
Sequencing Illumina libraries at high accuracy on the ONT MinION using R2C2
- Alexander Zee1,4,
- Dori Z.Q. Deng2,4,
- Matthew Adams2,4,
- Kayla D. Schimke1,4,
- Russell Corbett-Detig1,
- Shelbi L. Russell2,
- Xuan Zhang3,
- Robert J. Schmitz3 and
- Christopher Vollmers1
- 1Department of Biomolecular Engineering, University of California Santa Cruz, Santa Cruz, California 95064, USA;
- 2Department of Molecular, Cellular, and Developmental Biology, University of California Santa Cruz, Santa Cruz, California 95064, USA;
- 3Department of Genetics, University of Georgia, Athens, Georgia 30602, USA
-
↵4 These authors contributed equally to this work.
Abstract
High-throughput short-read sequencing has taken on a central role in research and diagnostics. Hundreds of different assays take advantage of Illumina short-read sequencers, the predominant short-read sequencing technology available today. Although other short-read sequencing technologies exist, the ubiquity of Illumina sequencers in sequencing core facilities and the high capital costs of these technologies have limited their adoption. Among a new generation of sequencing technologies, Oxford Nanopore Technologies (ONT) holds a unique position because the ONT MinION, an error-prone long-read sequencer, is associated with little to no capital cost. Here we show that we can make short-read Illumina libraries compatible with the ONT MinION by using the rolling circle to concatemeric consensus (R2C2) method to circularize and amplify the short library molecules. This results in longer DNA molecules containing tandem repeats of the original short library molecules. This longer DNA is ideally suited for the ONT MinION, and after sequencing, the tandem repeats in the resulting raw reads can be converted into high-accuracy consensus reads with similar error rates to that of the Illumina MiSeq. We highlight this capability by producing and benchmarking RNA-seq, ChIP-seq, and regular and target-enriched Tn5 libraries. We also explore the use of this approach for rapid evaluation of sequencing library metrics by implementing a real-time analysis workflow.
Over the past 15 years, high-throughput short-read sequencing technology has revolutionized biological, biomedical, and clinical research. Hundreds of sequencing-based methods exist today to query gene expression (RNA-seq) (Mortazavi et al. 2008), chromatin state (chromatin immunoprecipitation [ChIP]-seq and ATAC-seq) (Barski et al. 2007; Buenrostro et al. 2013), protein abundance (Stoeckius et al. 2017) and, of course, to aid the assembly of genomes (Burton et al. 2013)—among many other applications. All of these methods produce a final sequencing library that contains ∼200- to 600-bp double-stranded DNA molecules with ends of a known sequence. In the vast majority of cases, these ends are Illumina sequencing adapters.
Despite the existence of other sequencing technologies, Illumina has been the dominating short-read sequencing technology over the past decade. However, because of the high capital cost of Illumina short-read instruments, all but the most well equipped laboratories outsource their Illumina sequencing to core facilities. Although this provides access to the most recent sequencing technology, this outsourcing can lead to long delays between running an experiment and receiving results. Therefore, placing a benchtop sequencer with capabilities comparable to an Illumina sequencer in most molecular biology and diagnostic laboratories could be truly transformative by accelerating as well as fully integrating genomics assays into standard laboratory workflows. In a molecular biology laboratory, it would speed up developing or establishing new types of sequencing libraries. In a diagnostic laboratory, it could enable fast sample turn-around as well as encourage the transition away from diagnostic methods like fluorescence in situ hybridization (FISH), which is still routinely used for the detection of gene fusions in certain cancers despite having a >20% false-negative rate and the availability of more accurate sequencing-based replacements (Ali et al. 2016; Nohr et al. 2019).
Over the past few years Oxford Nanopore Technologies (ONT) sequencers have rapidly matured. Currently, the ONT MinION sequencer's base throughput (up to 30 Gb per flowcell) can exceed that of the Illumina MiSeq sequencer (18 Gb for a 2 × 300-bp run). Additionally, this throughput comes with tunable read length, so a successful MinION run can in theory produce 10 million 3-kb reads or 5 million 6-kb reads. Further, the MinION sequencer is only a fraction of the cost of other high-throughput sequencers. However, standard per-base sequencing accuracy of the newest base-calling software guppy5 is only ∼96% and is dominated by insertion and deletion errors, which are almost absent in Illumina data. Furthermore, ONT MinION's sequencing accuracy declines with shorter reads (Thirunavukarasu et al. 2021).
Here, we implemented a simple workflow that converts almost any Illumina sequencing library into DNA of lengths optimal for the ONT MinION and generates data at similar cost and accuracy as the Illumina MiSeq. We made this possible by using the previously published and optimized rolling circle to concatemeric consensus (R2C2) method (Volden et al. 2018; Byrne et al. 2019; Adams et al. 2020; Cole et al. 2020; Vollmers et al. 2021; Volden and Vollmers 2022). R2C2 circularizes dsDNA libraries and amplifies those circles using rolling circle amplification to create long molecules with multiple tandem repeats of the original molecule's sequence. These long molecules can then be sequenced on ONT instruments to generate long raw reads, which are then computationally processed into accurate consensus reads. In previous studies focused on full-length cDNA molecules, we have achieved median read accuracies of 99.5% with this method (Vollmers et al. 2021). Because Illumina libraries are shorter than full-length cDNA, we modified the R2C2 protocol to generate a large number of shorter MinION raw reads while maintaining consensus accuracy levels on par with the Illumina MiSeq sequencer.
We benchmark this extension of the R2C2 method by converting and sequencing RNA-seq and ChIP-seq, as well as regular and target-enriched genomic DNA Tn5 Illumina libraries. We implemented a computational workflow for demultiplexing Illumina library indexes from R2C2 data and have, where possible, relied on established analysis workflows for downstream analysis originally developed for Illumina data. If R2C2 and Illumina data required different computational approaches, that is, assembly and variant calling, we chose the optimal tool for either data type.
To take advantage of the real-time data generation of ONT sequencers, we also developed Processing Live Nanopore Experiments (PLNK), for monitoring and rapid evaluation of sequencing runs. PLNK uses several tools to base-call, demultiplex, and map reads as they are generated. PLNK then reports, in real-time, run features like what percentages of reads belong to each library in a library pool, what percentages of reads in each library map to a list of target regions, and what the read coverage of these target regions is for each library.
This work was performed with the aim of evaluating whether R2C2 and the associated computational methods C3POa and PLNK could be used to replace and potentially even improve on dedicated Illumina sequencers for the analysis of short-read libraries.
Results
To generate R2C2 data for a diverse selection of Illumina libraries, we processed and sequenced (1) Illumina RNA-seq libraries of the human A549 cancer cell line, (2) Illumina ChIP-seq and input libraries of soybean samples, (3) Illumina Tn5-based genomic DNA libraries of a Wolbachia-containing Drosophila melanogaster cell line, and (4) Illumina Tn5-based genomic DNA libraries generated from the lung cancer cell lines NCI-H1650 and NCI-H1975, which we enriched for the protein-coding regions of approximately 100 cancer-relevant genes (Fig. 1).

Experiment overview. Illumina RNA-seq, ChIP-seq, and Tn5-based genomic libraries (regular and enriched) were generated from different samples. The Illumina libraries were then circularized and amplified using rolling circle amplification (RCA). The resulting DNA, containing tandem repeats of Illumina library molecules, was then prepared for sequencing on the ONT MinION sequencer.
To convert these Illumina libraries into R2C2 libraries, we circularized them using Gibson assembly (NEBuilder/NEB) with DNA splints compatible with Illumina p5 and p7 sequences (Supplemental Table S1). After the DNA circles are amplified with rolling circle amplification using Phi29 polymerase, we fragmented and size-selected the resulting high-molecular-weight DNA. We then sequenced this DNA on the ONT MinION using the LSK-110 ligation chemistry and 9.4.1 flowcells. We generated between 4 and 9.5 million raw reads per MinION flowcell (Table 1). All data were then base-called with the guppy5 dna_r9.4.1_450bps_sup.cfg model and consensus called using C3POa (v.2.2.3; https://github.com/rvolden/C3POa).
R2C2 sequencing run characteristics
To benchmark the R2C2 data for the Illumina libraries, we sequenced the same libraries with regular ONT 1D reads and on different Illumina sequencers. We then compared the metrics most relevant to the different library types.
Evaluating R2C2 for the sequencing of Illumina RNA-seq libraries
First, we benchmarked the ONT-based R2C2 method for the generation of RNA-seq data from Illumina libraries. We prepared four technical replicate libraries from a single RNA sample in the form of dual indexed paired-end Illumina libraries using the NEBNext Ultra II directional RNA kit with RNA of the human lung carcinoma cell line A549. We pooled and sequenced these libraries with the ONT MinION both directly (1D) and after R2C2 conversion (R2C2), as well as with the Illumina MiSeq.
To establish the effect of R2C2 conversion on the throughput of the ONT MinION when sequencing short Illumina libraries, we processed the raw reads generated by both 1D and R2C2 sequencing runs. Raw read numbers for 1D and R2C2 runs generated from one ONT MinION flowcell were similar at around 11.8 million reads. However, 1D reads were less likely than R2C2 reads to (1) pass filter during base-calling, (2) contain both p5 and p7 Illumina adapter sequences, and (3) be successfully demultiplexed. After preprocessing, only 2.5 million 1D reads (21%) remained compared with around 8 million R2C2 reads (Table 2).
R2C2 and 1D read numbers throughout processing steps
The 1D read numbers we generated for the RNA-seq libraries are similar to published 1D read numbers generated for libraries of similar lengths. A recent large-scale study on GTEx samples (Glinos et al. 2022) sequenced ∼600-nt-long cDNA molecules across dozens of flowcells and generated about 6 million reads per MinION flowcell. The Long-read RNA-seq Genome Annotation Assessment Project (LRGASP) Consortium (Pardo-Palacios et al. 2021) sequenced ∼520-nt-long cDNA molecules and generated about 18 million reads per flowcell. Even the most productive 1D run in these studies, potentially generating up to 20 million raw reads for molecules of this length (Pardo-Palacios et al. 2021), would still generate fewer demultiplexed reads (21% of 20 million or fewer than 5 million) than the R2C2 run we performed here.
To validate the demultiplexing of Illumina library pools from R2C2 data, we compared the ratio of reads assigned to each library in Illumina MiSeq, R2C2, and ONT 1D data based on their combination of i5 and i7 indexes. For all three methods, three technical replicate libraries were pooled at a 4:2:1 ratio. The Illumina MiSeq produced a 4:2.03:1.58 read ratio after demultiplexing. R2C2 produced a 4:1.91:1.34 ratio, and ONT 1D produced a 4:2.5:1.82 ratio. With these results being quite similar, the differences are likely owing to pipetting variability when pooling the libraries for the different sequencing methods. Further, to evaluate our ability to quantitatively pool libraries at different points in the R2C2 workflow, we processed a fourth replicate in parallel and added it at a specific ratio after rolling circle amplification. The fourth replicate represented 40.5% of the R2C2 data, which is slightly more than the 30% of R2C2 DNA it represented in the MinION sequencing run. Finally, 9.71% of the R2C2 reads were not assigned to any index combination and 1.7% of the R2C2 reads were assigned to index combinations not present in the pool, implying only 0.0289% (1.7% × 1.7%) of the R2C2 reads were assigned to the wrong index combination owing to index hopping.
Next, we established the effect of R2C2 conversion on read accuracy compared with ONT 1D and Illumina MiSeq data sets. We aligned all complete p5- and p7-containing and demultiplexed R2C2 (8,066,704) and 1D reads (2,530,950) as well as Illumina MiSeq reads (20,830,560 2 × 300-bp paired-end reads) generated from these RNA-seq libraries using minimap2. We then calculated the median read accuracy, accuracy per base, and read position–dependent accuracy per base (Table 3).
Sequencing error rates of different methods based on minimap2 alignments of all demultiplex reads
Although median read accuracy is a useful and often reported metric to compare error-prone long-read sequencing technologies, it becomes less useful in this study. The sequencing reads we aim to compare are very short—either owing to the short length of the molecules sequenced (1D and R2C2) (Fig. 2A) or to technology limitations (Illumina MiSeq)—and often accurate enough to be unlikely to contain errors at that length, causing many individual sequencing reads to be 100% accurate. This is obvious with read 1 of the Illumina MiSeq having a median accuracy of 100%, which contains little information on the real Illumina MiSeq error rate. Accuracy per base (%), that is, (correct bases of all reads/all bases of all reads) × 100, is a more useful metric to compare accurate short reads. Using this metric, we see that 1D reads are the least accurate with an accuracy per base of 96.59%. R2C2 falls between Illumina MiSeq read 1 (99.47%) and read 2 (98.57%) with an accuracy per base of 98.87%. Further, although R2C2 reads contained more deletion and insertion errors, they contained fewer mismatch errors than both Illumina MiSeq read 1 and read 2.
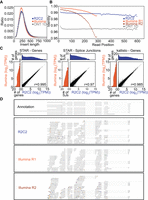
Sequencing Illumina RNA-seq libraries on the ONT MinION after R2C2 conversion. Insert length distribution (A) and read position–dependent identity to the reference genome (B) of R2C2 and Illumina MiSeq reads generated from the same Illumina library. (C) Comparisons of R2C2 and Illumina MiSeq read-based gene expression and splice junction usage quantification by STAR and kallisto are shown as scatter plots with marginal distributions (log2 normalized) shown as histograms. (D) Genome browser-style visualization of read alignments to the Actb locus. Mismatches are marked by lines colored by the read base (A, orange; T, green; C, blue; G, purple). Insertions are shown as gaps in the alignments, and deletions are shown as black lines.
Read position–dependent accuracy of 1D, R2C2, and Illumina MiSeq read 1 and read 2 adds further detail to this comparison. In contrast to 1D and R2C2 data, Illumina MiSeq base accuracy decreased with increasing read cycles, particularly in read 2, with R2C2 surpassing Illumina MiSeq accuracy for read 2 lengths over ∼175 bp (Fig. 2B,D). To ensure that our Illumina MiSeq run was not an outlier in terms of accuracy typical of Illumina benchtop sequencers, we performed the same position-dependent accuracy analysis on publicly available Illumina MiSeq, iSeq, and MiniSeq data, which showed the same overall trends (Supplemental Fig. S1).
Next, we aimed to establish whether R2C2 RNA-seq and ONT 1D data could be analyzed using computational tools designed and established for Illumina RNA-seq data. To quantify gene expression levels, we aligned and evaluated the entire demultiplexed R2C2 (8,066,704 reads) and ONT 1D (2,530,950 reads) data sets as well as our Illumina MiSeq data set (20,830,560 read pairs) using the STAR aligner (Dobin et al. 2013) (STARlong executable for R2C2 and ONT1D data), which is routinely used for standard Illumina RNA-seq analysis; 7,365,398 R2C2 reads (91.66%), 1,834,065 ONT 1D reads (72.48%), and 18,649,031 Illumina MiSeq reads (90.08%) mapped uniquely to the human genome. The STAR log files indicated that compared with R2C2 (1) the low overall accuracy of ONT 1D reads and (2) the declining quality of MiSeq reads means STAR aligner is less likely to align them. Although more forgiving aligners like minimap2 exist, they are not intended for spliced short-read alignments and therefore not optimized for this use-case.
Based on these read alignments, STAR determined normalized gene counts for the Illumina MiSeq, R2C2, and ONT 1D data sets. Illumina MiSeq gene counts showed Pearson's r-values of 0.995 and 0.987 compared with R2C2 (Fig. 2C) and ONT 1D, respectively. Additionally, STAR also determined normalized splice junction counts for the three data sets, which provide a higher-resolution view of the transcriptome. Illumina MiSeq splice junction counts showed Pearson's r-values of 0.974 and 0.929 compared with R2C2 (Fig. 2C) and ONT 1D. Finally, we also tested whether ultrafast pseudoalignment-based tools will generate reliable gene expression levels based on R2C2 and ONT 1D reads that feature more insertion and deletion rates compared with standard Illumina data. We used one such tool, kallisto (Bray et al. 2016), and found that gene expression values as determined for Illumina MiSeq had Pearson's r-values of 0.985 and 0.973 compared with R2C2 (Fig. 2C) and ONT 1D.
Overall, this comparison showed that using R2C2 we can convert Illumina RNA-seq libraries into DNA ideally suited for the ONT MinION. Not only does R2C2 generate more reads than regular ONT 1D ligation protocols, but R2C2 reads are also much more accurate. Because they are more accurate, R2C2 reads are also more efficiently demultiplexed and aligned than ONT 1D reads. Further, because they are similar in accuracy to Illumina reads, standard Illumina tools, like STAR and kallisto, can be used to analyze them. The gene expression and splice junction values generated by R2C2 are highly similar to those generated by Illumina MiSeq data from the same libraries.
Evaluating R2C2 for the sequencing of Illumina ChIP-seq libraries
Next, we tested the ability of R2C2 for the quality control of Illumina ChIP-seq libraries. To do this, we converted a previously generated ChIP-seq library targeting the H3K4me3 histone modification in a Glycine max (soybean) sample. The H3K4me3 library and its corresponding control Input library have previously been sequenced on an Illumina NovaSeq 6000 to a depth of 8,413,865 and 32,377,813 2 × 150-bp paired-end reads, respectively (Table 4). Based on their alignment, the sequenced molecule libraries had an insert length of 390 bp (H3K4me3) and 312 bp (Input) (Table 4).
ChIP-seq read characteristics
Because the H3K4me3 and Input libraries were prepared with only a single index distinguishing them, we converted the libraries separately with R2C2 using distinct DNA splints that contained unique index sequences. This added an extra level of indexing to minimize concerns of potential index cross talk. We splint-indexed and pooled the H3K4me3 and Input ChIP-seq Illumina libraries and sequenced the pool on a single ONT MinION flowcell. We then demultiplexed the resulting R2C2 reads, assigning 2,493,021 and 1,530,914 reads (1.6:1) to the H3K4me3 and Input libraries (Table 4), respectively, a ratio that corresponded well with the 1.35:1 ratio at which they were pooled before sequencing. Importantly, the demultiplexing script scored only 163,489 (3.9%) reads as “undetermined” and assigned only 4014 (0.1%) reads to a combination of indexes not present in the library. This indicated that the extra level of indexing was highly successful in minimizing index hopping.
The demultiplexed R2C2 reads showed median read accuracy of 99.23% (H3K4me3) and 98.8% (Input) as well as median read length of 556 bp (H3K4me3) and 459 bp (Input) (Table 4). Molecules sequenced by R2C2 were therefore longer than molecules sequenced by the Illumina NovaSeq 6000 (Fig. 3A). The difference between the technologies is likely owing to the bias of the Illumina NovaSeq toward shorter molecules.
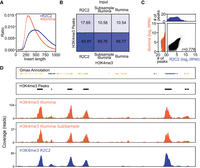
Sequencing ChIP-seq libraries on the ONT MinION after R2C2 conversion. (A) Insert length distribution of R2C2 and Illumina NovaSeq 6000 reads generated from the same Illumina library. (B) Percentage of reads in the R2C2, subsampled Illumina, and full Illumina data sets overlapping with H3K4me3 peaks generated from the full Illumina H3K4me3 data set using MACS2. (C) The comparison of the number of R2C2 and subsampled Illumina reads overlapping with H3K4me3 peaks is shown as scatter plots with marginal distributions shown as histograms. Pearson's r is shown at the bottom right. (D) Genome annotation, H3K4me3 peak areas, and read coverage histograms are shown for a section of the Gmax genome.
To test whether R2C2 reads could replace the same number of Illumina reads, we subsampled the Illumina sequencing data to the depth of the R2C2 data for both samples. We then aligned both Illumina NovaSeq 6000, subsampled Illumina NovaSeq 6000, and R2C2 reads to the G. max genome (Gmax_508_v4.0) (Valliyodan et al. 2019). For alignment, we chose the short-read preset of the minimap2 (Li 2018) aligner for both the Illumina and R2C2 data. We then called peaks on the full H3K4me3 Illumina NovaSeq 6000 data set using MACS2 and tested whether both the subsampled Illumina NovaSeq 6000 and R2C2 data could be used to evaluate the success of a ChIP experiment. Visual inspection of the data using the Phytozome JBrowse genome browser (Goodstein et al. 2012) and our own tools (Fig. 3D) showed that the subsampled Illumina NovaSeq 6000 and R2C2 data both showed the same enrichment patterns as the full Illumina NovaSeq 6000 data. A systematic analysis showed that 84% of the R2C2 reads and 69% of the subsampled Illumina reads overlap with an H3K4me3 peak identified on the full Illumina data, whereas only 18% and 11% of the respective Input reads do so (Fig. 3B).
To investigate this discrepancy in percentage of reads overlapping with H3K4me3 peaks, especially for the H3K4me3 library, we focused on differences between the R2C2 and Illumina sequencing reads. The most obvious difference is the read length with the Illumina reads originating from much shorter molecules (or library inserts). Indeed, when we recalculated this read percentage for Illumina reads originating from inserts >450 nt, it increased to 76%. Next, we analyzed the GC content of Illumina and R2C2 reads and found that—in contrast to all other experiments in this paper (Supplemental Fig. S2)—Illumina reads had a lower GC content than R2C2 reads (39% vs. 42%). To see whether the difference in insert length and GC content together would explain the discrepancy in percentage of reads overlapping with H3K4me3, we again recalculated this read percentage only for Illumina reads originating from inserts >450 nt and with a GC content >39%, that is, reads derived from long and GC-rich molecules. Here, we found that this read percentage increased to 83.2%, virtually matching the R2C2 percentage. Ultimately, this suggested that R2C2 sampled longer and slightly more GC-rich molecules from the ChIP-seq libraries. Although it is not clear why the longer molecules are more likely to overlap with H3K4me3 peaks, these peaks happen to be more GC-rich than the rest of the genome (40% vs. 30%), explaining why more GC-rich molecules are more likely to overlap with H3K4me3 peaks.
To compare whether the R2C2 and subsampled Illumina NovaSeq 6000 data sets are also similar quantitatively, we counted how many reads for each of the data sets fell into each H3K4me3 peak we identified using the full Illumina NovaSeq 6000 data set and MACS2. We found that the peak depths are correlated (Pearson's r = 0.776) (Fig. 3C). This correlation is increased to r = 0.866 when this analysis was performed with the longer/more GC-rich subsample of Illumina reads but remained lower than what we observed with the RNA-seq data. This means that although R2C2 can be used to evaluate whether a ChIP-seq experiment successfully enriched targeted chromatin, in this particular experiment, R2C2 sampled a different population of molecules than the Illumina NovaSeq 6000, thereby complicating quantitative comparisons.
Evaluating R2C2 for the sequencing of size-selected Illumina Tn5 libraries
In contrast to the other parts of the paper, which represent head-to-head comparisons between R2C2- and Illumina-based sequencing of the same short-read libraries, here, we tested whether the ability of R2C2 to sequence “medium-length” molecules >600 nt could aid in small genome assembly tasks. Illumina library preparation methods like Tn5-based tagmentation can generate library molecules >600 nt, which are too long to be sequenced efficiently by Illumina sequencers but can be efficiently processed and sequenced using R2C2. To generate these medium-length molecules for the purpose of genome assembly, we chose to size-select a Tn5-based Illumina library for molecules between 800 and 1200 bp in length, corresponding to genomic DNA inserts of ∼600–1000 bp. We then R2C2-converted and sequenced this size-selected library on the ONT MinION.
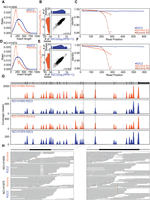
For this test, we chose to sequence the 1.2-Mb genome of the Wolbachia bacterial endosymbiont of D. melanogaster and prepared Tn5 libraries from DNA extracted from Wolbachia-containing D. melanogaster S2 cells. We generated a total of 3,338,280 R2C2 consensus reads with a median length of 680 bp. Out of these reads, we assembled 879,303 reads that did not align to the D. melanogaster genome. We used miniasm (Li 2016) for this assembly task and polished the resulting assembly using Medaka (v.1.4.4; https://github.com/nanoporetech/medaka). The resulting assembly contained 95 contigs, which covered 97.2% of the Wolbachia genome (Fig. 4A,B), and had a NGA50 of 29,963 bp and 8.5/5.6 mismatches/indels per 100 kb of sequence.

Comparing R2C2 and Illumina based assemblies of a small genome. Illumina 2 × 150 reads were assembled in 134 contigs using Meraculous. R2C2 reads were assembled using miniasm into 95 contigs. The alignments of the contigs of both assemblies—(A) Illumina and (B) R2C2—are shown as dot plots generated by MUMmer (Kurtz et al. 2004). Both approaches failed to assemble a section of the Wolbachia genome that contains pseudogenes and a transposable element near coordinate 500,000.
We also generated an assembly from Illumina NextSeq 2 × 150 bp reads from a non-size-selected Tn5 library of the same cell line. From 2,552,018 2 × 150-bp Illumina reads, we extracted 779,206 reads that did not align to the D. melanogaster genome and assembled those reads using Meraculous (Chapman et al. 2011). The resulting assembly contained 136 contigs that covered 91.6% of the Wolbachia genome (Fig. 4) and had a NGA50 of 23,217 bp and 0.5/0.6 mismatches/indels per 100 kb of sequence. Neither assembly had misassemblies as determined by QUAST (Gurevich et al. 2013).
Comparing Illumina and R2C2 assemblies of the Wolbachia genome (NC_002978.6) showed R2C2 can generate more contiguous and complete assemblies from the same library type (Fig. 4A,B). However, systematic errors produced by the ONT MinION cannot be fully removed by the R2C2 consensus process or Medaka polishing. The assembly we generated, therefore, does have more mismatches and indel errors than its Illumina counterpart. This ultimately suggests that when limited to a single Tn5 library owing to sample constraints, R2C2 can be a valuable addition to an assembly effort, but depending on use-case, further polishing with Illumina data might be required to achieve the desired base accuracy.
Evaluating R2C2 for the sequencing of target-enriched Illumina Tn5 libraries
We tested the ability of R2C2 to evaluate target-enriched Tn5 libraries and benchmark our ability to detect germline variants in the resulting data. To this end, we generated dual-indexed Tn5 libraries from genomic DNA of two cancer cell lines (NCI-H1650 and NCI-H1975) with known mutations in the EGFR gene. We pooled these libraries and enriched the pool for a panel of cancer genes based on the Stanford solid tumor STAMP panel (Newman et al. 2014) using a Twist Bioscience oligos panel and reagents (Supplemental Table S2). We performed this enrichment experiment once, without optimization, and using custom blocking oligos, therefore expecting enrichment to be far from optimal. To compare R2C2 and Illumina MiSeq, we sequenced these enriched Tn5 libraries on (1) a multiplexed Illumina MiSeq 2 × 300-bp paired-end run and (2) an ONT MinION after R2C2 conversion.
The multiplexed MiSeq run generated 7,430,624 read pairs for the NCI-H1650 library and 1,142,187 read pairs for the NCI-H1975 library. The ONT MinION run generated 3,825,657 R2C2 reads after C3POa processing. Demultiplexing then assigned 2,057,155 (53.7%) R2C2 reads to the NCI-H1650 library and 1,021,758 (26.7%) R2C2 reads to NCI-H1975. Although 537,997 (14.1%) R2C2 reads were not assigned to any sample, only 5.4% of reads were assigned to one of the two combinations of Illumina indexes not included in the pool, implying that only 0.29% (5.4% × 5.4%) of reads were assigned to the wrong sample in our dual indexed library.
After demultiplexing, we compared the insert length and target enrichment across samples and methods. We did so by merging the Illumina MiSeq read pairs using bbmerge (Bushnell et al. 2017). As with the ChIP-seq experiment, R2C2 data showed longer insert lengths than the Illumina MiSeq data, with the R2C2 insert length more closely resembling the actual length of the input library (Fig. 5A,D; Supplemental Fig. S3). We aligned the reads of different samples and methods to the human genome using the short-read preset of minimap2 and determined the percentage of reads overlapped with a target region and the coverage for each region. For NCI-H1650, 15.8% of R2C2 reads and 14.4% of Illumina MiSeq reads overlapped with a target region, producing a median coverage of 128 (fifth percentile: 28; 95th percentile: 310) for R2C2 and 558 (fifth percentile: 134; 95th percentile: 1220) for Illumina MiSeq. For NCI-H1975, 18.5% of R2C2 reads and 16.8% of Illumina MiSeq reads overlapped with a target region with a median coverage of 69 (fifth percentile: 13; 95th percentile: 166) for R2C2 and 110 (fifth percentile: 23; 95th percentile: 225) for Illumina MiSeq. The per-base coverage of the R2C2 and Illumina MiSeq data sets was very well correlated within samples, with NCI-H1650 showing a Pearson's r = 0.91 and NCI-H1975 showing a Pearson's r = 0.89 (Fig. 5B,E).
Evaluating target-enriched Tn5 libraries with R2C2. (A,D) Insert length of library molecules sequenced by Illumina or R2C2 approaches. (B,E) Comparison of per-base coverage in the Illumina and R2C2 data sets. Marginal distributions are log2 normalized. (C,F) Alignment-based read position–dependent accuracy shown for the indicated sequencing reads and methods. (G,H) Sequencing coverage plot of the target-enriched Tn5 libraries for R2C2 and Illumina results at Chromosome 7: 55,134,584–55,211,629, which covers a part of the EGFR gene. Top panel shows the annotation of one EGFR isoform. The x-axis of the coverage plot is the base pair position, and the y-axis is the total number of reads at each position. The dotted lines indicate zoomed-in views of exons that contain the 15-bp deletion in NCI-H1650 (left) and the C-to-T and T-to-G point mutations in NCI-H1975 (right). Both samples’ Illumina reads and the R2C2 read alignments of the selected regions are shown. The mismatches are colored based on the read base (A, orange; T, green; C, blue; G, purple).
Next, we used the read alignments to determine per-base accuracy levels for all samples and method combinations. The NCI-H1975 sample, which also produced fewer reads than expected on the Illumina MiSeq, produced reads at a lower-than-expected accuracy. Read alignments suggested that the average per-base accuracies for read 1 and read 2 in NCI-H1975 were 96.81% and 98.26% compared with 98.37% and 97.88% for NCI-H1650. As expected, the per-base accuracy was highly position dependent and declined with increasing sequencing cycle number (Fig. 5C,F). Furthermore, the actual accuracy of the MiSeq reads is likely even lower owing to alignments not being extended once the read and genome are too dissimilar. The accuracy of the R2C2 reads in both NCI-H1975 and NCI-H1650 was similar and stable throughout the reads at 98.40% and 98.28%, meaning that, in this case, the R2C2 reads had a higher per-base accuracy than the combined MiSeq reads.
Visualizing Illumina MiSeq and the R2C2 read alignments showed that both methods successfully enriched for (Fig. 5G) and detected the 15-bp heterozygous deletion in the EGFR gene in the NCI-H1650 cell line and the C-to-T heterozygous variants in the EGFR gene in the NCI-H1975 cell line (Fig. 5H). To systematically evaluate the germline variant detection ability of Illumina MiSeq and R2C2 reads, we used DeepVariant (Poplin et al. 2018) for calling germline variants based on the Illumina MiSeq data and used PEPPER-Margin-DeepVariant (Shafin et al. 2021), a variant caller designed for ONT data sets, for calling germline variants in the R2C2 sequencing results. Because of the poor sequencing performance of the Illumina MiSeq for the NCI-H1975 library, we only performed this analysis on NCI-H1650. For NCI-H1650, Illumina/DeepVariant detected 119 variants in the enriched genomic regions when using a QUAL cut-off of 33.3 or more. R2C2/PEPPER-Margin-DeepVariant detected 122 variants in the enriched genomic regions when using a QUAL score of 3.8 or more, including 117 of the 119 Illumina/DeepVariant calls. When we used Illumina/DeepVariant variants as a ground truth, the R2C2/PEPPER-Margin-DeepVariant method achieved 95.9% precision and 98.3% recall.
When we visualized the reads on which the false-positive and false-negative R2C2/PEPPER-Margin-DeepVariant variant calls were made (Supplemental Fig. S4), we found that the false-positive variants were supported by fewer than half of the R2C2 reads. Moreover, when we colored the reads based on the direction of their raw reads, we found that false-positive variants were supported only by reads originating from one raw read direction. We hypothesized that if we oriented reads using the direction of their raw reads—instead of using the p5 and p7 adapters on their ends—before variant calling, it would more closely resemble regular ONT reads and provide more useful information to PEPPER-Margin-DeepVariant. Indeed, when reanalyzing the reoriented reads and using a QUAL score of nine or more, PEPPER-Margin-DeepVariant detected 116 variants that were all present in the Illumina/DeepVariant calls. This means that reorienting the reads before variant calling eliminated all false positives in the R2C2/PEPPER-Margin-DeepVariant variant calls. Reflecting known systematic errors of ONT sequencers, two of the three false negatives missing from the R2C2/PEPPER-Margin-DeepVariant variant calls were a deletion (TA → T) next to a 13-nt A homopolymer at Chr 17: 7,667,260 and a variant (G → C) next to an 8-nt C homopolymer at Chr 12: 120,994,314. The third missing variant, a G → A call at Chr 5: 112,839,666, had a 46% frequency in both Illumina and R2C2 reads and was initially identified as a candidate by PEPPER-Margin-DeepVariant but was ultimately scored as a “RefCall,” not a variant. Overall, reorienting the reads by raw read direction before running PEPPER-Margin-DeepVariant increased precision to 100% while achieving a recall of 97.4%.
This showed that R2C2 can accurately quantify what percentage of molecules in an enriched Tn5 Illumina library overlap with a target region. Despite showing longer insert lengths than the Illumina MiSeq data set, the R2C2 data set showed per-base coverage that was highly correlated with the Illumina MiSeq data. In this experiment, R2C2 actually showed a higher average per-base accuracy than the Illumina MiSeq. After reorienting R2C2 reads, variants called based on the R2C2 and Illumina MiSeq data were very similar. This shows the promise of variant calling based on ONT data but also highlights that extra care has to be taken when preparing data for use in neural network–based variant callers like DeepVariant.
Real-time analysis of Illumina library metrics using PLNK
To enable the real-time monitoring of sequencing runs and the rapid evaluation of metrics of libraries sequenced in those runs, we created the computational pipeline Processing Live Nanopore Experiments (PLNK). PLNK controls real-time base-calling, raw read processing into R2C2 consensus reads, demultiplexing of R2C2 reads, and the alignment of demultiplexed R2C2 reads to a genome. Based on the resulting alignments and the user-defined regions of interest, PLNK then determines the on-target percentage and resulting target coverage for each demultiplexed sample. PLNK runs alongside a MinION sequencing run, tracking the creation of new FAST5 files and processing them individually in the order they are generated. To do this, PLNK controls several external tools: guppy5 for base-calling, C3POa for R2C2 consensus generation, a separate Python script for demultiplexing (based on splint sequences and Illumina indexes), and mappy (minimap2 Python library) for aligning reads to a provided genome (Fig. 6A).
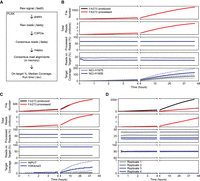
Real-time characterization of Illumina sequencing libraries. (A) Diagram of PLNK functionality; FAST5 files processed in the order they are produced. PLNK controls guppy5 for base-calling, C3POa for consensus calling, and mappy for alignment, as well as calculates metrics based on those alignments. (B–D) Simulation of real-time analysis for enriched Tn5 (B), ChIP-seq (C), and RNA-seq (D) libraries. For each time point, panels from top to bottom show (1) the number of FAST5 files that are produced and processed, (2) the number of demultiplexed reads produced by guppy5/C3POa/demultiplexing, (3) the percentage of reads associated with each library in the sequenced pool, (4) the percentage of reads overlapping with target regions, and (5) the median read coverage of bases in the target regions.
To test whether our pipeline could keep up with ONT MinION data generation and provide real-time analysis, we simulated ONT MinION runs using FAST5 files from previously completed sequencing experiments, our Tn5, ChIP-seq, and RNA-seq data. We used the FAST5 files’ metadata to determine the time intervals at which files were generated by the MinKnow software and copied the FAST5 files to a new output directory at those intervals. We then started PLNK to monitor the generation and control the processing of FAST5 files in this new output directory. First, we simulated the real-time analysis of the target-enriched Tn5 data. Using a desktop computer and limiting PLNK to the use of eight CPU threads and two Nvidia RTX2070 GPUs, the pipeline processed sequencing data at the same rate that a single MinION produced FAST5 files. Importantly, both the library composition (percentage of demultiplexed reads assigned to either sample; NCI-H1650 and NCI-1975), as well as the percentage of reads on-target, stabilized after less than an hour and agreed very well with the numbers generated from the whole data set (Fig. 6B). Additionally, throughout the run, PLNK reported the overall coverage of target regions in real-time.
When we simulated the analysis of ChIP-seq and RNA-seq experiments, PLNK kept up with ChIP-seq but not with the RNA-seq experiment (Fig. 6C,D). Because the RNA-seq experiment produced the largest amount of data in the study, this was not unexpected. In both cases, however, library composition and on-target percent both stabilized within the first hour of sequencing and reflected the number derived from the complete data set. This means that the library composition and quality of target-enriched Tn5 libraries (as measured by reads overlapping target areas), ChIP-seq libraries (as measured by reads overlapping with peak areas, promoters, or gene bodies—depending on targeted histone mark), and RNA-seq libraries (as measured by reads overlapping with exons) can be determined with minimal sequencing time.
The bottleneck for analysis in our desktop computer setup seemed to be the guppy5-based base-calling using the slower yet most accurate “sup” base-calling configuration. Although we could use a faster, less accurate setting to keep up with even the fastest data-producing experiments, using the most accurate model means the data can be used for in-depth analysis once the run has completed and PLNK has processed all the files, without the need to re-base-call the raw data.
Overall, this suggests that PLNK can be used to monitor ONT sequencing runs in real-time. This makes it possible to stop ONT sequencing runs when the goal of an experiment is achieved. For the rapid evaluation of library pools, this could be 1 h into a run once library composition and quality metrics have stabilized. For run monitoring, this could be several hours into a run once a specific coverage of defined target regions is reached. In both cases, a run can be stopped, allowing the ONT MinION flowcell to be flushed, stored, and ultimately reused.
Discussion
The capabilities of the dominant Illumina sequencing technology—producing massive numbers of short reads—have shaped the development of sequencing-based assays more than any other single factor.
Although long-read sequencers by Pacific Biosciences (PacBio) and ONT have now superseded Illumina instruments as the gold standard technology for genome assembly, producing libraries for these long-read sequencers requires relatively large amounts of high-quality DNA material. In many cases, both DNA input amount and/or quality of a sample may not match these requirements, leaving amplification-based short-read sequencing as the only option to extract large amounts of sequencing data from that sample.
Beyond the sequencing and assembly of genomes, there are hundreds of assays adapted for short reads. These assays are highly diverse and require different levels of read numbers and accuracy, and many, like standard RNA-seq, ChIP-seq, or targeted sequencing of PCR amplified genomic DNA, are unlikely to ever take advantage of the raw read length ONT and PacBio sequencers provide. However, there have been several studies to take advantage of long-read sequencing instruments in sequencing shorter molecules. Some assays (OCEAN, MAS-Iso-Seq) work by either concatenating (Al'Khafaji et al. 2021; Thirunavukarasu et al. 2021) or otherwise preparing (Baslan et al. 2021) short molecules for sequencing on the PacBio or ONT instrument. Although these assays can generate more short reads, they have to contend either with the high cost of the PacBio Sequel IIe sequencer or with the low per-base accuracy of raw ONT reads, which even with the latest guppy5 algorithm is only 96% in our hands. Even at 96%, this ONT raw accuracy is likely sufficient for certain applications like ChIP-seq, where reads simply have to be aligned to a genome and counted. For these applications, preparing and sequencing short-read libraries directly on an ONT sequencer is a straightforward option. This approach would also allow the usage of native ONT barcoding strategies, which are more robust at low accuracy. However, sequencing short-read libraries directly on ONT sequencers has the downside that these sequencers have reduced output when sequencing short molecules <1 kb. Specifically, when sequencing molecules that are ∼500 nt in length, the overall base-output of an ONT MinION flowcells seems to vary between 3 Gb (Glinos et al. 2022), 4 Gb (this study), and 9 Gb (Pardo-Palacios et al. 2021)—far below the 30 Gb maximum output these flowcells can achieve when sequencing longer molecules. There is therefore room to optimize ONT library preparations for short-read sequencing.
Taking inspiration from the highly accurate but throughput-limited PacBio Iso-Seq and HiFi workflows, circularizing-based (R2C2, INC-seq, and HiFRe) (Li et al. 2016; Volden et al. 2018; Wilson et al. 2019) methods have been developed to trade throughput for accuracy on ONT MinION and PromethION sequencers. Using a modified R2C2 method we present here, we show that we can convert any Illumina sequencing library with double-stranded adapters—PCR-free “crocodile adapter”–style libraries will not work—into an R2C2 library that is several kilobases long and therefore takes full advantage of the ONT MinION's throughput. The close to optimal base-output of up to 24 Gb (9.5 million raw reads × 2.5-kb average read length) when sequencing R2C2 libraries allowed us to produce not only more accurate reads but also a higher number of total reads than regular ONT 1D libraries of the same short-insert Illumina libraries. In fact, the throughput and accuracy of R2C2 were comparable to Illumina MiSeq 2 × 300-bp runs.
By generating up to 8.99 million reads (8.1 million demultiplexed) with a per-base accuracy of 98.87% (Illumina MiSeq read 1: 99.47%; read 2: 98.57%) from a single ONT MinION flowcell, this approach can compete with the Illumina MiSeq and other benchtop Illumina sequencers on accuracy and cost—even without taking instrument cost into account (Supplemental Table S3). Improved consensus tools (Silvestre-Ryan and Holmes 2021), the consistently improving ONT sequencing chemistry and base-callers, and the imminent release of a much cheaper ONT PromethION variant (P2Solo) all have the potential to further skew both accuracy and throughput comparison in R2C2's favor in the near future. Not only might improving ONT sequencing chemistry improve throughput, but it might also mitigate the considerable variability in throughput we see in R2C2 read output (4 to 9 million reads).
We have shown the capabilities and limitations of this approach here by evaluating the conversion of RNA-seq, ChIP-seq, genomic Tn5, and target-enriched genomic Tn5 libraries. The R2C2 data were more than accurate enough to demultiplex Illumina libraries based on their i5 and i7 indexes. Furthermore, RNA-seq data produced with R2C2 were almost entirely interchangeable with data produced by the Illumina MiSeq. Library metrics derived from R2C2 data generated from ChIP-seq and target-enriched Tn5 libraries showed library metrics very similar to those determined from data generated by Illumina sequencers. One notable exception to this were insert length distributions of Illumina libraries, where R2C2 produced longer insert distributions than Illumina sequencers, which are known to prefer shorter molecules enough to affect analysis outcomes (Gohl et al. 2019). For the ChIP-seq experiment, but no other experiment in this paper (Supplemental Fig. S3), R2C2 reads also had a slightly higher GC content, which made the Illumina/R2C2 comparison less quantitative than it was, for example, in the RNA-seq experiment. For germline variant calling, R2C2 reads analyzed with PEPPER-Margin-DeepVariant produced variant calls highly similar to Illumina/DeepVariant variant calls, with no false positives (precision 100%) and only three false negatives (recall 97.4%), two of which were next to homopolymers, which are known to be a challenge for ONT sequencers.
Taken together, we have established that R2C2 can be used as a drop-in replacement for many sequencing-based applications that would usually demand a dedicated short-read Illumina sequencer. One important thing to note is that R2C2 adds complexity to an ONT experiment. Sequencing a short-read library using regular 1D ONT sequencing at most requires the library to be PCR amplified to reach the 1 μg input requirement of ONT library preparations. In contrast, R2C2 is a multistep protocol that, while requiring little hands-on time, is composed of circularization (1 h), linear DNA removal (1–6 h), rolling circle amplification (overnight), and debranching (2 h) followed by size-selection (1 h). However, R2C2 uses only off-the-shelf reagents and requires no special equipment, meaning that performing a pilot experiment to establish whether R2C2 would be superior to 1D reads and a good replacement for any particular short-read assay should be possible for the vast majority of molecular biology laboratories.
Pilot experiments might be required for new library types because converting short-read libraries with R2C2 and sequencing them on an ONT sequencer may change what molecules in a pool will be sequenced. This is a consequence of R2C2 requiring several processing steps and ONT sequencers featuring a unique underlying technology that is totally distinct from Illumina or any other short-read sequencing technology. For example, in some experiments, R2C2/ONT sampled longer molecules than Illumina sequencers. Further, in the ChIP-seq experiment alone, those longer reads were also more GC-rich. Additionally, applications in which very high read and/or consensus accuracy is required, for example, somatic variant calling, will pose a challenge for R2C2. In essence, before R2C2 is used for a short-read experiment, the requirements for this experiment should be carefully considered.
In addition to Illumina libraries, the R2C2 method can also be easily adapted to libraries generated for one of several other sequencing instruments now entering the market, simply by modifying the splint used to circularize the library. As part of our C3POa tool, we now provide a script that designs splints and the oligos needed to make them for any amplified sequencing library based on the primers used to amplify it.
Beyond simply competing with benchtop sequencers like the Illumina MiSeq, R2C2 can be used for a new group of assays around “medium-length” 600- to 2000-nt reads. Libraries with insert lengths of this size can be size-selected from standard Illumina library preparations, and R2C2 is easily adapted to libraries with different insert lengths by modifying the size-selection of its rolling circle amplification product to include only molecules bigger than three to four times the original library size. We provided one example of the resulting “medium-length” R2C2 reads by analyzing size-selected Tn5 libraries. We showed that these reads can, for example, provide an advantage for the sequencing of small genomes. Among many other potential applications, “medium-length” reads could be applied to standard fragmentation-based RNA-seq libraries to provide more contiguous splicing information for very long transcripts (>15 kb) where full-length cDNA-based approaches fail.
One of the unique strengths of ONT-based sequencing methods is that, beyond the standard approach of analyzing sequencing runs once they are completed, many library metrics can be derived in real-time. This is starting to get exploited in clinical and metagenomics assays with tools like SURPIrt (Gu et al. 2021) or with more powerful tools like MinoTour (Munro et al. 2021). The PLNK tool we developed here is therefore a powerful tool to monitor sequencing runs and can be used for the rapid evaluation of library metrics. This makes it possible to stop a run once a predetermined target coverage is reached or once it is clear whether a library construction and pooling was successful. For example, using PLNK, we showed that key metrics of the RNA-seq, ChIP-seq, and enriched Tn5 libraries can be evaluated in under 1 h of sequencing, making it possible to flush, store, and reuse the flowcells used for these experiments.
In summary, we have shown that, using R2C2, the ONT MinION can—with some limitations—be used as an accurate short-read sequencer with several advantages over dedicated short-read sequencers. Because the ONT MinION comes with minimal instrument cost, R2C2 allows standard short-read genomic assays to be performed in any laboratory immediately after a library is produced. The use-cases for this, just as the many use-cases for Illumina benchtop sequencers, will vary from laboratory to laboratory. For laboratories performing small-scale experiments—like RNA-seq of a few samples—the R2C2/ONT MinION combination should be entirely sufficient. For laboratories performing large-scale experiments—like ChIP-seq of dozens of samples—the R2C2/ONT MinION combination should be useful to rapidly evaluate library pool compositions and metrics before committing to the cost and turnaround time that deeply sequencing a library pool at a core facility on an Illumina HiSeq or NovaSeq 6000 requires.
In either case, the presence of a capable short-read sequencer in most molecular biology or clinical laboratories could be truly disruptive by eliminating long turnaround times and therefore accelerating experiments.
Methods
Library preparation
RNA-seq
Four RNA-seq libraries were prepared with the NEBNext Ultra II directional RNA library prep kit for Illumina (NEB E7760) following the manufacturer's protocol. For each library, 100 ng of poly(A) selected RNA from the human lung carcinoma cell line A549 (Takara 636141) was used as input. The RNA fragmentation step was performed for 5 min at 94°C. PCR enrichment of adaptor ligated DNA was performed for nine cycles using the NEBNext multiplex oligos for Illumina (NEB E7600S) kit to add Illumina dual index sequences. Three libraries were pooled at a 4 ng, 2 ng, and 1 ng before sequencing on an Illumina MiSeq instrument for paired-end 2 × 300-bp sequencing. The same three RNA-seq libraries were pooled again at the same ratio for further R2C2 library preparation. For the 1D and R2C2 runs, the fourth RNA-seq library was prepared and added right before ONT library preparation.
ChIP-seq
ChIP was performed following the detailed protocol of Ricci et al. (2020) with minor modification. In brief, approximately 30 developing seeds at the cotyledon stage were used for chromatin extraction. Immediately after harvesting, the tissue was crosslinked as described in the referenced protocol and immediately flash-frozen in liquid nitrogen. To make antibody-coated beads, 25 μL Dynabeads Protein A (Thermo Fisher Scientific 10002D) were washed with ChIP dilution buffer and then incubated with 2 μg antibodies (anti-H3K4me3, Millipore-Sigma 07-473) for at least 3 h at 4°C. After the nuclei extraction, the lysed nuclei suspension was sonicated to 200–500 bp on a Diagenode Bioruptor on the high setting for 30 min. Tubes were centrifuged at 12,000g for 5 min at 4°C, and the supernatant was transferred to new tubes. At this point, 10 μL of ChIP input aliquots was collected. Sonicated chromatin was diluted 10-fold in the ChIP dilution buffer to bring the SDS buffer concentration down to 0.1%. The diluted chromatin was incubated with antibody-coated beads overnight at 4°C, washed, and reverse-crosslinked. The library was prepared in accordance with the referenced protocol.
Tn5
Genomic DNA from a Wolbachia-containing D. melanogaster cell line was extracted using a lysis-buffer plus SPRI-bead purification. The Tn5 reaction was then performed using 1 μL (22 ng) of this genomic DNA, 1 μL of the loaded Tn5-AR, 1 μL of the loaded Tn5-BR, 13 μL of H2O, and 4 μL of 5× TAPS-PEG buffer and incubated for 8 min at 55°C (Supplemental Table S1). The Tn5 reaction was inactivated by cooling down to 4°C and the addition of 5 µL of 0.2% sodium dodecyl sulfate and then incubated for 10 min. Five microliters of the resulting product was nick-translated for 5 min at 72°C and further amplified using KAPA Hifi Polymerase (KAPA) using Nextera Index primers with an incubation for 30 sec of 98°C, followed by 16 cycles of (20 sec at 98°C, 15 sec at 65°C, 30 sec at 72°C) with a final extension of 5 min at 72°C. Before R2C2 conversion, the resulting Tn5 library was size-selected for molecules between 800 and 1200 bp on a 1% low-melt agarose gel.
Target-enriched Tn5
The Tn5 library was prepared using genomic DNA from cell lines NCI-H1650 (ATCC CRL-5883D) and NCI-H1975 (ATCC CRL-5908DQ). A total of 100 ng genomic DNA of each sample was treated with Tn5 enzyme loaded with Tn5ME-A/R and Tn5ME-B/R. The Tn5 reaction was performed using 1 μL of the gDNA, 1 μL of the loaded Tn5-AR, 1 μL of the loaded Tn5-BR, 13 μL of H2O, and 4 μL of 5× TAPS-PEG buffer and incubated for 8 min at 55°C. The Tn5 reaction was inactivated by cooling down to 4°C and the addition of 5 µL of 0.2% sodium dodecyl sulfate and then incubated for 10 min. Five microliters of the resulting product was nick-translated for 5 min at 72°C and further amplified using KAPA Hifi Polymerase (KAPA) using Nextera_Primer_B_Universal and Nextera_Primer_A_Universal (Smart-seq2) with an incubation for 30 sec at 98°C, followed by 16 cycles of (20 sec at 98°C, 15 sec at 65°C, 30 sec at 72°C) with a final extension of 5 min at 72°C.
The resulting Tn5 library was then enriched with Twist fast hybridization reagents and customized oligo panels that were designed based on the Stanford STAMP panel. The hybridization reaction of the panel and the Tn5 libraries was performed using 294 ng of NCI-H1975 Tn5 library, 360 ng of NCI-H1650 Tn5 library, 8 μL of blocking oligo pool (100 μM), 8 μL of universal blockers, 5 μL of blocker solution, and 4 μL of the custom panel. The mix was dehydrated using a SpeedVac and was resuspended in 20 μL fast hybridization mix at 65°C. After the addition of 30 μL of hybridization enhancer, the mixture was incubated for 5 min at 95°C and for 4 h at 60°C. After hybridization, the reaction mix was incubated with prewashed streptavidin binding beads and washed using the fast wash buffer one and fast wash buffer two for six times. The streptavidin beads and the DNA mixture were used directly for reamplification with universal primers and Equinox library amp mix. The mixture was incubated for 45 sec at 98°C, followed by 16 cycles of (15 sec at 98°C, 30 sec at 65°C, 30 sec at 72°C) with a final extension of 1 min at 72°C. The final enriched Tn5 library DNA product was cleaned up using SPRI beads at 1.8:1 (beads:sample) ratio.
R2C2 conversion
Pooled Illumina libraries were first circularized by Gibson assembly with a DNA splint containing end sequences complementary to ends of Illumina libraries (Supplemental Table S1). Illumina libraries and DNA splint were mixed at a 1:1 ng ratio using NEBuilder HiFi DNA assembly master mix (NEB E2621). Any noncircularized DNA was digested overnight using ExoI, ExoIII, and Lambda exonuclease (all NEB). The reaction was then cleaned up using SPRI beads at a 0.85:1 (bead:sample) ratio. The circularized library was then used for an overnight RCA reaction using Phi29 (NEB) with random hexamer primers. The RCA product was debranched with T7 endonuclease (NEB) for 2 h at 37°C and then cleaned using a Zymo DNA Clean and Concentrator column-5 (Zymo D4013). The cleaned RCA product was digested using NEBNext dsDNA fragmentase (NEB M0348) following the manufacturer's protocol with a 10-min incubation. For the regular Tn5 library, digested RCA product was cleaned using SPRI beads. For all other libraries, the digested RCA product was size-selected using a 1% low-melt agarose gel: DNA between 2 and 10 kb was excised from the gel, which was then digested using NEB beta-agarase. DNA was then cleaned using SPRI beads.
ONT sequencing
ONT libraries were prepared from R2C2 DNA or directly from Illumina libraries using the ONT ligation sequencing kit (ONT SQK-LSK110) following the manufacturer's protocol and then sequenced on an ONT MinION flowcell (R9.4.1). When preparing ONT libraries from Illumina libraries, SPRI bead purifications throughout the protocol were adjusted to accommodate for their short length. Additional library was loaded on the same flowcell after nuclease flush.
Illumina sequencing
Library pools were sequenced either on the Illumina MiSeq using 2 × 300 (RNA-seq and target enriched Tn5 libraries), the Illumina NextSeq 500 2 × 150 (Tn5 library), or the Illumina NovaSeq 6000 (ChIP-seq).
Analysis
R2C2 and 1D
Raw nanopore sequencing data in the FAST5 file format were base-called using the “sup” setting of guppy5 to generate FASTQ files. R2C2 raw reads in FASTQ format were then processed by C3POa (v.2.2.3; https://github.com/rvolden/C3POa) to generate accurate consensus reads. R2C2 consensus reads and ONT 1D reads were further processed with C3POa (C3POa_postprocessing.py), using the ‐‐trim setting and the following p5/p7 adapter sequences:
>3Prime_adapter
CAAGCAGAAGACGGCATACG
>5Prime_adapter
AATGATACGGCGACCACCGAGATCT
Custom scripts (available at https://github.com/kschimke/PLNK) were used to demultiplex reads based on the sequences of their DNA splints and Illumina indexes and to trim the rest of the Illumina sequencing adapters.
RNA-seq
To determine accuracy levels R2C2, 1D, Illumina MiSeq reads were aligned to the human genome reference (hg38) using minimap2 (v2.18-r1015) (Li 2018).
minimap2 -ax splice ‐‐cs=long ‐‐MD ‐‐secondary=no
Position-dependent accuracy was determined after converting SAM files with the sam2pairwise tool (v.1.0.0; https://zenodo.org/record/11377).
Illumina reads were adapter-trimmed using cutadapt (v3.2) (Martin 2011).
cutadapt -m 30 -j 50 -a AGATCGGAAGAGC -A AGATCGGAAGAGC
Illumina and R2C2 reads were aligned to the human genome (hg38) using STAR and STARlong (v2.7.3a) (Dobin et al. 2013).
STAR ‐‐quantMode GeneCounts ‐‐outSAMattributes NH HI NM MD AS nM jM jI XS
To determine insert length, Illumina read pairs were merged using bbmerge (v38.92) with default settings.
ChIP-seq
Illumina reads were subsampled using a custom script (https:// github.com/alexanderkzee/BWN) to match the total reads from the corresponding R2C2 library.
Illumina and R2C2 reads were aligned to the G. max genome (Gmax_508_v4.0) using minimap2 (v2.18-r1015) (Li 2018).
minimap2 -ax sr ‐‐cs=long ‐‐MD ‐‐secondary=no
Peaks in H3K4me3 Illumina data were called using MACS2 (Zhang et al. 2008).
macs2 callpeak -t K4.bam -c INPUT.bam -f BAM -n K4_Illumina ‐‐nomodel ‐‐extsize 200
Tn5
R2C2 reads were aligned to the D. melanogaster genome (dm6) using minimap2 (v2.18-r1015).
minimap2 -ax sr ‐‐cs=long ‐‐MD ‐‐secondary=no
R2C2 reads that did not align to the Drosophila genome were then assembled using miniasm.
minimap2 -x ava-ont [dehosted r2c2 file] [dehosted r2c2 file] > [ava paf file]
miniasm -f [dehosted r2c2 file] [ava paf file] -m 450 -s 250 > [gfa raw assembly]
We aligned Illumina reads to the D. melanogaster genome (dm6) using BWA-MEM (Li 2013) under default parameters. We then extracted the sample IDs for reads that did not map to the host genome and extract that set from the raw FASTQ files.
Illumina reads that did not align to the Drosophila genome were then assembled using Meraculous, setting the minimum contig depth to 10, expected genome size to 0.013, and using a k-mer of 51 and otherwise default parameters.
Target-enriched Tn5
Illumina reads were adapter trimmed using cutadapt (v3.2).
cutadapt -m 30 -j 50 -a AGATCGGAAGAGC -A AGATCGGAAGAGC
Trimmed Illumina and R2C2 reads were aligned to the human genome (hg38) using minimap2 (v2.18-r1015).
minimap2 -ax sr ‐‐cs=long ‐‐MD ‐‐secondary=no
Germline variants in Illumina data of NCI-H1650 were called using DeepVariant (Poplin et al. 2018). Germline variant in R2C2 data of NCI-H1650 were called using PEPPER-Margin-DeepVariant (Shafin et al. 2021).
Real-time analysis with PLNK
RNA-seq, ChIP-seq, and Enriched Tn5 MinION runs were simulated by reading the mtime metadata entry of FAST5 files in the output folder of the completed runs and then calculating the time intervals at which files were created by the MinKNOW software. Files created during the first 48 h or until the first library reload were then copied into a new folder at those intervals. PLNK (https://github.com/kschimke/PLNK) was started after the simulation and was given key information about the run (splint and Illumina indexes in the format of a sample sheet, target regions in BED format, genome sequence in FASTA format) and a config file containing paths to tools used by PLNK.
Analysis of public MiniSeq, iSeq, and MiSeq data
Sequencing runs of genomic Escherichia coli DNA were downloaded from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra). We selected three runs each for MiniSeq (SRR20643069, SRR20643071, SRR20643072; generated by the GenomeTrakr project), iSeq (SRR14617007, SRR14617041, SRR14617075) (Mitchell et al. 2022), and MiSeq (SRR19575967, SRR19575968, SRR19575973; generated by the National Microbiology Laboratory).
To generate accuracy-by-position data, reads for each run were processed separately. First reads were aligned to an E. coli reference genome (CP014314; downloaded from the NCBI GenBank database [https://www.ncbi.nlm.nih.gov/genbank/]) using minimap2. Then the genome was polished using these alignments with Pilon (Walker et al. 2014). Reads were then realigned to the polished genome using minimap2, and position-dependent accuracy was calculated after converting the resulting SAM files using the sam2pairwise tool.
General analysis
SAMtools (v1.11-18-gc17e914) (Li et al. 2009) was used extensively during analysis for SAM file processing. Python (Oliphant 2007), Matplotlib (Hunter 2007), Numpy (Harris et al. 2020), and SciPy (Virtanen et al. 2020) were all used to analyze and visualize the data.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA775962. All codes used for analysis are available at GitHub (https://github.com/kschimke/PLNK; https://github.com/alexanderkzee/BWN; https://github.com/rvolden/C3POa) and as Supplemental Code.
Competing interest statement
Oxford Nanopore Technologies (ONT) has paid for travel and accommodations for C.V. to their London Calling conference twice between 2016 and 2018. In 2020, ONT supplied sequencing reagents to the laboratory of C.V. for work in the LRGASP Consortium on an unrelated project.
Acknowledgments
We thank the UCSC Paleogenomics Laboratory sequencing facility for sequencing the RNA-seq, Tn5, and enriched Tn5 libraries. We also thank Kishwar Shafin for his support with running PEPPER-Margin-DeepVariant. We acknowledge funding by the National Institute of General Medical Sciences/National Institutes of Health grant R35GM133569 (to C.V.) and R35GM128932 (to R.C.-D.). This study was funded with support from the National Science Foundation (IOS-1856627) and the United Soybean Board to R.J.S.
Author contributions: A.Z. led the R2C2 conversion of ChIP-seq libraries and Tn5 libraries, analyzed ChIP-seq data, and wrote and edited the manuscript. D.Z.Q.D. led the generation and R2C2 conversion of target-enriched Tn5 libraries, analyzed the target-enriched Tn5 data, and wrote and edited the manuscript. M.A. optimized the R2C2 method for the conversion of Illumina libraries, led the generation and R2C2 conversion of RNA-seq libraries, analyzed the RNA-seq data, and wrote and edited the manuscript. K.D.S. led the development of the PLNK real-time analysis tool, performed real-time analysis simulations, and wrote and edited the manuscript. R.C.-D. conceptualized and supervised the generation of Tn5 libraries, analyzed the Tn5 library–based genome assemblies, and edited the manuscript. S.L.R. conceptualized the generation of Tn5 libraries, generated Wolbachia-containing D. melanogaster cell lines, and edited the manuscript. X.Z. generated and supervised the Illumina sequencing of ChIP-seq libraries and edited the manuscript. R.J.S. designed and supervised the generation of ChIP-seq libraries and data and edited the manuscript. C.V. conceptualized and designed the project, supervised the generation of data, supervised Illumina and R2C2 data analysis, performed germline variant calling on target-enriched Tn5 Illumina and R2C2 data, and wrote and edited the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277031.122.
- Received June 15, 2022.
- Accepted October 21, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








