To visualize the ChIP-seq signal, files containing Z-score-normalized FPKM values on 100-bp bins were loaded into the UCSC Genome Browser. Average FPKM values were then summarized within each 2-kb window across the whole genome, and the Pearson's correlations between replicates were calculated with cor function in the R environment (v4.0.3) (R Core Team 2020).
Public histone modification ChIP-seq data for mouse and human oocytes and embryos were downloaded from NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database under accession number GSE72784 (Dahl et al. 2016), GSE73952 (Liu et al. 2016), and GSE124718 (Xia et al. 2019). Same analyses were then performed as described for porcine ChIP-seq data.
ChIP-seq peak calling
Two ChIP-seq replicates were pooled for peak calling using MACS2 (v2.2.5) (Zhang et al. 2008) with the parameters “‐‐broad -f BAMPE -g 2e9 ‐‐nolambda ‐‐keep-dup all”. Noisy peaks (Z-score-transformed FPKM values below the bottom 1% of total peaks) were removed for further analyses. Called peaks were annotated to the nearest genes within 3 kb around transcription start sites (TSSs) using the ChIPseeker package (v1.26.2) (Yu et al. 2015) in the R environment.
RNA-seq data processing
Trimmed RNA-seq reads were aligned to the porcine genome (SusScr11) with Subjunc (v2.0.1) (Liao et al. 2013) using default parameters. The featureCount program (v2.0.1) (Liao et al. 2014) was used to quantify gene expression according to the RefSeq genes annotation (NCBI annotation release 106). Gene expression differences between two samples were evaluated with the DESeq2 package (v1.22.1) (Love et al. 2014) in the R environment, and raw P values were further corrected by false discovery rate (FDR). Finally, genes with fold change > 2 and FDR < 0.05 were determined as differentially expressed genes. ZGA genes in porcine embryos were defined as the FPKM ratios of 4C/2C, 4C/MII, 8C/4C, 8C/2C, 8C/MII > 2, when the FPKM values of 4C and 8C > 5, respectively; ZGA genes in mouse embryos were defined as the FPKM ratios of late 2C/early 2C and late 2C/MII > 2, when the FPKM values of late 2C > 5; ZGA genes in human embryos were defined as the FPKM ratios of 8C/4C and 8C/MII > 2, when the FPKM values of 8C > 5.
Analyzed RNA-seq results for mouse and human oocytes and embryos were downloaded from NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database under accession numbers GSE71434 (Zhang et al. 2016) and GSE36552 (Yan et al. 2013). For human, gene expression values from a single cell were averaged for the same developmental stage.
Gene Ontology analysis
Gene Ontology enrichment for gene lists generated from ChIP-seq and RNA-seq analyses was performed with clusterProfiler (v3.18.1) (Yu et al. 2012) package in the R environment. GO enrichment was restricted to biological process.
DNA methylation analysis
DNA methylation sequencing data of porcine and mouse oocytes and sperm were downloaded from the GEO database under accession number GSE143850 (Ivanova et al. 2020) and GSE56697 (Wang et al. 2014). Adapters and low quality bases were removed by Trimmomatic and were analyzed by MethPipe (v4.1.1) (Song et al. 2013). PMDs in porcine and mouse MII oocytes were identified with the “pmd” program in the MethPipe package.
Statistical analysis
All experiments were performed at least three times, except for ULI-NChIP-seq in two replicates. All middle lines in the box-whisker plots indicate the median, the edges indicate the 25th/75th percentiles and the whiskers indicate the 2.5th/97.5th percentiles. P-values were determined by Wilcoxon test or two-tailed Student's t-test. Significant differences were shown with *, **, *** for indicating P < 0.05, 0.01, 0.001, respectively. * refers to Supplemental Figures S5F and S9I; ** refers to Figure 3B,E and Supplemental Figures S4B,D and S9B,E,I; *** refers to Figures 3F and 5B and Supplemental Figures S4F and S9F,G,H.
Data access
ChIP-seq data generated in this study have been submitted to Genome Sequence Archive (GSA; https://bigd.big.ac.cn/) under accession number CRA003606 and are also available at the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE163709. RNA-seq data generated in this study have been submitted to GSA under accession number CRA004237.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. 31941006, 31970822, 31902161), Huazhong Agricultural University-Agricultural Genomics Institute at Shenzhen Cooperation Fund (no. SZYJY2021009), Key Research and Development Program of Hubei Province (no. 2021BBA221), Fundamental Research Funds for the Central Universities (no. 2662022DKPY001), Major Project of Hubei Hongshan Laboratory (no. 2021hszd003), and the National Key R&D Program of China, Stem Cell and Translational Research (2016YFA0100203). We thank Dr. Heide Schatten (University of Missouri–Columbia) for language editing and Prof. Carmen Williams (National Institutes of Health/National Institute of Environmental Health Sciences), Prof. Yawei Gao (Tongji University), and Prof. Wei Xie (Tsinghua University) for providing advice for this study. The computations in this paper were run on the bioinformatics computing platform of the National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University.
Author contributions: Y.-L.M. conceived and supervised this study. G.B., W.Z., X.L., L.Y., K.Z., S.W., Z.K.L., Z.F., T.T.W., T.H., R.H., Z.T.L., and T.W. performed the experiments. W.Z., J.Z., and L.W. analyzed the sequencing data. W.Z., X.L., and G.B. wrote the manuscript. Y.-L.M., X.Z., S.Z., G.B., W.Z., X.L., and J.Z. revised the manuscript. All authors have read and approved the final manuscript for publication.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276207.121.
- Received September 16, 2021.
- Accepted July 19, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








