
Somatic retrotransposition in the developing rhesus macaque brain
- Victor Billon1,2,11,
- Francisco J. Sanchez-Luque3,4,5,11,
- Jay Rasmussen1,11,
- Gabriela O. Bodea1,6,11,
- Daniel J. Gerhardt6,
- Patricia Gerdes6,
- Seth W. Cheetham6,
- Stephanie N. Schauer6,
- Prabha Ajjikuttira1,
- Thomas J. Meyer7,
- Cora E. Layman8,
- Kimberly A. Nevonen8,
- Natasha Jansz6,
- Jose L. Garcia-Perez3,4,
- Sandra R. Richardson6,
- Adam D. Ewing6,
- Lucia Carbone7,8,9,10 and
- Geoffrey J. Faulkner1,6
- 1Queensland Brain Institute, University of Queensland, St. Lucia, Queensland 4067, Australia;
- 2Biology Department, École Normale Supérieure Paris-Saclay, 91190 Gif-sur-Yvette, France;
- 3GENYO. Pfizer-University of Granada-Andalusian Government Centre for Genomics and Oncological Research, PTS Granada 18016, Spain;
- 4MRC Human Genetics Unit, Institute of Genetics and Cancer, University of Edinburgh, Western General Hospital, Edinburgh EH4 2XU, United Kingdom;
- 5Institute of Parasitology and Biomedicine “Lopez-Neyra”–Spanish National Research Council, PTS Granada 18016, Spain;
- 6Mater Research Institute–University of Queensland, Woolloongabba, Queensland 4102, Australia;
- 7Division of Genetics, Oregon National Primate Research Center, Beaverton, Oregon 97006, USA;
- 8Department of Medicine, Knight Cardiovascular Institute, Oregon Health and Science University, Portland, Oregon 97239, USA;
- 9Department of Molecular and Medical Genetics, Oregon Health and Science University, Portland, Oregon 97239, USA;
- 10Department of Medical Informatics and Clinical Epidemiology, Oregon Health and Science University, Portland, Oregon 97239, USA
-
↵11 These authors contributed equally to this work.
Abstract
The retrotransposon LINE-1 (L1) is central to the recent evolutionary history of the human genome and continues to drive genetic diversity and germline pathogenesis. However, the spatiotemporal extent and biological significance of somatic L1 activity are poorly defined and are virtually unexplored in other primates. From a single L1 lineage active at the divergence of apes and Old World monkeys, successive L1 subfamilies have emerged in each descendant primate germline. As revealed by case studies, the presently active human L1 subfamily can also mobilize during embryonic and brain development in vivo. It is unknown whether nonhuman primate L1s can similarly generate somatic insertions in the brain. Here we applied approximately 40× single-cell whole-genome sequencing (scWGS), as well as retrotransposon capture sequencing (RC-seq), to 20 hippocampal neurons from two rhesus macaques (Macaca mulatta). In one animal, we detected and PCR-validated a somatic L1 insertion that generated target site duplications, carried a short 5′ transduction, and was present in ∼7% of hippocampal neurons but absent from cerebellum and nonbrain tissues. The corresponding donor L1 allele was exceptionally mobile in vitro and was embedded in PRDM4, a gene expressed throughout development and in neural stem cells. Nanopore long-read methylome and RNA-seq transcriptome analyses indicated young retrotransposon subfamily activation in the early embryo, followed by repression in adult tissues. These data highlight endogenous macaque L1 retrotransposition potential, provide prototypical evidence of L1-mediated somatic mosaicism in a nonhuman primate, and allude to L1 mobility in the brain over the past 30 million years of human evolution.
Neurons can acquire mutations during brain development and later life. Human single-cell and bulk tissue genomic analyses have revealed neuronal somatic copy number variants (McConnell et al. 2013; Chronister et al. 2019), single-nucleotide variants (Chen et al. 2017; Abascal et al. 2021; Xing et al. 2021), and transposable element (TE) insertions (Evrony et al. 2015; Erwin et al. 2016; Sanchez-Luque et al. 2019). The estimated frequency of each of these mutations depends on the detection method and brain region assayed (McConnell et al. 2017; Chronister et al. 2019; Abascal et al. 2021; Xing et al. 2021). The spatial distribution of a given somatic variant is influenced by its type and genomic location, the cell in which it arose, and, potentially, postmutational selection. As a result, a mutation may be present throughout the brain or in only one cell and, in either case, contribute to a wider mosaic genome landscape. Although somatic variants can drive neuronal pathogenesis, such as MTOR mutations leading to focal cortical dysplasia (King et al. 2015; Lim et al. 2015; Nakashima et al. 2015), it is unclear whether they influence normal brain function or phenotype (Faulkner and Garcia-Perez 2017; McConnell et al. 2017).
The retrotransposon long interspersed element 1 (LINE-1, or L1) constitutes 17% of the human genome (International Human Genome Sequencing Consortium 2001). Despite their prevalence, only a small subset of L1 copies are mobile in the germline or in neuronal lineage cells (Brouha et al. 2003; Muotri et al. 2005; Coufal et al. 2009; Beck et al. 2010; Evrony et al. 2015; Erwin et al. 2016; Faulkner and Garcia-Perez 2017; Macia et al. 2017; Sanchez-Luque et al. 2019). To retrotranspose, L1 generates a bicistronic mRNA encoding two proteins (ORF1p and ORF2p) that, among several key activities, catalyze genomic DNA nicking and reverse transcription of the L1 mRNA (Feng et al. 1996; Moran et al. 1996; Kazazian and Moran 2017; Scott and Devine 2017). New L1 insertions typically carry retrotransposition hallmarks, including target site duplications (TSDs) and a long 3′ poly(A) tract, and integrate at a degenerate 5′-TTTT/AA-3′ motif (Moran et al. 1996; Jurka 1997; Doucet et al. 2015). Numerous factors restrict L1 retrotransposition in somatic cells (Goodier 2016), including transcriptional silencing complexes recruited by DNA methylation (Thayer et al. 1993; Muotri et al. 2010; Castro-Diaz et al. 2014; de la Rica et al. 2016; Scott et al. 2016; Robbez-Masson et al. 2018; Deniz et al. 2019; Greenberg and Bourc'his 2019; Ewing et al. 2020). Embryonic L1 insertions can nonetheless arise before gastrulation (van den Hurk et al. 2007; Kano et al. 2009; Richardson et al. 2017; Feusier et al. 2019) and before the complete establishment of L1 methylation (Macia et al. 2017; Sanchez-Luque et al. 2019). Single-cell whole-genome sequencing (scWGS) of postmitotic neurons (Evrony et al. 2015; Sanchez-Luque et al. 2019) has uncovered somatic retrotransposition events traced to unusual donor (source) L1 loci that evade methylation in mature tissues (Faulkner and Billon 2018; Sanchez-Luque et al. 2019; Ewing et al. 2020). Despite relevant studies of endogenous retrotransposition in the mouse and fly (Li et al. 2013; Hazen et al. 2016; Richardson et al. 2017; Keegan et al. 2021; Siudeja et al. 2021), scWGS has to date been applied only to human neurons (Evrony et al. 2015; Sanchez-Luque et al. 2019), leaving the capacity of L1 to retrotranspose in nonhuman primate neuronal lineages an important open question. Starting with Haig Kazazian's first report of germline L1 mutagenesis (Kazazian et al. 1988), case studies have largely underpinned efforts to define the spatiotemporal extent of L1 mobility (Miki et al. 1992; Brouha et al. 2002; van den Hurk et al. 2007; de Boer et al. 2014; Evrony et al. 2015; Scott et al. 2016; Sanchez-Luque et al. 2019), founded on the principle that one robustly verified insertion is sufficient to show L1 retrotransposition can occur in a given context.
Because of its neuroanatomical, cognitive, social, and genetic similarities with humans, the rhesus macaque (Macaca mulatta) is a cornerstone model organism for biomedical and neuroscience research (Phillips et al. 2014; Feng et al. 2020). L1 copies comprise 16% of the macaque reference genome, including about 44,000 annotated as rhesus-specific L1 (L1RS) sequences (Tang and Liang 2019; Warren et al. 2020). L1RS2 is the youngest and most active macaque L1 subfamily and incorporates 2235 full-length (>6-kbp) elements, far more than the equivalent human-specific L1 (L1HS) subfamily (329 full-length copies) (Warren et al. 2020). Several Alu short interspersed element (SINE) subfamilies, presumably retrotransposed in trans by L1 proteins (Dewannieux et al. 2003), are mobile in the macaque germline, as may be endogenous retroviruses (ERVs) (Han et al. 2007; Liu et al. 2009; Tang and Liang 2019; Warren et al. 2020). It is, however, unknown whether TEs retrotranspose in macaque neuronal lineage cells. Here, we exploited an updated macaque reference genome assembly, which greatly improved TE annotations (Warren et al. 2020), to profile retrotransposition in individual hippocampal neurons and, more broadly, survey macaque TE transcription and repression in vivo.
Results
Genomic analyses of macaque retrotransposition
To explore TE mobilization in the macaque brain, we isolated RBFOX3+ (also known as NeuN+) neuronal nuclei from the postmortem hippocampi of two animals (ON22212 and ON22213; both 7-yr-old male adults) and performed multiple displacement amplification (MDA) on each nucleus (Sanchez-Luque et al. 2019). We applied Illumina scWGS (average 40× genome-wide depth) to 20 neurons (seven from ON22212 and 13 from ON22213) that passed quality control, and performed Illumina WGS (average 41× depth) on matched bulk liver tissues (Fig. 1A; Supplemental Table S1). To further increase coverage of TE–genome junctions, we synthesized a new retrotransposon capture sequencing (RC-seq) (Baillie et al. 2011) probe pool targeting young macaque TE subfamilies (Han et al. 2007; Warren et al. 2020). Conceptually, this design most closely resembled one we previously developed in the mouse (Richardson et al. 2017), and involved 80 densely overlapping probes targeting the 5′ and 3′ termini of eight TE subfamily consensus sequences (Supplemental Table S1). Barcoded Illumina libraries generated from each of the 20 MDA-amplified neuronal nuclei were hybridized to this probe pool, eluted, and then subjected to paired-end 2 × 150 mer sequencing to maximize the number of RC-seq reads spanning a TE–genome junction (Fig. 1A).
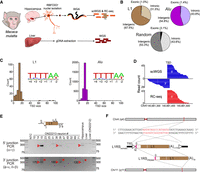
Characterization of germline and somatic macaque TE insertions. (A) Genomics experimental design. Individual hippocampal neuron (RBFOX3+) nuclei from two rhesus macaques (ON22212 and ON22213) were subjected to whole-genome amplification (WGA), followed by Illumina scWGS and RC-seq, to identify somatic TE insertions. Bulk liver DNA was analyzed with Illumina WGS to discriminate germline and somatic variants. (B) Percentages of exonic, intronic, and intergenic nonreference L1 (top left) and Alu (top right) insertions. Genomic features were annotated according to RefSeq coordinates, with the underlying proportions of each feature (random expectation) shown at bottom. (C) Target site duplication (TSD) size distributions for nonreference L1 (left) and Alu (right) insertions, as annotated by TEBreak. Inset sequence logos (Crooks et al. 2004) display the observed integration site nucleotide composition for each TE family. These resembled the L1 endonuclease motif. (D) A somatic L1RS2 insertion (L1RSsomatic) was detected on Chromosome 4 of animal ON22213 hippocampal neuron #15. Reads spanning the 5′ or 3′ L1–genome junctions of this event are shown, as is the corresponding TSD. (E) PCR validation of L1RSsomatic. Primer (symbols α, ε, δ, γ, β, and Φ) positions relative to the L1 insertion are indicated in the schematic provided at top. The 5′ L1–genome junction was amplified by combining primers α and γ, whereas nested PCR (ε + Φ then δ + β) was used to amplify the 3′ L1–genome junction. Reaction input in each case consisted of nontemplate control (NTC), 13 ON22213 hippocampal neurons analyzed with scWGS and RC-seq, bulk ON22213 hippocampus and liver DNA, and bulk ON22212 liver. Red arrowheads and crosses indicate amplicons confirmed as on-target and off-target, respectively, by capillary sequencing. Numbers next to confirmed 3′ L1–genome junction bands indicate the L1 poly(A) tract length for that amplicon. (F) Complete sequence characterization of L1RSsomatic. TSD nucleotides are highlighted in red. The intergenic L1 was full length (L1RS2 subfamily consensus start position 0), carried a 4-bp 5′ transduction (pink rectangle) with an untemplated guanine (underlined G), and was followed by a long, pure 3′ poly(A) tract. The transduction indicated a putative donor L1 intronic to the PRDM4 gene on Chromosome 11 (L1RSPRDM4).
Using the TEBreak computational pipeline (Carreira et al. 2016), we identified 194 L1, 3348 Alu and no ERV nonreference insertions in the two liver WGS data sets (Supplemental Table S2). The Alu insertion count and frequency relative to L1 were each higher than what would be predicted based on prior human analyses, as expected (Tang and Liang 2019; Ewing et al. 2020). Of the 3542 total events, 2781 were present in one or the other animal, but not both. One hundred eighty-nine of 194 (97.4%) nonreference L1s were annotated as belonging to the L1RS2 subfamily, and 44/194 (22.7%) were 5′ inverted (Ostertag and Kazazian 2001). L1 and Alu insertions were depleted from annotated protein-coding exons (Fig. 1B). Intronic L1 insertions were significantly (P < 0.04, binomial test) less abundant (61/194, 31.4%) than random expectation (43.6%) (Fig. 1B), and disproportionately few (23/61, 37.7%) of these were sense-oriented to their host gene. Given modest genome-wide L1 integration site preferences, which mainly reflect the underlying distribution of AT-rich sequences, these patterns were likely dominated by postintegration selection and are concordant with prior results obtained by human analyses (Smit 1999; Ewing and Kazazian 2010; Attig et al. 2018; Flasch et al. 2019; Sultana et al. 2019; Smits et al. 2021). Consistent with L1-mediated retrotransposition in humans and other mammals (Moran et al. 1996; Jurka 1997; Richardson et al. 2017; Tang and Liang 2019; Ewing et al. 2020; Smits et al. 2021), the L1 and Alu insertions generated TSDs with a median length of 15 bp (Fig. 1C) and integrated at a motif strongly resembling the preferred L1 endonuclease motif (Fig. 1C). These analyses highlighted TE-driven genetic polymorphism among macaques, as well as the capacity of TEBreak to identify and characterize nonreference TE insertions in this model organism.
Next, we used the reference and nonreference L1RS2 insertions found in the bulk liver WGS data sets to estimate the detection sensitivity for potential somatic L1 insertions present in the 20 MDA-amplified neuronal genomes. For the reference analysis, we joined adjacent L1RS annotations, the majority of which represent 5′ inverted L1s that are often annotated as two oppositely oriented elements sharing a breakpoint, reducing the number of L1RS2 copies from 6492 to 5221. Of these, 3200 (61.3%) and 3113 (60.0%) were present in ON22212 and ON22213, respectively. Sensitivity was then recorded as a range, with the lower, more stringent, bound based on insertions being found by five or more reads at each of their 5′ and 3′ genome TE–genome junctions in a given neuron, and the upper bound only requiring at least one read at either junction. On average, 19.0%–63.6% of the reference L1RS2 copies were detected in the corresponding MDA-amplified neurons, including 10.4%–50.8% of heterozygous insertions. Of the aforementioned 189 nonreference L1RS2 insertions, 12.0%–44.9% were on average identified in the MDA-amplified neurons, including 11.8%–42.7% of heterozygous elements (Supplemental Table S2). These results anticipated the false-negative rate of our single-cell genomic analysis when applied to the discovery of somatic TE insertions.
A somatic L1RS insertion arising during brain development
Candidate TE insertions detected by scWGS or RC-seq, called stringently by TEBreak in at least one neuron from only one animal, and absent from the liver WGS were considered provisional somatic events (Fig. 1A). With these parameters, we identified an intergenic somatic L1RS insertion, which we called L1RSsomatic, on Chromosome 4 of animal ON22213 neuron #15 (Fig. 1D; Supplemental Table S2). PCR followed by capillary sequencing recovered the 5′ and 3′ junctions of L1RSsomatic in two of the ON22213 neurons analyzed by scWGS (#15 and #39) and in the matched bulk hippocampus (Fig. 1E). L1RSsomatic was not detected by PCR in the liver of either ON22212 or ON22213. Among 59 additional MDA-amplified RBFOX3+ neuronal nuclei, junction-specific PCRs identified L1RSsomatic in neurons #29, #55, and #57 (Supplemental Fig. S1A,B). Hence, five out of 72 (6.9%) of the tested ON22213 neurons, which were isolated from three separate hippocampus samples (Supplemental Fig. S1A,B), harbored L1RSsomatic. Junction PCRs amplified L1RSsomatic in two out of four additional ON22213 bulk hippocampus samples and not in skeletal muscle, sciatic nerve, spinal cord, or cerebellum (Supplemental Fig. S2). L1RSsomatic therefore arose during central nervous system development, most likely in a neural progenitor cell located in the ventricular zone of the anterior (rostral) neural tube.
L1RSsomatic was full length, belonged to the L1RS2 subfamily, integrated at a 5′-TATT/AT-3′ motif, and was flanked by a 16-bp TSD (Fig. 1F). These features were consistent with a bona fide L1 retrotransposition event (Moran et al. 1996; Jurka 1997). Capillary sequencing of the 3′ junction PCR products revealed a very long (>170 nt) poly(A) tract at the 3′ end of L1RSsomatic in neuron #15 (Supplemental Fig. S1C). As observed for somatic L1 insertions found by scWGS of human neurons (Evrony et al. 2015; Sanchez-Luque et al. 2019), the poly(A) tract of L1RSsomatic varied substantially in length (∼110 bp to ∼170 bp) from neuron to neuron (Fig. 1E; Supplemental Fig. S1B). L1RSsomatic was preceded by a 5′ untemplated guanine, as potentially associated with reverse transcription of an mRNA 5′ cap structure (Lavie et al. 2004; Gilbert et al. 2005), as well as a 4-bp 5′ transduction (Fig. 1F).
We traced the 5′ transduced sequence (AGAG) to a putative L1RS2 donor element positioned in sense to intron 10 of the PRDM4 gene on Chromosome 11 (Fig. 1F). We termed this element L1RSPRDM4. To characterize L1RSsomatic and L1RSPRDM4, we PCR-amplified and fully capillary-sequenced each element using template DNA from animal ON22213 (Fig. 2A; Supplemental Table S2). L1RSsomatic and L1RSPRDM4 were identical, apart from the much shorter 3′ poly(A) tract carried by L1RSPRDM4 (Fig. 2B). Another candidate donor L1 (Chr 4: 107,868,275–107,874,430) closely matched L1RSsomatic but lacked the adjacent 5′ AGAG sequence. Moreover, visual inspection of the aligned bulk liver WGS data indicated this element on Chromosome 4 was absent from ON22213. None of the nonreference full-length L1s detected by the ON22213 liver WGS (Supplemental Table S2) were preceded by a 5′ AGAG. These analyses strongly linked L1RSsomatic to L1RSPRDM4 or, with lower probability, to a closely related but undetected nonreference donor L1 located perhaps in an unassembled genomic region (Ewing et al. 2020; Zhou et al. 2020).
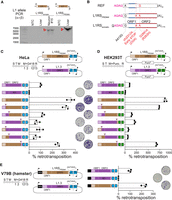
An endogenous L1 that is mobile in the macaque brain and in vitro. (A) The complete sequence of L1RSsomatic and its homozygous donor element, L1RSPRDM4, was amplified by PCR reactions (primers α + β) with input template DNA from ON22213 neuron #15 and bulk liver, respectively. Note that primer α spanned the 5′ junction of L1RSsomatic to more efficiently amplify the L1 allele. Red arrowheads indicate amplicons confirmed as on-target by capillary sequencing. (B) L1RSsomatic and L1RSPRDM4 were cloned and completely capillary-sequenced. Nucleotide variants among the reference (REF) genome L1RSPRDM4 sequence, the two identical L1RSPRDM4 alleles carried by animal ON22213, and the L1RSsomatic sequence are shown. Nonsynonymous mutations are highlighted in red. The 4-bp 5′ transduction (AGAG) carried by L1RSsomatic is colored in pink. (C) Engineered L1 retrotransposition efficiency measured in cultured HeLa cells (Moran et al. 1996). The assay design (top) shows either L1RSPRDM4 (brown) or L1.3 (purple), a highly mobile human L1 (Dombroski et al. 1993), tagged with a neomycin (G418)-resistance cassette activated only upon retrotransposition. (S) Seeding, (T) transfection, (M) change of media, (R) result analysis, (filled lollipop) polyadenylation signal. Numbers represent days of treatment with G418. AA(T)AAA indicates where a thymine base was removed to ablate the natural L1RSPRDM4 and L1.3 polyadenylation signals. Tested elements (bottom) included, in order, positive (L1.3) and negative (L1.3 RT−, D702A mutant) controls (Moran et al. 1996; Sassaman et al. 1997); L1RSPRDM4; a set of three chimeric elements where L1.3 was fused to L1RSPRDM4 at the 3′ end of the L1.3 5′ UTR, ORF1, and ORF2; and a set of three reciprocal elements where L1RSPRDM4 and L1.3 were joined at the 3′ end of the L1RSPRDM4 5′ UTR, ORF1, and ORF2 sequences. L1 expression was driven by native promoters only. Chimeric element fusion points are marked by inverted triangles. Representative well pictures are shown. Histogram values are normalized to L1.3 (100%). Data consist of three technical replicates and their mean ± SD, obtained from one representative experiment of three independent biological replicates. (D) As per C, except assayed in HEK293T cells using an EGFP-based L1 reporter system (Ostertag et al., 2000) in which cells are selected for puromycin resistance, and retrotransposition efficiency is measured as the percentage of EGFP+ sorted cells. (E) As per C, except with the inclusion of a cytomegalovirus promoter (CMVp) to additionally drive L1RSPRDM4 and L1.3 expression, as well as testing in the Chinese hamster fibroblast V79B cell line.
WGS-based genotyping indicated that, as expected, L1RSsomatic was heterozygous in ON22213 neuron #15, whereas L1RSPRDM4 was homozygous in ON22213 liver. L1RSPRDM4 was heterozygous in ON22212 liver. An analysis of primate reference genome assemblies indicated L1RSPRDM4 was present in the closely related crab-eating macaque (Macaca fascicularis) and absent from the more evolutionarily distant southern pig-tailed macaque (Macaca nemestrina) and green monkey (Chlorocebus sabaeus), suggesting L1RSPRDM4 entered the macaque germline 3–5 million years ago (Kent et al. 2002; Springer et al. 2012; Kumar et al. 2017). The two L1RSPRDM4 alleles carried by ON22213 were identical and deviated from the macaque reference genome L1RSPRDM4 element at a single 5′ UTR position (A413G) and two ORF2 nucleotide positions: (A)2312A, which introduced to the reference sequence a premature ORF2p stop codon, and G2891A, a nonsynonymous mutation of unclear significance for ORF2p activity (Fig. 2B). These analyses confirmed L1RSPRDM4 and L1RSsomatic encoded intact ORFs, whereas the reference L1RSPRDM4 sequence did not, indicating L1RSPRDM4 may be present and retrotransposition competent in only some macaques.
Exceptional L1RSPRDM4 retrotransposition in cultured cells
To assess the retrotransposition efficiency of L1RSPRDM4, and therefore L1RSsomatic, we used a quantitative cell culture-based retrotransposition assay (Moran et al. 1996; Kopera et al. 2016) in which an L1 is tagged with an antibiotic selectable marker cassette only activated upon retrotransposition (Fig. 2C). Using this assay in HeLa cells, we found L1RSPRDM4 mobilized more than threefold more efficiently than a highly active human L1HS element (L1.3) carrying the same marker cassette and used as a positive control (Dombroski et al. 1993; Sassaman et al. 1997). No retrotransposition was detected for a negative control L1.3 disabled by an ORF2p reverse transcriptase mutation (D702A) (Moran et al. 1996).
We next tested L1RSPRDM4 in cultured HEK293T cells using a related assay in which, instead of an antibiotic selectable marker cassette, retrotransposition activates an enhanced green fluorescent protein (EGFP) marker, and L1 mobility is quantified via flow cytometry (Ostertag et al. 2000; Kopera et al. 2016). In HEK293T cells, L1RSPRDM4 reproducibly mobilized more than eightfold more efficiently than L1.3, whereas the L1.3 ORF2p reverse transcriptase mutant did not retrotranspose (Fig. 2D). An alignment of the L1RS2 and L1HS consensus sequences (Supplemental Fig. S3A) indicated amino acid substitutions in both ORFs (Supplemental Fig. S3B), particularly ORF1, as noted previously (Taylor et al. 2013; Khazina and Weichenrieder 2018). To explore the disparate retrotransposition efficiencies of L1RSPRDM4 and L1.3, we generated and tested a series of chimeric L1.3-L1RSPRDM4 elements in the HeLa- and HEK293T-based experimental assays. Although interchanging either the 5′ or 3′ UTR of L1.3 and L1RSPRDM4 in each system minimally impacted their mobility, replacing either L1RSPRDM4 ORF with the corresponding L1.3 ORF severely reduced retrotransposition compared with L1RSPRDM4 (Fig. 2C,D). Next, we used the antibiotic-resistance cassette-based retrotransposition assay to test L1RSPRDM4 and L1.3 activity in Chinese hamster V79B cells, where the expression of each L1 was ensured by a cytomegalovirus promoter (CMVp) element (Fig. 2E). L1RSPRDM4 mobilized more than 2.2-fold more efficiently than L1.3. These results alluded to a functional interplay between L1RSPRDM4 ORF1p and ORF2p that may be less relevant to human L1s, as L1RSPRDM4 retrotransposed most efficiently when both of its native ORFs were present and was far more mobile than L1.3 in human cells and in the more evolutionarily distant context of a rodent cell line. The retrotransposition competence of L1RSPRDM4 was also consistent with its mobilization in vivo.
L1PRDM4 methylation in adult tissues
The developmental origins of somatic mutations, including L1 insertions, can be inferred from their spatial distribution in adult tissues (Evrony et al. 2015; Richardson et al. 2017; Sanchez-Luque et al. 2019). Detection of L1RSsomatic in bulk ON22213 hippocampus and a substantial fraction (∼7%) of MDA-amplified hippocampal neurons, but not in other tissue samples, pointed to its integration at the outset of brain development but after formation of the neural tube (Stiles and Jernigan 2010). DNA methylation mediates L1 transcriptional silencing (Thayer et al. 1993; Deniz et al. 2019; Greenberg and Bourc'his 2019) and is relaxed among L1 promoters during early embryogenesis (Coufal et al. 2009; Macia et al. 2017; Sanchez-Luque et al. 2019). Notably, neuronal and nonneuronal L1 insertions occurring later in human development have been traced to donor L1s escaping methylation even in mature somatic cells (Evrony et al. 2015; Scott et al. 2016; Sanchez-Luque et al. 2019; Ewing et al. 2020). On this basis, we hypothesized L1RSPRDM4 was aberrantly demethylated in the hippocampus. To test this possibility and evaluate TE methylation genome-wide, we applied Oxford Nanopore Technologies (ONT) long-read sequencing to ON22213 bulk hippocampus and liver tissue (Supplemental Table S1). Examining the PRDM4 locus, we found the PRDM4 promoter was fully unmethylated, whereas the promoter and body of L1RSPRDM4 were nearly completely methylated (Fig. 3A). We confirmed these results with locus-specific bisulfite sequencing (Fig. 3B,C) and concluded L1RSPRDM4 did not escape methylation in adult tissues.
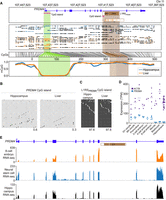
Regulation and embryonic expression of the PRDM4 locus. (A) Methylation profile of the PRDM4 locus obtained from ONT long-read sequencing (Ewing et al. 2020; Cheetham et al. 2022). The first panel shows L1RSPRDM4 oriented in sense to intron 10 of PRDM4, with genomic coordinates (rheMac10) provided, as well as a magnified view of the L1RSPRDM4 5′ UTR displaying CpG dinucleotides (orange lines) forming a CpG island (pink bar). The positions of primers used to assess L1RSPRDM4 methylation via locus-specific bisulfite sequencing in panel C are shown. The second panel displays animal ON22213 ONT read alignments, with unmethylated CpGs colored in blue (hippocampus) and orange (liver), and methylated CpGs colored black. The third panel indicates the relationship between CpG positions in genome space and CpG space, including those corresponding to the PRDM4 CpG island (shaded light green) and the L1RSPRDM4 5′ UTR and body (shaded light and dark brown, respectively). The fourth panel indicates the fraction of methylated CpGs for each tissue across CpG space. (B) Targeted bisulfite sequencing of the PRDM4 CpG island, as indicated in panel A, in animal ON22213 hippocampus and liver tissue. Each cartoon displays 50 nonidentical randomly selected sequences, where methylated CpGs (mCpGs) and unmethylated CpGs are represented by black and white circles, respectively, as well as the overall mCpG percentage. (C) As per B, except for the L1RSPRDM4 5′ UTR CpG island. (D) PRDM4 expression (blue circles) measured in RNA-seq tags per million (TPM) compared with that of the housekeeping gene ACTB (purple squares). Data were obtained from prior analyses of germinal vesicle (GV) and metaphase II (MII) oocytes, preimplantation embryo development stages (Wang et al. 2017), and adult hippocampus (Yin et al. 2020). Horizontal bars represent the mean of biological replicates. (E) Examples of PRDM4 expression during rhesus macaque development, showing WIG coverage tracks generated from published eight-cell embryo, neural stem cell, and hippocampus RNA-seq data sets (Zhao et al. 2014; Wang et al. 2017; Yin et al. 2020).
The expression of an intronic donor L1 may be influenced by the activity of its host gene (Philippe et al. 2016), as per an L1HS element located in the human TTC28 gene that is highly mobile in epithelial cancers (Tubio et al. 2014; Sanchez-Luque et al. 2019). PRDM4 is strongly expressed in mammalian embryonic cells and later down-regulated as a catalyst for neuronal differentiation (Chittka et al. 2012; Bogani et al. 2013). We therefore compiled published RNA-seq transcriptome profiling data from various stages of early macaque development, including metaphase I and II oocytes, six stages of preimplantation embryogenesis, and adult hippocampus (Wang et al. 2017; Yin et al. 2020). This analysis indicated high PRDM4 expression was maintained until the eight-cell stage and was followed by an 85% reduction at the morula (16-cell) stage (Fig. 3D). PRDM4 nonetheless was expressed in the hippocampus (Fig. 3D,E), and in neural stem cells generated in vitro (Fig. 3E; Zhao et al. 2014). We concluded L1RSPRDM4 was positioned in a genomic locus likely transcribed throughout embryogenesis and brain development, when L1RSsomatic arose, despite near-complete methylation of the L1RSPRDM4 promoter in the mature hippocampus.
Dynamic TE expression during macaque development
Human pluripotent cells support endogenous L1 demethylation, transcription, and mobilization (Garcia-Perez et al. 2007; Klawitter et al. 2016; Macia et al. 2017; Sanchez-Luque et al. 2019). Although accurate TE locus-specific measurement of transcription with short-read RNA-seq is extremely challenging (Lanciano and Cristofari 2020), this approach can be used to quantify expression of TE subfamilies genome-wide (Faulkner et al. 2008, 2009; Hashimoto et al. 2009). We therefore used the RNA-seq data sets described above to temporally profile the transcript abundance of a focused cohort of TE subfamilies, selected to represent the LINE, SINE, and ERV superfamilies. These were L1RS2, L1PA5 (mobile in the last common macaque–human ancestor and now immobile), AluYRa1 (the most numerous macaque AluY element), and MacERV1 (a young, horizontally transferred macaque ERV) (Han et al. 2007; Warren et al. 2020). As controls, we reanalyzed published human (Zhang et al. 2019) and mouse (Macfarlan et al. 2012) RNA-seq data sets and faithfully recapitulated the associated conclusions of abundant human endogenous retrovirus-H (HERVH) and murine endogenous retrovirus-L (MERVL) expression, respectively, in preimplantation embryonic cells (Supplemental Fig. S4; Peaston et al. 2004; Svoboda et al. 2004; Macfarlan et al. 2012; Lu et al. 2014; Grow et al. 2015; Zhang et al. 2019). Examining macaque TE subfamily expression with two computational pipelines (Faulkner et al. 2008; Hashimoto et al. 2009; Jin et al. 2015), we noted L1RS2 was consistently more highly expressed than L1PA5 throughout development, particularly at the eight-cell and morula stages, whereas AluYRa1 expression lagged slightly behind, peaking at the morula and blastocyst stages (Fig. 4A; Supplemental Fig. S5). In contrast, MacERV1 displayed a 17-fold increase in expression between metaphase II oocytes and the two-cell stage, as seen for MERVL in the mouse (Macfarlan et al. 2012), and was lowly expressed from the morula stage onward (Fig. 4A). The widespread occurrence of TEs within introns and immediately downstream from protein-coding genes can make readthrough and independent TE transcription difficult to distinguish with short-read RNA-seq (Lanciano and Cristofari 2020). However, closely examining individual L1RS2 and AluYRa1 loci, we found the most strongly expressed elements tended to be intergenic (Supplemental Fig. S6A) or, if adjacent to a protein-coding gene, show more temporally restricted expression than that gene (Supplemental Fig. S6B,C). These RNA-seq analyses altogether highlighted L1RS2 and AluYRa1 transcriptional activation in the macaque embryo, distinct to that of other TEs and consistent with the in vivo timing of endogenous retrotransposition events traced elsewhere to human and mouse embryogenesis (van den Hurk et al. 2007; Richardson et al. 2017; Feusier et al. 2019).
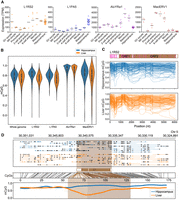
Genome-wide analyses of young TE subfamily transcription and methylation. (A) Subfamily-wide TE expression measured by RNA-seq (TPM) and an existing strategy to account for multimapping reads (Faulkner et al. 2008, 2009; Hashimoto et al. 2009). Data were obtained from prior studies (Wang et al. 2017; Yin et al. 2020) and encompassed GV and MII oocytes, early embryonic development, and adult hippocampus. Horizontal bars represent the mean of biological replicates. (B) Violin plots showing CpG methylation ascertained by ONT sequencing upon animal ON22213 hippocampus and liver. Results are shown for the whole genome (6-kbp windows), L1RS2 and L1PA5 copies >6 kbp, AluYRa1 copies >300 bp, and MacERV1 long terminal repeats >300 bp. (C) Composite L1RS subfamily methylation profiles. Each graph displays 100 profiles. A schematic of the L1RS2 consensus sequence is provided at top, with CpG positions indicated by pink bars. (D) Exemplar methylation profile of an L1RS2 element located on Chromosome 5 and hypomethylated in the liver. The panel is composed as described for Figure 3A.
Macaque TE methylome landscapes
Exceptions to DNA methylation at specific donor L1 loci appear to facilitate somatic retrotransposition in humans (Faulkner and Billon 2018). However, it was unclear whether similar “escapee” L1s reside in macaque methylomes, especially as L1RSPRDM4 was here heavily methylated in the hippocampus. We therefore analyzed our macaque ONT sequencing data with MethylArtist (Cheetham et al. 2022) to survey TE subfamily methylation genome-wide and at individual TE loci. We observed median CpG methylation values of 83.3% and 75.0% for L1RS2 copies in the hippocampus and liver, respectively, with these values substantially lower than those for AluYRa1 (94.0% and 92.9%) (Fig. 4B). L1RS2 was modestly (∼3%) more methylated than L1PA5 in each tissue. Of the TE subfamilies analyzed, MacERV1 elements were the most variably methylated (Fig. 4B). These trends largely aligned with those observed for the approximately equivalent TE subfamilies in human tissues, in which TE methylation in the hippocampus was also generally higher than in liver (Ewing et al. 2020). Profiling methylation across full-length (>6-kbp) L1RS2 copies, we observed a trough within the 5′ UTR (Fig. 4C). This trough was, however, less pronounced than the one identified for the human L1HS subfamily (Ewing et al. 2020), perhaps owing to the differing 5′ UTR CpG densities of L1RS2 (2.7 CpGs per 100 bp) and L1HS (4.3 CpGs per 100 bp). We found 88 L1RS2, 22 L1PA5, two MacERV1, and 176 AluYRa1 copies differentially methylated (P < 0.05, Fisher's exact test with Bonferroni correction) in the hippocampus compared with the liver, with most being less methylated in the latter tissue (Fig. 4B,D; Supplemental Fig. S7A; Supplemental Table S3). As well, seven L1RS2, two L1PA5, one MacERV1, and 76 AluYRa1 copies were <50% methylated in both the hippocampus and liver (Supplemental Fig. S7B; Supplemental Table S3). These results indicated that, although the vast majority of young TEs were repressed, a handful were unmethylated in a subset of macaque brain or liver cells.
Discussion
Endogenous L1 retrotransposition requires a complex series of molecular steps to be completed amidst the host genome defenses maintained by somatic and germ cells (Goodier 2016; Scott and Devine 2017). We show here that the cellular circumstances leading to L1 mobility can nonetheless come about during macaque brain development, as in humans (Evrony et al. 2015; Erwin et al. 2016; Sanchez-Luque et al. 2019). That this mechanism may be evolutionarily conserved is notable, given these species diverged nearly 30 million years ago (Kumar et al. 2017), as did their mobile L1 subfamilies and host defense pathways. We speculate the L1PA5 common ancestor of the youngest macaque (L1RS2) and human (L1HS) subfamilies (Warren et al. 2020) was similarly able to retrotranspose in the neuronal lineage, and this potential was inherited by other primate L1 subfamilies.
L1RSsomatic bore a striking resemblance to the three somatic L1 insertions characterized to date via scWGS of human neurons (Evrony et al. 2015; Sanchez-Luque et al. 2019). Each of these four events generated TSDs of 13–20 bp, incorporated 3′ poly(A) tracts of ∼90 bp to ∼170 bp, integrated at a degenerate L1 endonuclease motif, and, via transductions, was traced to mobile donor L1s. These sequence features are congruent with the mechanistic model of L1 target-primed reverse transcription (Moran et al. 1996; Jurka 1997). As per the three human insertions, L1RSsomatic was detected in multiple postmitotic neurons, where its poly(A) tract varied considerably in length, consistent with asymmetric poly(A) microsatellite shortening in the clonal lineages giving rise to the hippocampus (Grandi et al. 2013; Evrony et al. 2015; Sanchez-Luque et al. 2019). The detection of multiple L1RSPRDM4 alleles reinforces prior findings relating to retrotransposition-competent L1 alleles in the human brain (Sanchez-Luque et al. 2019) and germline (Lutz et al. 2003; del Carmen Seleme et al. 2006). These data indicate neurodevelopmentally active primate donor L1s can be polymorphic, and include both mobile and immobile alleles.
5′ and 3′ transductions are carried by <1% and <10%, respectively, of new human germline L1 insertions (International Human Genome Sequencing Consortium 2001; Gardner et al. 2017; Ewing et al. 2020). In contrast, all four human and macaque somatic L1s identified to date with scWGS incorporated a 5′ (two) or 3′ (two) transduction. The reasons for this apparent bias, however, remain unresolved. The untemplated guanine preceding the 4-bp L1RSsomatic 5′ transduction (Lavie et al. 2004) and the presence of a pyrimidine/purine initiator dinucleotide (Sandelin et al. 2007) at the corresponding transcription start site upstream of L1RSPRDM4 together indicate the mRNA template for L1RSsomatic could have been transcribed at the direction of the canonical L1RSPRDM4 5′ UTR promoter and then capped and reverse-transcribed without 5′ truncation. The L1RSPRDM4 promoter nonetheless provides the main difference with the three human insertions; these were each traced (Sanchez-Luque et al. 2019) to a donor L1, or an upstream promoter, demethylated in brain tissue, whereas L1RSPRDM4 was nearly completely methylated. We propose the following scenarios, in order of decreasing likelihood, to explain L1RSsomatic in the face of L1RSPRDM4 promoter methylation in hippocampus: (1) L1RSPRDM4 was hypomethylated and transcribed in the neural progenitor cell giving rise to L1RSsomatic; (2) the requisite L1RSPRDM4 mRNA was carried forward from earlier embryonic development; (3) L1RSPRDM4 was transcribed as part of a chimeric mRNA initiated by the demethylated PRDM4 promoter and 5′ truncated to remove almost all of the upstream PRDM4 exons; or (4) DNA methylation does not as strongly underpin macaque L1RS2 transcriptional repression as it does human L1HS repression.
L1RSPRDM4 is the first endogenously mobile nonhuman primate L1, to our knowledge, to be tested in a cultured cell retrotransposition assay. Its natural mobility in vitro was high: greater than threefold, greater than eightfold, and greater than 2.2-fold more than the positive control L1.3 in HeLa, HEK293T, and V79B cells, respectively. Adaptive evolution involving strong positive selection of amino acid substitutions has been observed among primate L1 protein domains (Boissinot and Furano 2001; Khan et al. 2006; Wagstaff et al. 2013; Khazina and Weichenrieder 2018; Furano et al. 2020). However, as opposed to its individual ORF1p or ORF2p activities or 5′ UTR promoter strength, the efficiency of L1RSPRDM4 appeared because of ORF1p-ORF2p synergy. We speculate that evolutionary changes in ORF1p–ORF2p epistatic interactions (Wagstaff et al. 2011) occurred after the divergence of humans and the macaque and, as a result, increased L1RS2 retrotransposition efficiency. Another possible explanation is that these interactions were supported by the ancestral L1PA5 subfamily and weakened, or were lost, during the later evolution of the L1HS lineage. Either rationale is supported by the relative retrotransposition efficiencies of L1RSPRDM4 and L1.3 in cultured rodent V79B cells, in which host–factor interactions specific to the RNA or proteins of either element are presumably absent. Of the young macaque L1 subfamilies, L1RS2 has the lowest average sequence divergence (1.1%) and the highest proportion of full-length (>6-kbp) elements (Warren et al. 2020). Nearly sevenfold more full-length L1RS2 copies than L1HS copies are annotated in the respective reference genomes, and these also make up a higher proportion of the elements in that subfamily (L1RS2: 34.4%, L1HS: 19.3%), despite L1RS2 modestly predating the emergence of L1HS (Khan et al. 2006; Warren et al. 2020). These differences, as well as the exceptional in vitro mobility of L1RSPRDM4, imply endogenous L1 retrotransposition potential may presently be higher in macaque than in humans.
Single-cell analyses now provide conclusive evidence of L1-mediated somatic mosaicism in the macaque and human brain. Endogenous retrotransposition is also likely encountered in mouse and fly neuronal lineages (Muotri et al. 2005; Coufal et al. 2009; Li et al. 2013; Hazen et al. 2016; Keegan et al. 2021; Siudeja et al. 2021). A major limitation of scWGS is the generation of MDA and Illumina library preparation false positives (Faulkner and Garcia-Perez 2017; Treiber and Waddell 2017; Abascal et al. 2021). The stringent approach adopted here and elsewhere (Evrony et al. 2015; Sanchez-Luque et al. 2019), requiring complete resolution of L1RSsomatic and its associated retrotransposition hallmarks, excludes false positives with near certainty but also raises the prospect of false negatives. On average, only 11.8% of the heterozygous nonreference L1RS2 insertions present in our bulk liver WGS data sets were found, using robust thresholds, in the corresponding neuronal genomes. Although L1RSsomatic was detected by PCR at its 5′ or 3′ genomic junction in 5/72 MDA-amplified neurons from ON22213, and in bulk hippocampus, the complete insertion could only be PCR-amplified in neuron #15. Furthermore, we analyzed a pan-neuronal (RBFOX3+) hippocampal population, which could obscure potential sublineage-specific L1 activity (Faulkner and Garcia-Perez 2017; Bodea et al. 2022). These considerations in our view prohibit an accurate calculation of L1 mobilization frequency in the macaque brain, extrapolated from one bona fide somatic L1RS2 insertion. We did not identify any somatic Alu insertions, concordant with prior human neuron scWGS analyses (Evrony et al. 2015; Sanchez-Luque et al. 2019). We have also not included an analysis of somatic single-nucleotide variants, because of the potential difficulties in distinguishing these from MDA artifacts present in scWGS data (Abascal et al. 2021). Finally, although the impact of somatic retrotransposition upon brain development remains hypothesized (Muotri and Gage 2006; Erwin et al. 2014), its apparent occurrence among multiple primate species, and likely other animals, may inform future studies testing the association of L1 mobility or expression with cellular or cognitive functions.
Methods
Macaque samples
Snap frozen hippocampus and liver tissue from two postmortem macaques (identifiers ON22212 and ON22213) without evidence of pathology was provided by the Monkey Alcohol Tissue Research Resource (MATRR) biobank (https://gleek.ecs.baylor.edu/) to L.C. with ethical approval to be used as described previously (Daunais et al. 2014). ON22212 and ON22213 were 7-yr-old male adults, bred in the same animal research facility without parents or grandparents in common.
Isolation and whole-genome amplification of neuronal nuclei
Single neurons were isolated from hippocampal tissue and genomic DNA amplified via MDA as previously described (Sanchez-Luque et al. 2019). Reagents were prechilled and the entire procedure performed on ice. Frozen hippocampus samples were first gently Dounce homogenized for 2 min in 2 mL cold nucleus extraction buffer composed of 10 mM Tris (pH 7.4), 10 mM NaCl, 3 mM MgCl2, and 0.1% IGEPAL CA-630 (Sigma-Aldrich). Tissue homogenates were then filtered into a 5-mL tube with a 40-µm cell-strainer cap and centrifuged at 500g for 2 min at 4°C. Following centrifugation, pellets were resuspended in a wash buffer of 1% bovine serum albumin (BSA; Sigma-Aldrich A2153) in PBS. To tag neuronal nuclei, anti-RBFOX3 (Merck-Millipore MAB377X) antibodies and DAPI (Sigma-Aldrich D9542) were added to the solution and incubated for 15 min at 4°C. Nuclei were spun down as above and resuspended in 1 × PBS. DAPI+/RBFOX3+ nuclei were sorted using a BD FACSAria cell sorter (Becton Dickinson) in a block buffer (10% goat serum and 5% BSA). Purified nuclei were then picked using an Olympus IX71 inverted microscope, with an Eppendorf TransferMan 2 micromanipulator and Eppendorf CellTram. During picking, single nuclei were washed in PBS and transferred to individual UV-sterilized 0.2-mL PCR tubes. MDA was then performed upon each nucleus using a REPLI-g single cell kit (Qiagen 150345). First, nuclei were incubated for 10 min at 65°C in 3 µL buffer D2 and then placed on ice with 3 µL stop solution. DNA was amplified for 8 h at 30°C with 1× sc reaction buffer, phi29 DNA polymerase, and nuclease-free UV-treated water for a final volume of 40 µL. The polymerase was then inactivated for 3 min at 65°C. MDA-amplified DNA clean-up was performed with 1:1.3 (v/v) ratio AMPure XP beads (Beckman Coulter A63881) immediately before the de-branching step. To screen nuclei for those with the most even genome-wide amplification, multiplexed PCR primers were designed for a panel of 12 nonrepetitive loci (Supplemental Table S1). For each nucleus, three reactions were undertaken on a DNA engine tetrad 2 thermal cycler (Bio-Rad), with MyTaq HS DNA polymerase (Bioline BIO-21105), 5× MyTaq reaction buffer, 10 µL of a 25 mM primer mix containing four primer pairs, 12 ng template DNA, and 0.25 U of enzyme in a 25 μL final volume. PCR cycling conditions were as follows: (1 min at 95°C) × 1; (15 sec at 95°C; 15 sec at 58°C; 15 sec at 72°C) × 35; (5 min at 72°C; hold at 4°C) × 1. Amplicons were visualized via GelDoc (Bio-Rad) on a 1.5% agarose gel stained with SYBR Safe (Invitrogen S33102). Twenty nuclei (seven from ON22212 and 13 from ON22213) where at least nine of 12 genomic loci amplified were selected for further analysis.
Illumina sequencing
Genomic DNA was extracted from ON22212 and ON22213 bulk liver tissue. Libraries were prepared for these samples, as well the material from the 20 MDA-amplified neuronal nuclei, using a TruSeq DNA PCR-free kit (Illumina 20015963) and an insert size of 550 bp. Each library was subjected separately to paired-end 2 × 150 mer WGS using an Illumina HiSeq X platform (Macrogen).
For RC-seq, DNA from the MDA-amplified neuronal nuclei was used to prepare barcoded libraries with a TruSeq nano DNA kit (Illumina FC-121-4001/2). Briefly, 4 µg of MDA-amplified DNA was diluted to 130 µL final volume and sheared in a Covaris M220 focused-ultrasonicator (peak power 50, duty factor 20, pulses per burst 200) for 110 sec in MicroTube AFA snap-cap tubes (Covaris 520045). DNA was purified via AMPure XP bead clean-up using a 1:1 volume of beads and eluting in 60 µL of resuspension buffer. The TruSeq Nano protocol was then followed as indicated by the manufacturer until the tandem clean-up after the adaptor ligation step. At this stage, samples were instead suspended in 20 µL of resuspension buffer and loaded on a 2% high-resolution agarose gel (Sigma-Aldrich) for visualization via electrophoresis. Size-selection was achieved by purifying gel cuts of 600–650 bp size, which were eluted using a MinElute gel extraction kit (Qiagen 28604). QG buffer was added at a ratio of 600 µL per 0.1 mg of gel cut and the agarose dissolved at room temperature. Elution was performed using 12.5 µL of 60°C preheated EB buffer twice, for a final 25 µL elution volume. Library amplification was performed using 1× phusion high-fidelity PCR master mix (New England Biolabs) with 100 pmol of each Illumina primer in a 100 µL final volume. Cycling conditions were as follows: (45 sec at 98°C) × 1; (15 sec at 98°C; 30 sec at 60°C; 30 sec at 72°C) × 7; (5 min at 72°C; hold at 4°C) × 1. Samples were purified by AMPure XP beads clean up using 1:1 ratio of DNA to beads, eluted in 30 µL of molecular grade water, and quantified using a Bioanalyzer DNA 1000 chip (Agilent Technologies) according to the manufacturer's instructions. Libraries were pooled in equimolar amounts and the pool hybridized as per a prior study (Richardson et al. 2017) to a custom sequence capture probe pool (Roche NimbleGen) targeting the L1RS2, L1RS37, AluYRa4, AluYRb4, AluYRc2, AluYRd4, LTR14 (HERVK14), and LTR4 (MacERV1) subfamilies (Supplemental Table S1). The posthybridization library pool was then sequenced with paired-end 2 × 150 mer reads, using two flow cells of an Illumina HiSeq X platform (Macrogen). This was intended to achieve a similar postenrichment sequencing depth per targeted element as the WGS, while minimizing read duplicates caused by saturating the comparatively narrower TE–junction target windows generated by the TE enrichment probes.
Nonreference TE insertion detection
Bulk liver WGS data were aligned to the rheMac10 reference genome with BWA-MEM (optional parameters -M -Y) (Li 2013). Alignments were then analyzed with TEBreak (Carreira et al. 2016) using default parameters, where the Repbase consensus sequences for the L1RS2, L1RS37, L1PA5, AluYRa4, AluYRb4, AluYRc2, AluYRd4, and LTR4 (MacERV1) subfamilies were used to annotated potential insertions caused by young TEs. The TEBreak output table was then parsed to retain only putative nonreference insertions detected by at least five reads spanning each of their (5′ and 3′) TE–genome junctions and to remove insertions that were outside of canonical assembled macaque chromosomes (Chr 1–20, X, Y), or were 3′ truncated or, for Alu, were 5′ inverted or 5′ truncated by more than 1 nucleotide. Insertions were further stratified as homozygous (variant allele fraction ≥ 0.8) or heterozygous (variant allele fraction < 0.8) with the number of reads spanning the annotated (empty) insertion point providing the denominator. To identify putative somatic TE insertions, the neuron scWGS and RC-seq data sets were similarly aligned, added to, and processed together with the bulk liver WGS with TEBreak. The TEBreak output table was filtered as before, except with the additional requirement that any events called in either liver sample, or in neurons from more than one animal, were removed. The resulting filtered tables listed 3543 nonreference TE insertions, including L1RSsomatic, and are presented as Supplemental Table S2.
L1RSsomatic junction PCR validation experiments
We designed PCR primers to amplify the 5′ and 3′ L1–genome junctions of L1RSsomatic (Supplemental Table S2) with Primer3 (Untergasser et al. 2012). Reactions were undertaken on a DNA engine tetrad 2 thermal cycler (Bio-Rad), with MyTaq HS DNA polymerase, 5× MyTaq reaction buffer, 10 pmol of each primer, 5 ng of template DNA, and 2.5 U of enzyme in a 25 μL final volume. 5′ Junction PCR cycling conditions were as follows: (1 min at 95°C) × 1; (15 sec at 95°C; 15 sec at 59°C; 15 sec at 72°C) × 40; (5 min at 72°C; hold at 4°C) × 1. 3′ Junction nested PCR involved two reactions, the first with cycling conditions of (1 min at 95°C) × 1; (15 sec at 95°C; 15 sec at 59°C; 15 sec at 72°C) × 15; (5 min at 72°C; hold at 4°C) × 1, followed by sample treatment with ExoSAP-IT PCR product cleanup (Thermo Fisher Scientific 75001.1.ML; 15 min at 37°C; 15 min at 80°C), and a second reaction with cycling conditions of (1 min at 95°C) × 1; (15 sec at 95°C; 15 sec at 59°C; 15 sec at 72°C) × 30; (5 min at 72°C; hold at 4°C) × 1. All PCRs were performed with nontemplate control (NTC), each MDA-amplified DNA from animal ON22213 hippocampal neurons, as well as DNA extracted from animals ON22212 and ON22213 bulk tissues. Amplicons were visualized via GelDoc (Bio-Rad) on a 1.5% agarose gel stained with SYBR safe. GeneRuler 1 kb plus (Thermo Fisher Scientific SM1331) was used as the ladder. Amplicons of the correct size were gel-extracted using a Qiagen MinElute gel extraction kit and cloned with a pGEM-T easy vector system (Promega A1360) using One Shot TOP10 chemically competent Escherichia coli cells (Thermo Fisher Scientific C404010) before capillary sequencing by Macrogen. For each L1RSsomatic 3′ junction amplicon, the L1 poly(A) size was estimated by taking the average of the pure poly(A) (or poly(T)) tract lengths observed by capillary sequencing in both directions, as per Supplemental Figure S1C. Note that this approach provided a lower bound estimate for bulk or pooled input DNA, owing to polymerase slippage and the varying template poly(A) tract lengths among the neurons carrying L1RSsomatic.
PCR amplification of L1RSsomatic and L1RSPRDM4
To test the retrotransposition efficiency of L1RSPRDM4, we designed PCR primers specific to L1RSsomatic (Supplemental Table S2) by placing a forward primer, incorporating a NotI restriction site to facilitate later cloning, across the 5′ L1–genome junction and a reverse primer in the 3′ L1–genome flanking region. PCR was performed using an expand long-range dNTPack kit (Sigma-Aldrich 11681834001) in 1× reaction buffer with MgCl2, 0.5 mM of each dNTP, 3% DMSO, 10 pmol of each primer, 1.75 U of enzyme, and 10 ng of ON22213 hippocampal neuron #15 MDA-amplified DNA template in a 25 μL final volume. PCR cycling conditions were as follows: (2 min at 92°C) × 1; (10 sec at 92°C; 15 sec at 58°C; 6 min at 68°C) × 10; (10 sec at 92°C; 15 sec at 58°C; 6 min at 68°C + 20 sec/cycle) × 30; (10 min at 68°C; hold at 4°C) × 1. The L1RSPRDM4 donor element for L1RSsomatic was amplified by forward (5′-GGACAGTAGGCGGAGTTGAG-3′) and reverse (5′-CCACCATGCCCAGTCTACTT-3′) primers placed in the 5′ and 3′ genomic flanks of L1RSPRDM4, respectively, with the same reaction conditions as used to amplify L1RSsomatic, except using 10 ng of animal ON22213 liver DNA template. PCR products were resolved by electrophoresis on a 1% agarose gel and imaged with a Typhoon FLA 9500 scanner (GE Healthcare Life Sciences). PCR bands of the appropriate size were excised and purified via conventional phenol:chloroform DNA extraction followed by ethanol precipitation. PCR products containing L1RSsomatic and L1RSPRDM4 were cloned in a TOPO XL PCR cloning kit (Invitrogen K8050-10) using One Shot TOP10 electrocomp E. coli cells (Thermo Fisher Scientific C404050). PCR products and TOPO XL clones were capillary-sequenced using stepping primers to resolve the complete sequence of each L1 and identify potential allelic variants within the two L1RSPRDM4 alleles, using a previous approach (Sanchez-Luque et al. 2019). The two alleles of L1RSPRDM4 and the sequence of L1RSsomatic were found to be identical (Supplemental Table S2).
Cultured cell retrotransposition assays
The L1RSsomatic sequence was cloned into three pCEP4-derived vectors to assay its retrotransposition efficiency, based on a prior strategy applied to human L1s (Sanchez-Luque et al. 2019). The first and second vectors contained a neomycin resistance cassette (mneoI) driven by a simian virus 40 early promoter (SV40p) and terminated by a herpes simplex virus (HSV)–thymidine kinase polyadenylation signal and positioned downstream from, and in reverse orientation to, the L1 but was interrupted by an intron in the same orientation as the L1, meaning the cassette was only activated by retrotransposition (Moran et al. 1996). The second vector included a CMVp upstream of the L1 to ensure its transcription (Moran et al. 1996). The third vector was similar in structure but lacked the upstream CMVp and, instead of the mneoI cassette, contained an EGFP retrotransposition reporter cassette (mEGFPI) driven by CMVp. Also, in this vector, the original pCEP4 hygromycin-resistance marker was replaced by a puromycin-resistance gene for selecting transfected cells (Ostertag et al. 2000). Each of these vectors was originally designed to clone L1 sequences between the NotI and BstZ17I restriction sites, lacking a small fragment downstream from the BstZ17I site in the L1 3′ UTR (Moran et al. 1996; Ostertag et al. 2000). We previously restored the L1 3′UTR, only deleting the thymine base within the natural polyadenylation signal to still allow the retrotransposition cassette to be incorporated into the L1 mRNA (Sanchez-Luque et al. 2019). The cloning strategy here broadly involved rebuilding the L1RSsomatic sequence into the vector from several TOPO XL clone segments, avoiding clone-specific PCR mutations, and similarly altering the L1 polyadenylation signal as for the human L1.3 controls. We took advantage of the BstZ17I site in the L1RSsomatic 3′ UTR (conserved with human L1HS) to first engineer the 3′ end of L1RSsomatic downstream from the BstZ17I site into the vector, without the polyadenylation signal. We amplified the ∼80 bp fragment downstream from the BstZ17I site from a TOPO XL clone without PCR mutations by using the primers (5′-GGAAGATCTCTAGCGGCCGCATGTATACATATGTAACAAACCTGCACGTTATGCACA-3′) and (5′-GAGATTTAAATTTTTTTTTTTTTTTATACTTTAAGTTGTAGGGTACATG-3′). This reaction generated a 104-bp amplicon containing a NotI restriction site upstream of the BstZ17I site and a SwaI restriction site downstream from a 15-bp polyadenine tract and lacking the polyadenylation signal. PCR was performed using the Q5 high-fidelity DNA polymerase (New England Biolabs M0491S) in a reaction containing 1 × Q5 reaction buffer, 0.2 mM of each dNTP, 20 pmol of each primer, 1 U of enzyme, and ∼50 ng of input DNA in a 25 μL final volume. Cycling conditions were as follows: (30 sec at 98°C) × 1; (10 sec at 98°C; 15 sec at 58°C; 30 sec at 72°C) × 30; (2 min at 72°C; hold at 4°C) × 1. The resulting fragment was digested with NotI and SwaI restriction enzymes and cloned into the NotI and BstZ17I sites of the three aforementioned original vectors lacking the human L1 3′ UTR downstream from the BstZ17I site (Moran et al. 1996; Ostertag et al. 2000). The remaining L1RSsomatic sequence was reconstructed between the NotI site and the new BstZ17I site. Plasmid DNA vectors were produced using a Qiagen plasmid midi kit (12143).
Engineered L1 retrotransposition experiments were performed in HeLa, HEK293T, and V79B Chinese hamster lung fibroblast cells, following previously described guidelines (Sanchez-Luque et al. 2019). For the HeLa neomycin-resistance cassette-based reporter assay (Moran et al. 1996), HeLa JVM cells were seeded into six-well plates at a density of 5 × 103 cells/well in 2 mL of Dulbecco's Modified Eagle Medium (DMEM; Gibco 11965092), 10% fetal bovine serum (FBS; Gibco 10082147), 2 mM L-glutamine (Gibco 25030081), and 100 U/mL penicillin–streptomycin solution (Pen-Strep, Gibco 15140122) per well. Cells were incubated at 37°C, 5% CO2, and ∼95% humidity for the course of the experiment. Transfection was performed ∼14 h after seeding by adding 100 µL of transfection mix to each well, which contained 1 µg of plasmid DNA, 96 µL of Opti-MEM (Gibco 31985070), and 4 µL of FuGENE-HD (Promega E2311). Plates were shaken gently to homogenize the transfection mix. Technical replicates were plated from the same cell suspension and transfected with the same transfection master mix. Media were replaced with 2 mL of complete media 24 h after transfection and then replaced by complete media supplemented with 400 µg/mL of G418 sulphate (Geneticin Selective Antibiotic, Gibco 10131035) every 48 h for a total of 12 d. On day 14, media were aspirated and each well washed with 1–2 mL of Dulbecco's phosphate buffered saline (DPBS; Gibco 14190144). After aspirating the plates, cells were fixed by adding 1 mL of 1× DPBS, 0.2% glutaraldehyde, and 2% formaldehyde solution and incubating at room temperature for 20 min. The fixing solution was discarded and the wells carefully washed with reverse osmosis–purified (RO) H2O. Cell colonies were stained by adding 1 mL of 0.1% crystal violet solution to each well and incubating at room temperature for 10 min. The dying solution was discarded and the plates washed with RO H2O and air-dried before scanning. Plates were imaged using a Canon EOS Rebel T3 camera and a white light transilluminator.
The neomycin-resistance cassette-based reporter assay in the V79B cells was adapted from conditions used for similar experiments using Chinese hamster ovary cells (Morrish et al. 2007). This was performed as described above for HeLa cells but using a seeding density of 2 × 104 cells/well and DMEM with 1 g/L glucose (DMEM low glucose, Gibco 11885084), 10% FBS (Gibco 10082147), 2 mM L-glutamine (Gibco 25030081), 1× nonessential amino acids (100× NEAA; Gibco 11140050), and 100 U/mL penicillin–streptomycin solution (Pen-Strep, Gibco 15140122) as culture media for the course of the experiment. Media changes and antibiotic selection, respectively, were performed with the timing and G418 sulphate concentration (400 µg/mL) used for HeLa cells.
For the EGFP cassette-based reporter assay (Ostertag et al. 2000), experiments were performed using HEK293T cells. In six-well plates, 2 × 105 cells were seeded per well in media composed of 2 mL DMEM, 10% FBS, 2 mM L-glutamine, and 100 U/mL penicillin–streptomycin solution. Transfection was performed ∼14 h after seeding by adding a similar transfection mix as for HeLa cells above. Again, technical replicates were seeded from the same cell suspension and transfected with the same transfection master mix. Twenty-four hours after transfection, media were replaced and supplemented with 0.5 µg/mL puromycin (puromycin dihydrochloride, Gibco A1113803). This was then repeated daily for four more days but with media supplemented with 1 µg/mL of puromycin. On day 6, cells were washed with DPBS and incubated with 0.5 mL of trypsin-EDTA 0.25% for 5 min at 37°C. Trypsinization was stopped by adding 1 mL of DPBS with 10% FBS to each well. Cells were resuspended by pipetting, transferred to a 1.5-mL tube, and centrifuged at 450g for 5 min at 4°C. Supernatant was aspirated and cell pellets resuspended in 300 µL of 4°C 1× DPBS. Cells were analyzed in an Accuri flow cytometer (Becton Dickinson) with the assistance of the Institute of Genetics and Cancer flow cytometry facility.
Plasmid transfection efficiencies were calculated by cotransfecting with pCEP-EGFP into each cell line (Alisch et al. 2006). Briefly, 2 × 104 of HeLa, HEK293T, or V79B cells were seeded in 2 mL of the corresponding media in six-well plates. Cells were then transfected 14 h after seeding, as described above except with the addition of 0.5 µg pCEP-EGFP (Alisch et al. 2006; Garcia-Perez et al. 2007) alongside 0.5 µg of each L1 plasmid to each well. Media were replaced 24 h posttransfection without antibiotic supplementation and analyzed by flow cytometry on day 5 posttransfection. Untransfected cells were used to set the boundary in flow cytometry between EGFP− and EGFP+ events. Transfection efficiency assays for HeLa cells were performed in technical duplicates and used to normalize colony counts by the corresponding transfection efficiency. For HEK293T cells, transfected cells were selected through supplementing media with puromycin, and therefore, no correction by transfection efficiency was necessary. Thus, transfection efficiency analysis was performed only as a quality check.
For the retrotransposition assays, untransfected HeLa and V79B cells were selected with G418 as a negative control to confirm that neomycin-resistant colonies were owing to retrotransposition events. Untransfected HEK293T cells were selected with puromycin as a control to ensure the EGFP+ cell percentages for each tested construct were obtained from wells with no untransfected cells. No HeLa, HEK293T, or V79B cells survived antibiotic treatment. For the HEK293T cell-based assay, untransfected cells untreated with puromycin were used to set the EGFP− signal level in flow cytometry.
Bisulfite sequencing methylation analysis
Locus-specific bisulfite sequencing was performed as previously described for individual human L1 copies and protein-coding genes (Nguyen et al. 2018; Schauer et al. 2018; Salvador-Palomeque et al. 2019; Sanchez-Luque et al. 2019). Briefly, this involved first treating genomic DNA with an EZ DNA methylation lightning kit (Zymo Research D5030). Primers were then designed against a CpG island flanking the PRDM4 transcription start site (5′-TGTTATGAAGATTGAAATTTTGAG-3′ and 5′-CAACCCACCTAACAACTAC-3′) and the L1RSPRDM4 5′ end (5′-TGATAGTAAAGGTTTTGTAGAG-3′ and 5′-ACTACTATAAACTCCACCCAAT-3′). PCR reactions involved MyTaq HS DNA polymerase (Bioline) and the following cycling conditions for the PRDM4 assay: (2 min at 95°C) × 1; (30 sec at 95°C; 30 sec at 55°C; 30 sec at 72°C) × 40; (5 min at 72°C; hold at 4°C) × 1. The same conditions were used for the L1RSPRDM4 assay, apart from an annealing temperature of 52°C. Amplicons from each sample were then pooled and prepared for sequencing with a NEBNext Ultra II DNA library prep kit (New England Biolabs E7645S). Paired-end 2 × 300 mer sequencing was performed on a MiSeq platform (Illumina). Paired-end reads were assembled into contigs via FLASH (Magoč and Salzberg 2011) and assessed against target amplicons as previously described (Sanchez-Luque et al. 2019). Methylation cartoons were then generated for 50 randomly chosen reads for each amplicon and sample via the quantification tool for methylation analysis (QUMA) (Kumaki et al. 2008) with default parameters, plus requiring strict CpG recognition and excluding identical bisulfite sequences.
RNA-seq analyses
To quantify TE subfamily expression during macaque development, we assembled published RNA-seq data generated from single oocytes and preimplantation embryos (Wang et al. 2017) and hippocampus tissue (Yin et al. 2020). Oocyte and embryo sequencing data were obtained from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP089891 and encompassed germinal vesicle (GV) oocyte (n = 3), metaphase II stage oocyte (n = 3), pronucleus (one-cell embryo; n = 3), two-cell embryo (n = 3), four-cell embryo (n = 2), eight-cell embryo (n = 5), morula (n = 3), and blastocyst (n = 4) stages. Bulk hippocampus tissue RNA-seq data (SRA; SRP188855) were included from the dentate gyrus, CA1, and CA3 regions of eight animals, making a total of 24 samples. For each library, we aligned reads to the rheMac10 genome assembly with STAR (Dobin et al. 2013) version 2.6 (parameters ‐‐twopassMode Basic ‐‐outSAMprimaryFlag AllBestScore ‐‐winAnchorMultimapNmax 1000 ‐‐outFilterMultimapNmax 1000) and marked duplicate reads with Picard MarkDuplicates (http://broadinstitute.github.io/picard). To profile protein-coding gene expression, we considered only uniquely mapped reads overlapping RefSeq exon coordinates and built WIG plots (such as for PRDM4) in the Integrative Genomics Viewer (Robinson et al. 2011).
High copy number sequences with limited divergence, such as young TE subfamilies, present a significant mappability issue in which genuine signal is lost owing to reads mapping to multiple genomic loci (multimap reads) (Faulkner et al. 2008; Lanciano and Cristofari 2020). We therefore followed an existing strategy to, when possible, assign multimap reads a weighting at each position based on the relative abundance of uniquely mapping reads nearby (Faulkner et al. 2008, 2009; Hashimoto et al. 2009). Specifically, for each multimap read, we counted the number of uniquely mapped reads within 100 bp of the aligned multimap read at each of its potential best map genomic locations. We then assigned a weighting to each position in proportion to the fraction of uniquely mapped reads found at that position out of the total number of uniquely mapped reads found at any position for the given multimap read. If no uniquely mapped reads were found at any of the n multimap positions, each position was assigned a weighting of 1/n. Uniquely mapped reads were assigned a weighting of one. To produce estimates of transcript abundance for TE subfamilies, we intersected weighted alignments with RepeatMasker (Smit et al. 1996) coordinates, produced totals for each individual TE, and then summed these to produce a value for each TE subfamily genome-wide. Values were normalized by the total number of weighted mapped reads (tags per million). For display in histograms, L1RS2 was represented by the “L1_RS2” RepeatMasker subfamily, AluYRa1 and L1PA5 were eponymous, and MacERV1 was quantified as the sum of “MacERV1_int-int” and “MacERV1_LTR4” values.
As an orthogonal computational approach, we quantified transcript abundance across TE subfamilies using the TEtranscripts package (Jin et al. 2015). Again, we mapped the RNA-seq data described above to the rheMac10 reference genome using STAR (Dobin et al. 2013) as recommended (parameters ‐‐winAnchorMultimapNmax 100 ‐‐outFilterMultimapNmax 100) and marked duplicate reads with Picard MarkDuplicates. TEtranscripts version 2.2.1 was then used to generate read counts for protein-coding genes and repetitive elements, using annotations sourced from the ncbiRefSeq gene model tables of the UCSC Genome Browser (Kent et al. 2002) and a custom GTF file generated using RepeatMasker (Smit et al. 1996) provided with TEtranscripts. The fpm function of DESeq2 (Love et al. 2014) version 1.30.1 was used to normalize read counts (tags per million) for display histograms.
As positive controls, we used the same approaches to analyze RNA-seq data from prior studies reporting specific expression of MERVL (Macfarlan et al. 2012) and HERVH (Zhang et al. 2019) retrotransposons, respectively, in mouse two-cell embryo and human embryonic stem cell (hESC) samples. Mouse samples included triplicate two-cell embryo (SRA; SRP009468) and oocyte (SRA; SRP009469) experiments. MERVL expression was quantified as the sum of “MERVL-int” and “MT2_Mm” values, representing MERVL and its flanking LTR sequences. Human samples included duplicate hESC cardiomyocyte differentiation time courses (SRA; SRP152979), sampled at day 0 (hESC), day 2 (mesoderm), day 5 (cardiac mesoderm), day 7 (cardiac progenitor), day 15 (primitive cardiomyocyte), and day 80 (ventricular cardiomyocyte). HERVH expression was taken as the sum of “HERVH” and “LTR7” values. Reference genome assemblies mm10 and hg38, and their associated genome annotations, were used for mouse and human analyses, respectively.
ONT sequencing and methylation analysis
High-molecular-weight DNA was extracted from animal ON22213 hippocampus and liver tissue using a Nanobind CBB big DNA kit (Circulomics NB-900-001-01) and sheared to a ∼10-kb average size to improve sequencing yield. ONT sequencing libraries were prepared for each DNA sample using a ligation sequencing kit (ONT SQK-LSK109), pooled, and sequenced on an ONT PromethION platform. Bases were called with Guppy 4.0.11 (ONT) and reads aligned to the rheMac10 reference genome build using minimap2 version 2.20 (Li 2018) and SAMtools version 1.12 (Li et al. 2009). Reads were indexed and per-CpG methylation calls generated using nanopolish version 0.13.2 (Simpson et al. 2017). Methylation likelihood data were sorted by position and indexed using tabix version 1.12 (Li 2011). Methylation statistics for the genome divided into 6-kbp bins, as well as reference TEs defined by RepeatMasker coordinates (http://www.repeatmasker.org/), were generated using MethylArtist version 1.0.4 (Cheetham et al. 2022), using commands db-nanopolish, segmeth and segplot with default parameters. Methylation profiles for individual loci were generated using the MethylArtist command locus, where parameters specified a 30-bp sliding window with a 2-bp step, and smoothed with a window size of eight for the Hann function. The L1RS subfamily methylation profiles shown in Figure 4C were generated for elements >6 kbp with the MethylArtist composite command. To identify individual TEs showing differential methylation in the comparison of ON22213 hippocampus and liver ONT data (Supplemental Table S3), we required elements to have at least four reads and 20 methylation calls in each sample. Comparisons were performed via Fisher's exact test using methylated and nonmethylated call counts, with significance defined as a Bonferroni-corrected P-value of less than 0.05. The significance of observed versus expected intronic L1 insertions was calculated with a binomial test.
Data access
The Illumina and ONT sequencing data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB37719. Sanger trace files and unprocessed gel images for this study can be found in Supplemental File S1 and at Mendeley Data (https://dx.doi.org/10.17632/wpnv9ktv7p.2).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank John V. Moran for sharing L1.3 plasmids and the HeLa-JVM cell line, Margaret Z. Zdzienicka for sharing the V79B cell line, Jeffrey A. Jeddeloh for assistance with RC-seq probe design, and the QBI, TRI, and IGC flow cytometry facilities for technical advice. This study was funded by the following: Australian National Health and Medical Research Council (NHMRC) Investigator grants (GNT1161832 to S.W.C., GNT1176574 to N.J., GNT1173476 to S.R.R., GNT1173711 to G.J.F.), an NHMRC-ARC Dementia Research Development fellowship (GNT1108258 to G.O.B.), an Australian Government Research Training Program Scholarship awarded to P.G., the Australian Department of Health Medical Frontiers Future Fund (MRFF; MRF1175457 to A.D.E.), the Australian Research Council (DP200102919 to S.R.R. and G.J.F.), MINECO-FEDER (SAF2017-89745-R) and European Research Council (ERC-STG-2012-309433) funding and a private donation from Ms. Francisca Serrano (Trading y Bolsa para Torpes, Granada, Spain) to J.L.G-P., a National Institutes of Health (NIH) Office of Directors P51 grant (OD011092) to the Oregon National Primate Research Center to support L.C., an Andalusian Government EMERGIA grant (20_00225) to F.J.S-L., a CSL centenary fellowship to G.J.F., and the Mater Foundation. Rhesus macaque tissues were obtained from the Monkey Alcohol Tissue Research Resource (MATRR) biobank, supported by NIH grant 2R24 AA019431.
Author contributions: V.B., F.J.S-L., J.R., and G.O.B. are equal coauthors. V.B., F.J.S-L., J.R., G.O.B., D.J.G., P.G., S.N.S., P.A., C.E.L., and K.A.N. performed experiments. V.B., F.J.S-L., J.R., S.W.C., T.J.M., N.J., S.R.R., A.D.E., and G.J.F. analyzed the data. G.O.B., J.L.G-P., S.R.R., L.C., and G.J.F. provided resources. V.B., F.J.S-L., A.D.E., and G.J.F. prepared figures. V.B., L.C., and G.J.F. conceived and designed the project. G.J.F. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276451.121.
-
Freely available online through the Genome Research Open Access option.
- Received March 2, 2022.
- Accepted June 14, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








