
Gene essentiality in cancer cell lines is modified by the sex chromosomes
Abstract
Human sex differences arise from gonadal hormones and sex chromosomes. Studying the direct effects of sex chromosomes in humans is still challenging. Here we studied how the sex chromosomes can modulate gene expression and the outcome of mutations across the genome by exploiting the tendency of cancer cell lines to lose or gain sex chromosomes. We inferred the dosage of the sex chromosomes in 355 female and 408 male cancer cell lines and used it to dissect the contributions of the Y and X Chromosomes to sex-biased gene expression. Furthermore, based on genome-wide CRISPR screens, we identified genes whose essentiality is different between male and female cells depending on the sex chromosomes. The most significant genes were X-linked genes compensated by Y-linked paralogs. Our sex-based analysis identifies genes that, when mutated, can affect male and female cells differently and reinforces the roles of the X and Y Chromosomes in sex-specific cell function.
Males and females differ in many ways, among them the frequency of diseases (Cyranowski et al. 2000; Cook et al. 2011; Edgren et al. 2012; Ngo et al. 2014; May et al. 2019), the exhibition of symptoms for the same disease (Baba et al. 2005; Goldstein 2006), and the response to different drugs (Wang et al. 2016). For example, in cancer, the frequency of most nonreproductive cancers is higher in males (Cook et al. 2011; Tevfik Dorak and Karpuzoglu 2012). Some treatments have been shown to work differently in females and males for tumors with the same genetic characteristics (Pal and Hurria 2010).
The two main biological mechanisms that cause human sex differences are gonadal hormone secretions and genes located on the X and Y Chromosomes. There is considerable evidence for sex differences in diseases caused by gonadal hormone secretions (Baron-Cohen et al. 2005; Law et al. 2014; Fuseini and Newcomb 2017). However, it is challenging to show the direct effects of the sex chromosomes separated from the gonadal hormones, especially in humans (Snell and Turner 2018). Still, there are sex differences that are attributed directly to sex-specific genetic effects (Baron-Cohen et al. 2005; Chen et al. 2012; Wang et al. 2016). The effect of the sex chromosomes may not be only because of the expression of genes on the sex chromosomes, which may regulate other genes in the genome; sex differences can also arise from the concentration of heterochromatin factors on the inactive X Chromosome in females, resulting in their depletion from other regions in the genome, a phenomenon named heterochromatin sink (Francisco and Lemos 2014).
A key question of this study is to what extent the sex chromosome can contribute to sex differences in cell function. Our approach is to harness the tendency of cancer cells to lose or gain sex chromosomes (Richardson et al. 2006; Pageau et al. 2007; Bianchi 2009; Duijf et al. 2013; Kang et al. 2015). This enables us to compare male and female cell lines with different sets of sex chromosomes. However, using those cell lines has important limitations, especially owing to the differences from cells within a healthy tissue and the possibility that some of the results are specific to this system (Dvir et al. 2022).
To identify sex-specific mechanisms, we explored gene expression and the effect of loss-of-function mutations on cell survival and proliferation. Recent advances in CRISPR technology have enabled the systematic identification of essential genes necessary for the normal functioning of cells across the genome (Tzelepis et al. 2016; Yilmaz et al. 2018; Shohat and Shifman 2019). The Achilles project is the largest survey of human gene essentiality (Meyers et al. 2017). The project uses CRISPR loss-of-function screens to quantify the degree of gene essentiality in hundreds of human cancer cell lines. Our aim in this study was to develop an approach for studying the differences between male and female cells and the roles of the X and Y Chromosomes in cell function using gene expression data and the results of the CRISPR loss-of-function screens. The CRISPR screens can be used to identify mutations that affect the viability of male and female cells differently across the genome. Identifying those sex-specific effects has the potential to provide novel insights into the importance of sex chromosome dosage as a variable in experimental studies of cell function.
Results
Inferring the sex chromosome dosage of cancer cell lines
We began by inferring the dosage of the sex chromosomes in 843 cancer cell lines, including 371 cell lines that originated from females and 468 from males (we excluded four misclassified cell lines) (Supplemental Table S1).
To infer the presence of the Y Chromosome, we used the DNA copy number of all genes on the Y Chromosome (relative to all other genes) and the expression of Y-linked genes. Classification based on the Y Chromosome DNA copy number (Fig. 1A) was in high agreement with the classification based on gene expression (Fig. 1B,C). The number of X Chromosomes was inferred from the relative DNA copy number of X-linked genes (Fig. 1D). The X Chromosome dosage was validated based on the heterozygosity level of 17,463 single-nucleotide polymorphisms (SNPs) on the X Chromosome (SNP data were available for 60% of female cell lines) (Fig. 1E,F).
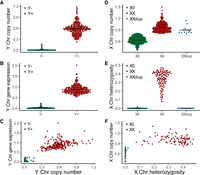
Identification of sex chromosome dosage in cancer cell lines. (A) The relative DNA copy number of Y-linked genes for the Y+ and Y− cell lines. (B) The expression of Y-linked genes for the Y+ and Y− cell lines. (C) The gene expression is plotted as a function of the relative DNA copy number of Y-linked genes. (D) The relative DNA copy number of X-linked genes for the XX, X0, and XXdup cell lines. (E) The proportion of heterozygote SNPs across the X Chromosomes for the female XX, X0, and XXdup cell lines (based on 17,463 high-confidence SNPs with minor allele frequency > 10%). (F) The proportion of heterozygote SNPs as a function of the relative DNA copy number of X Chromosome genes in female cell lines. DNA copy number values are log2(relative to ploidy + 1), and gene expression values are log2(TPM + 1). For the DNA copy number, a log ratio of one indicates that copy number is unchanged relative to a normal diploid sample (n = 2). The black lines are the mean value for each group. (Chr) Chromosome; (XXdup) cell lines with duplication of the active X Chromosome.
The analysis of SNP heterozygosity, together with the copy number of the X Chromosome (Fig. 1D–F), led to the identification of cell lines with two identical X Chromosomes (termed here “XXdup”). Loss of heterozygosity on the X Chromosome is known to occur in cancer lines as a result of losing the inactive copy followed by a duplication of the active copy (Kawakami et al. 2004; Sirchia et al. 2005; Richardson et al. 2006; Benoît et al. 2007; Pageau et al. 2007; Kang et al. 2015). To test the status of the female cell lines, we used the methylation level on the X Chromosome as an indication for X Chromosome inactivation (XCI) and the existence of two chromosomes. As expected, X Chromosome methylation levels were significantly higher in female XX cell lines than other cell lines, including female XXdup (Supplemental Fig. S1A,B).
Overall, we identified seven combinations of the sex chromosomes in 355 female and 408 male cell lines (Supplemental Fig. S1C). Most cell lines (92.5%) were assigned to four groups: XX female, XY male, X0 female, and X0 male cells.
Widespread effect of sex chromosome dosage on gene expression
The most obvious consequence of the different number of sex chromosomes is expression changes of genes located on the sex chromosomes, but autosomal genes may also be influenced. To determine the impact of the sex chromosomes on gene expression, we first performed differential expression analysis between XX and XY cell lines across 19,177 genes. Additionally, we tested the effect of the number of X and Y Chromosomes on gene expression. Both analyses were performed after excluding all the cell lines originating from sex-specific tissues (e.g., breast, ovary, and prostate) (for the remaining number of cell lines per group, see Supplemental Fig. S1D) and using a linear mixed-effect model accounting for the tissue of origin (as a random effect).
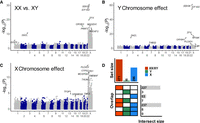
We found 50 differentially expressed genes (false discovery rate [FDR] < 0.05) between the XX and XY cell lines (Fig. 2A; Supplemental Table S2). Y-linked genes (n = 16) were among the most significant differentially expressed genes, followed by X-linked genes (n = 28). The majority of differentially expressed genes on the X Chromosome are known to escape from XCI (86%) (Fig. 2B). None of the differentially expressed genes are located in the pseudoautosomal regions (PARs) of the X and Y Chromosomes (Supplemental Fig. S2). In addition to genes on the sex chromosomes, six autosomal genes were differentially expressed between the XX and XY cell lines: MYCN, MFNG, NLRP2, HCLS1, ERICH6B, and ULK4.

Gene expression is influenced by sex chromosomes. Manhattan plots of the differential expression analysis, showing the −log10 of the P-values as a function of the chromosomal positions of genes. Results are shown for three tests: (A,B) differences between female XX and male XY cell lines, (C,D) association with the presence of the Y Chromosome, and (E,F) association with the number of X Chromosomes. The dashed line denotes a threshold of FDR = 0.05. The upper plots (A,C,E) show the results of genome-wide expression analysis, and the lower plots (B,D,F) show the results for the X Chromosome. The different colors indicate genes on the PARs and the X-inactivation status for the rest of the genes. The names of the 10 most significant genes are shown.
We next assessed if the sex-biased gene expression we observed in the cancer cell lines is replicated in an independent data set of human tissues. We compared the differentially expressed genes between the XX and XY cell lines to sex-biased genes reported based on the Genotype-Tissue Expression project (GTEx). For each differentially expressed gene that we discovered (excluding Y-linked genes), we counted in how many tissues they were among the 500 most significant genes in the GTEx. Out of our 34 differentially expressed genes (excluding the 16 Y-linked genes), 28 were among the most significant sex-biased genes in at least one tissue of the GTEx (P = 5.0 × 10−14, odds ratio [OR] = 17.3). The 28 genes included 26 X-linked genes and two autosomal genes (NLRP2 and HCLS1).
To identify genes influenced by the X and Y Chromosomes, independent of the sex status of the cell lines, we used a linear mixed-effect model that included the sex and the tissue of origin of the cell lines (as a random effect) (Supplemental Table S2). Using this model, we found 132 genes influenced by the presence of the Y Chromosome. The most significant genes were Y-linked (n = 24) and genes located in PAR1 (n = 9) (Fig. 2C,D). We also identified six genes on the X Chromosome and 93 autosomal genes.
The X Chromosome influenced the expression of 58 genes; the most significant genes were X-linked genes (n = 26): 24 of them are known to escape from XCI, and seven genes are located on PAR1 (Fig. 2E,F). Additionally, the expression of 25 autosomal genes was influenced by X Chromosome dosage. Genes located in PAR1 but not in PAR2 showed a similar decrease in expression owing to a lower dosage of X and Y Chromosomes, but no significant change was observed between XX and XY cell lines (Supplemental Fig. S2).
To validate the effect of the X and Y Chromosomes in external data sets, we compared our results with an expression microarray analysis performed on lymphoblastoid cell lines (LCLs) from individuals with diverse sex-chromosome aneuploidies (X0, XXX, XXY, XYY, and XXYY) (Raznahan et al. 2018). We compared the genes whose expression is predicted by the Y Chromosome to differential expression analysis between XY and X0 LCLs (n = 13 in each group). As expected, 10 Y-linked genes were significant (FDR < 0.05) in both data sets. Excluding the Y Chromosome, we found 15 genes to be significant in both data sets (out of 92 genes with available data; P = 2.2 × 10−7, OR = 6.0). This included the nine PAR1 genes and six autosomal genes. Comparing the results to the GTEx data, we found that out of 108 autosomal and X-linked genes whose expression is predicted by the Y Chromosome, 34 (including 24 autosomal genes) were among the 500 most significant sex-biased genes in the GTEx in at least one tissue (P = 0.018, OR = 1.70).
The findings of genes whose expression is predicted by the X Chromosome were compared to results obtained by analysis of LCLs with XX, X0, XY, and XXY. Out of 49 genes, 26 were significantly associated with the X Chromosome in both data sets (P = 1.0 × 10−21, OR = 22.9), including 22 genes located on the X Chromosome and four autosomal genes. In the GTEx data set, 39 genes (including seven autosomal genes) were in the top sex-biased genes in at least one tissue, significantly more than expected by chance (P = 9.7 × 10−14, OR = 7.5).
Thus, we identified changes in gene expression associated with the sex chromosomes that are consistent with changes identified in healthy tissues and LCLs from individuals with sex-chromosome aneuploidy, including the Turner (X0) and Klinefelter (XXY) syndromes.
The dosage of the sex chromosomes affects gene essentiality
The substantial sex differences in gene expression raise the question of whether those differences result in changes in cell phenotypes. To detect genes that show significant sexually dimorphic phenotypic effects, we studied differences in gene essentiality. We used a measure called essentiality score that quantifies how much a gene is essential for the viability of a specific cell line. The score is based on the change in the abundance of single-guide RNAs (sgRNAs) targeting the same gene during the screen. The average depletion level of sgRNAs targeting a gene at the end of the culture period (relative to the initial representation) indicates how essential the gene was for the proliferation and viability of the cells, with greater depletion indicating greater essentiality. The score we used was adjusted such that zero means the gene is not essential, and one is the median score of the common essential genes. Thus, the more positive the score, the more essential the gene is. We expect that a gene with a sexually dimorphic phenotype will have, on average, different scores in male and female cell lines or cell lines with different sex chromosome dosages.
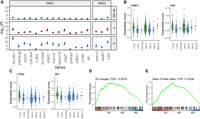
We first compared the degree of gene essentiality between XY male (n = 215) and XX female (n = 118) cell lines after excluding all cell lines originating from sex-specific tissues and controlling for the tissue of origin of the cell lines. We tested the differences in the essentiality score for 18,017 genes and identified 471 genes with significant (FDR < 0.05) sex-dependent essentiality (Supplemental Table S3). The majority of genes (n = 435) were X-linked genes more essential to female XX cell lines (Fig. 3A).
The sex chromosomes influence gene essentiality. (A–C) The Manhattan plots show the genome-wide significance for differences in essentiality score. Values are the −log10 of the P-values as a function of the chromosomal positions of genes. Results are shown for three tests: (A) differences in essentiality score between female XX and male XY cell lines, (B) association with the presence of the Y Chromosome, and (C) association with the number of X Chromosomes. The dashed line denotes a threshold of FDR = 0.05. The names of the six most significant X-linked genes and three autosomal genes are shown. (D) The level of overlap between significant genes in the three tests. The set size (top bar plot) shows the total number of significant genes in each test. The overlap (bottom left plot) shows different comparisons in each row. Genes uniquely identified as significant by a single test are represented as single-colored boxes, and overlaps are shown by two- or three-colored boxes. The intersect size (bottom right plot) shows the number of genes that overlap or are unique to each test.
We next tested the separate effects of the X and Y Chromosomes using a linear mixed-effect model that accounts for the sex and the tissue of origin of the cell lines (see Methods). In total, we found 306 genes whose essentiality is predicted significantly by the sex chromosomes (FDR < 0.05). Among them were 16 genes associated with the Y Chromosome (11 are genes located on the X Chromosome) (Fig. 3B) and 296 genes associated with the X Chromosome (255 are genes located on the X Chromosome) (Fig. 3C). Six genes were identified as influenced by both the X and Y Chromosomes (Fig. 3D).
A large proportion of the genes whose essentiality is predicted by the sex chromosomes (∼80%) also showed significant differences in the essentiality score between male and female cell lines without sex chromosome abnormalities (XX vs. XY) (Fig. 3D). To further eliminate the possibility that our results are caused by aneuploidies of other chromosomes that co-occur with changes in the sex chromosomes, we repeated the test for the effect of the Y or X Chromosomes on essentiality in mixed-effect models that included the dosage of each of the autosomes. The maximum P-value we obtained across the models, with different autosomal chromosomes as a covariance, was in 99% correlation with the original P-values.
Genes whose essentiality is associated with the X Chromosome are enriched with XCI escapers and testis genes
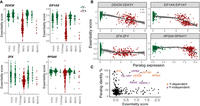
We wanted to characterize the genes whose essentiality is predicted by the dosage of the X Chromosome. First, we examined the six genes that are associated with both the X and Y Chromosomes but are not significant in comparing female XX and male XY cell lines. Five of those genes are located in the PAR, which could explain their similar association with both the X and Y Chromosomes. PAR genes showed a similar trend for association with the X and Y Chromosomes, but the significant levels were generally higher for the X Chromosome (Fig. 4A). The PAR genes were, on average, more essential to female XX, male XY, or male XXY cell lines relative to other cell lines (for an example of two genes, see Fig. 4B).
Characterization of genes associated with the X Chromosome. (A) Association of genes in the PARs with X and Y Chromosome dosage. Values are the −log10 of the P-values for the differences in essentiality score between female XX and male XY cell lines (top), the association of the essentiality score with the Y Chromosome (middle), and the association with the X Chromosome (bottom). The dashed line shows a P-value = 1. Genes with FDR < 0.05 are flagged with a star. (B,C) Distribution of essentiality scores across cell lines with different sex chromosome dosages. In green are groups that show a higher mean essentiality score. The gray line is the average across the cell lines. The black lines are the mean value for each group. (B) Example of two genes located in the PAR. (C) Example of two X-linked genes located outside the PAR. (D,E) Gene-set enrichment analysis (GSEA) plots. The position of genes in the gene set is marked with vertical bars. The genes are sorted based on the association significance with the X Chromosome. The green curve is the enrichment score based on a weighted running sum. (D) GSEA plot for escape and variable escape genes (as one group). (E) GSEA plot for genes expressed predominantly in the testis, based on data from the Human Protein Atlas.
We next examined all the genes whose essentiality is predicted by the X Chromosome (including genes predicted by both X and Y Chromosomes; n = 296). Almost all of those were genes located on the X Chromosome, whose essentiality is higher in female XX cell lines (251 out of 255 X-linked genes) (for an example of two genes, see Fig. 4C). Of the 240 significant X-linked genes with XCI information, 38 escape from XCI (15.8%), and 35 (14.6%) are variable escape genes (genes known to escape in 25%–75% of individuals) compared with 9% and 12%, respectively, in the nonsignificant genes. Gene-set enrichment analysis (GSEA) showed that the X-linked genes whose essentiality is predicted by the X Chromosome are significantly enriched with escape and variable escape genes (FDR = 0.0078) (Fig. 4D).
To further characterize the X-linked genes whose essentiality is predicted by the X Chromosome, we tested if they are predominantly expressed in any particular tissue, and found enrichment of genes selectively expressed in the testis (Supplemental Fig. S3A). Because the X Chromosome is enriched with genes expressed in male tissues (Lercher et al. 2003), we restricted the analysis to X-linked genes and found evidence for association with testis-specific genes even within the X Chromosome (Human Protein Atlas, FDR = 0.0036; TSEA, pSI threshold = 0.001, FDR = 0.069) (Fig. 4E; Supplemental Fig. S3B,C). There is no significant overlap between testis-specific genes and XCI escape genes (P = 0.88), which indicates that the enrichments are independent.
Paralogs on the Y Chromosome can modify gene essentiality
Among the 16 genes whose essentiality is predicted by the Y Chromosome, five are located on the PAR and were discussed above. We examine the remaining 11 genes influenced by the Y Chromosome. We noticed that among the 11 genes, seven have a paralog on the Y Chromosome, which is significantly more than expected (P = 6.2 × 10−11, OR = 113.5). It suggests that Y-linked genes may modulate the essentiality of their corresponding paralogs.
To study this option, we focused on the top four most significant genes whose essentiality is predicted by the Y Chromosome (EIF1AX, DDX3X, RPS4X, and ZFX). All are X-linked genes that have a paralog on the Y Chromosome (EIF1AY, DDX3Y, RPS4Y1, and ZFY). These four genes are more essential to cell lines not carrying a Y Chromosome (both male and female cell lines) and therefore are also significantly different between XX and XY cell lines (Fig. 5A). One possible explanation is that the Y-linked paralogs compensate for the loss of the X-linked genes. In support of this option, we found that the expression of the paralogs was significantly correlated with the levels of essentiality in a way that the X-linked genes are more essential for cell lines with lower expression of the Y-linked paralogs (all with P < 2.2 × 10−16) (Fig. 5B). To further show that the Y-linked paralogs are responsible for the effect, we analyzed 31 cell lines with partial deletions of the Y Chromosome (Supplemental Fig. S4A). We used those cell lines to identify the Y Chromosome regions more likely to be responsible for the changes in essentiality. We found that the predicted region contained the Y-linked paralog for all four genes (Supplemental Fig. S4B,C).
X-linked genes can be compensated by their Y-linked paralogs. (A) Distribution of essentiality scores for four X-linked genes that are more essential in cell lines without a Y Chromosome. The gray line is the average across the cell lines. The black lines are the mean value for each group. (B) Significant correlation between the essentiality score of the four X-linked genes and the expression of their Y-linked paralogs. The lines are locally estimated scatterplot smoothing (LOESS) curves ±95% confidence intervals. (C) Paralog sequence identity as a function of essentiality scores. Eight genes are highlighted with relatively high essentiality scores and sequence identity with the Y-linked paralog.
There are also five autosomal genes whose essentiality is predicted by the Y Chromosome, and all were more essential to cell lines carrying a Y Chromosome, suggesting another mechanism. For example, the DAZL gene, which is located on Chromosome 3 and has Y-linked paralogs, was significantly more essential in cell lines carrying a Y Chromosome (XY and XXY cell lines) (Supplemental Fig. S4D). These suggest that the Y Chromosome can influence the essentiality of the cells not only by compensation through paralogs.
The observation that four genes on the Y Chromosome can compensate for the loss of their X-linked paralog raises the question of why this is not true for other genes. We identified 279 genes with paralogs on the Y Chromosome; 272 of them were not significantly influenced by the Y Chromosome. We plotted the paralog sequence identity level against the essentiality scores of the genes (Fig. 5C). This plot revealed that the four X-linked genes we identified are characterized by a high similarity between the paralogs (median sequence identity = 92.7%) and by being highly essential (median essentiality score = 1.36). In contrast, most other genes are either not very essential (median essentiality score = 0.05) or have low similarity to their Y-linked paralog (median sequence identity = 19.2%). We detected only two genes (USP9X and TBL1XR1) with high similarity to their Y Chromosome paralogs (>85% sequence identity) that are relatively essential (essentiality scores = 0.68 and 0.34, respectively) (Fig. 5C). It is possible that the lack of compensation is owing to low expression of the paralog gene in the majority of Y+ cell lines, as in the case of TBL1Y (Supplemental Fig. S4E), or it may indicate functional divergence between the paralog pairs. There are two other essential genes (essentiality scores = 0.87 and 1.28) with ∼60% similarity to their Y Chromosome paralogs (RBMX and RBMXL1), whose essentiality is not predicted by the Y Chromosome. The Y Chromosome contains a cluster of RBMY paralogs expressed specifically in the testis (Mazeyrat et al. 1999).
Characterization of genes with sex-biased somatic mutations in cancer tumors
To study how our findings of gene essentiality modified by sex chromosomes relate to sex-dependent mutation abundance in vivo, we analyzed somatic mutations in cancer tumors. It is important to note that although the CRISPR screen in the cancer cell lines often results in a complete knockout of genes, most somatic mutations are in a heterozygote state with uncertain functional consequences.
We used the Catalogue of Somatic Mutations in Cancer (COSMIC) (Tate et al. 2019), including somatic mutations in tumor samples from 4755 females and 7489 males collected from 12 different tissues. We compared the rate of nonsynonymous mutations between male and female tumors and identified 21 genes with significant (FDR < 0.05) sex bias (genes with an excess of mutations in males or females) (Fig. 6A; Supplemental Table S4). All identified genes are located on the X Chromosome and none on the autosomes or the PAR regions. Three of the most significant genes (KDM6A, DDX3X, and KDM5C) are among six previously identified sex-biased genes (Dunford et al. 2017). None of the identified genes showed a significant difference in the mutation rate for synonymous mutations.
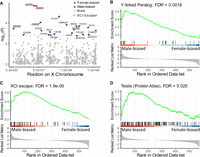
Genes with sex-biased somatic mutations in cancer tumors. (A) X-linked genes show sex bias in rates of somatic mutations. Values are the −log10 of the P-values for differences in mutation rates between males and females as a function of the positions of the genes on the X Chromosome. Genes that escape from XCI are highlighted. (B–D) GSEA plots. The vertical black lines indicate the position of genes in the sets relative to all X-linked genes that are ranked from the most significant male-biased genes to the most significant female-biased genes. The green curve is the enrichment score based on a weighted running sum. (B) GSEA plot for Y-linked paralogs. (C) GSEA plot for XCI escape genes. (D) GSEA plot for genes expressed predominantly in the testis, based on data from the Human Protein Atlas.
We next determined if the sex-biased genes in tumors share the same features as the genes we found to be influenced by the sex chromosomes. Using GSEA, we found that genes with Y-linked paralogs were underrepresented among female-biased genes (genes with an excess of mutations in females) (FDR = 0.0018) (Fig. 6B). This is consistent with the suggestion that genes with a paralog on the Y Chromosome can accumulate more deleterious somatic mutations in males because of male-specific redundancy. Likewise, genes that escape from XCI (Fig. 6C) and genes predominantly expressed in the testis (Fig. 6D; Supplemental Fig. S5) were also underrepresented among female-biased genes (FDR = 1.9 × 10−6, FDR = 0.025, respectively). These enrichments are consistent with the findings that those genes are more essential to female XX cell lines and, thus, should have fewer mutations in females.
Discussion
Multiple human disorders show sex differences in prevalence and symptoms, but the mechanisms remain unclear. Here, we inferred the sex chromosome dosage for 763 cancer cell lines and used it to study how the sex chromosomes influence gene expression and essentiality. Our study is unique in the ability to assign the observed sex differences to the influence of the X or Y Chromosomes. We found that the expression of 192 genes is associated with the cell's sex or the dosage of the sex chromosomes. As expected, the strongest effects were for Y-linked genes, followed by X-linked genes that escape from XCI. Moreover, we found that the dosage of the sex chromosomes can modify the phenotypic outcome of CRISPR screens for 533 genes (2.9% of all genes). Our findings show that the presence of X and Y Chromosomes can impact how the cell responds to perturbation.
By studying gene essentiality, we identified two groups of genes enriched among the genes associated with the X Chromosome: genes that escape from XCI and genes expressed predominantly in the testis. The study of somatic mutations in cancer showed a consistent enrichment of these groups in sex-biased genes. Functional experiments to assess the genes associated with the X Chromosome are needed to evaluate the role of escape genes and genes expressed in the testis in sex-dependent essentiality.
Compared to the large number of genes associated with the X Chromosome, there were only 16 genes whose essentiality was associated with the Y Chromosome. In contrast, the expression of many more genes (n = 132) was associated with the Y Chromosome. The most promising Y-linked genes that might underline this variability in expression are the eight known dosage-sensitive regulators of gene activity (UTY, EIF1AY, ZFY, RPS4Y1, KDM5D, DDX3Y, USP9Y, and TBL1Y) (Bellott et al. 2014). The eight genes all have X Chromosome paralogs that escape from XCI.
By integrating the essentiality scores with gene expression analysis and partial Y Chromosome deletions, we show that the most striking Y-dependent effects are explained by Y-linked genes that can compensate for the loss of an X-linked paralog. Among the four genes we found to be explained by Y-linked paralogs (DDX3X, EIF1AX, RPS4X, and ZFX), two (DDX3X and RPS4X) were previously reported to have redundant roles with their Y paralogs based on functional experiments (Watanabe et al. 1993; Venkataramanan et al. 2021). Moreover, a study in mice showed that complete knockout of Ddx3x causes microcephaly only in females because Ddx3y can compensate for its loss in the developing male cortex (Hoye et al. 2022).
Our study has several limitations that are mainly a result of the use of data from cancer cell lines and CRISPR screens. First, cancer cell lines have multiple genomic alterations (Mani and Chinnaiyan 2010) that might affect the results. However, our analysis shows that our results are not associated with the dosage of other autosomal chromosomes. Second, we used a measure of gene essentiality for the proliferation and survival of cancer cell lines in vitro, and some of the findings might be specific to cancer cell lines. Our results are based on hundreds of cancer cell lines from multiple tissues; therefore, they may be more general but, at the same time, fail to identify tissue-specific effects (Dvir et al. 2022). Our findings may be more relevant to other proliferating tissues, particularly developing tissues that share features with cancer cell lines (Ma et al. 2010). More generally, there may be phenotypic differences between male and female cells and between X and Y paralogs that are not fully captured by CRISPR screens that largely measure the proliferation and survival of cells. Third, some CRISPR screen findings may result from cell-cycle arrest caused by double-strand breaks (DSBs) induced by Cas9 (Aguirre et al. 2016). Future studies are needed with other methods and noncancer cells to fully characterize the phenomenon of the sex chromosomes as modifiers of mutations.
The results show that both the X and Y Chromosomes have a global influence on gene expression and the essentiality of genes. We show that comparing cell lines with different compositions of sex chromosomes enables better discovery of differential expression and essentiality than comparing male and female cell lines. Our approach can be extended to other phenotypes, specific cell types, and developmental stages. In addition to the implications of our results for studying the differences between males and females, they are relevant to understanding specific disorders and genetic alterations. This includes syndromes with sex chromosome abnormalities like Turner syndrome and Klinefelter syndrome and the mosaic loss of the Y Chromosome frequently observed both in cancer cells and during the normal aging process of male individuals (Guo et al. 2020). Our results also reflect the importance of knowing and reporting the sex of cell lines and the status of the sex chromosomes when studying cellular and molecular biology.
Methods
Classification of sex chromosome dosage
Gene expression and DNA copy number data (gene-level copy number) were obtained from the Dependency Map (DepMap) project (22Q1) (Tsherniak et al. 2017). The presence or absence of the Y Chromosome was inferred based on the relative DNA copy number of Y Chromosome genes (compared with all other genes) and gene expression of Y Chromosome genes. For each cell line, we calculated the mean relative DNA copy number of the Y Chromosome and the first principal component (PC) in a principal component analysis (PCA) with the expression of all Y-linked genes. This analysis divided the cell lines into two clusters, where >99% of females were in the Y− cell line cluster. Cell lines outside the main clusters were excluded from further analysis (N = 59). Four female cell lines classified as Y+ were also excluded from the analysis. Cell lines lacking DNA copy number data were classified only based on the gene expression of Y-linked genes (N = 7).
The dosage of the X Chromosome was based on the mean DNA copy number of X-linked genes and the heterozygosity level of SNPs on the X Chromosome (SNP array data; NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] accession: GSE36138) (Barretina et al. 2012). SNPs were called using the crlmm R package (Carvalho et al. 2010), and the percentage of heterozygote SNPs was calculated for common (minor allele frequency > 10%) high-confidence SNPs (mean confidence across samples > 0.95). The distribution of the mean DNA copy number of X-linked genes was bimodal, and the threshold for classification to XX or X0 was based on the local minimum between the two peaks (thresholds calculated separately for males and females). We excluded 10 female cell lines with conflicting results about the dosage of the X Chromosome based on the DNA copy number relative to the heterozygosity. Female cell lines lacking SNP array data (N = 179) were classified based only on the DNA copy number of X-linked genes.
Comparing X Chromosome methylation levels between samples
Data on gene-wise methylation levels (promoter 1 kb upstream TSS) were downloaded from the CCLE web portal (Ghandi et al. 2019; https://sites.broadinstitute.org/ccle). Methylation levels were available for 651 samples (82%). The mean X Chromosome methylation level was calculated for each sample based on 386 inactivated genes (excluding XCI escape genes). A comparison of mean methylation levels between the groups was performed using analysis of variance (ANOVA) followed by a Tukey's honestly significant difference (HSD) multiple comparison test.
Differential essentiality and differential expression analyses
Differential essentiality and differential expression analyses were performed similarly using the package dream (Hoffman and Roussos 2021) under R (R Core Team 2022). The dream package borrows information across genes to better estimate the variance while allowing random effects to be estimated separately for each gene. The first test was between XY male and XX female cell lines and included the tissue of origin as a random effect in the linear mixed-effect model. To study the effect of the sex chromosomes, we used all the cell lines, excluding female XXdup (cell lines with two identical X Chromosomes). The linear mixed-effect model included the sex, the number of Y and X Chromosomes (fixed effects), and the tissue of origin as a random effect. A gene was defined as significant based on FDR < 0.05.
The gene expression analysis results were compared with two external data sets: First, we compared the expression analysis results using the two models to sex-biased genes reported in the GTEx project (Aguet et al. 2020). We used a Fisher's exact test to test the association between our significant genes and genes at the top 500 most significant genes in at least one tissue of the GTEx. Second, we compared the genes significantly associated with the sex chromosomes also with expression microarray analysis performed on LCLs from individuals with diverse sex-chromosome aneuploidies (X0, XXX, XXY, XYY, and XXYY; GEO accession: GSE126712) (Raznahan et al. 2018). Genes associated with the Y Chromosome were tested for association with differentially expressed genes (FDR < 0.05) between XY and X0 LCLs, and the genes associated with the X Chromosome were tested for association with genes influenced by the X Chromosome in the expression analysis of LCLs with XX, X0, XY, and XXY.
To rule out the possibility that the association of X and Y Chromosomes with gene essentiality is a result of correlation with other chromosome abnormalities, we used models that included the dosage of each autosome as a covariance. The dosage of the autosomes was calculated based on the mean copy number of all genes in the chromosome. The P-values for the association with the sex chromosomes were compared between models with and without the autosomes.
Gene-set enrichment analysis
GSEA was performed using the clusterProfiler (Yu et al. 2012) and enrichplot (https://yulab-smu.top/biomedical-knowledge-mining-book/) packages in R. The data sets tested included the following: (1) genes that escape from XCI (a gene was defined as an escape or variable escape gene based on previously reported combined XCI status) (Tukiainen et al. 2017); (2) three different sets of testis-specific genes (the list of genes predominantly expressed in the testis was based on the definition in the Human Protein Atlas [Uhlén et al. 2015] or the Tissue-Specific Expression Analysis [TSEA] tool [Dougherty et al. 2010], with two different thresholds [pSI < 0.05, pSI < 0.001]); and (3) genes with Y-linked paralogs. We obtained the list of the paralogs of human protein-coding genes from Ensembl BioMart (biomaRt R package) (Durinck et al. 2005) along with protein sequence similarity information.
Fine mapping of the associated regions on the Y Chromosome
Thirty-one cell lines with partial Y Chromosome deletion were used (with 6% to 93% of Y-linked genes present). For each of
the 77 Y-linked genes with DNA copy number information, we calculated the difference in standard deviations between the observed
and the expected essentiality scores, assuming that each gene is the causal gene. The genes with the minimal difference between
the expected and observed scores were considered the most likely causal genes. The differences in essentiality scores between
cell lines with and without the candidate genes were tested using a Welch's t-test. The differences between the observed and the expected essentiality scores were calculated according to the following
equation: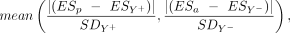 where ESp is the mean essentiality score for cell lines with a deletion that does not include the gene; ESa is the mean essentiality score of cell lines with a partial deletion that includes the gene; ESY+ is the mean essentiality score for Y+ cell lines; ESY− is the mean essentiality score for Y− cell lines; SDY+ is the standard deviation for the essentiality score of Y+ cell lines; and SDY− is the standard deviation for the essentiality score for Y− cell lines.
where ESp is the mean essentiality score for cell lines with a deletion that does not include the gene; ESa is the mean essentiality score of cell lines with a partial deletion that includes the gene; ESY+ is the mean essentiality score for Y+ cell lines; ESY− is the mean essentiality score for Y− cell lines; SDY+ is the standard deviation for the essentiality score of Y+ cell lines; and SDY− is the standard deviation for the essentiality score for Y− cell lines.
Somatic mutations in cancer tumors
Somatic mutations from whole-genome screens (not including targeted studies) were obtained from the COSMIC website (Tate et al. 2019). We excluded mutations in the mitochondria genome, non-PAR genes on the Y Chromosome, and noncoding regions. We also removed mutations with an unknown tissue of origin and from male- and female-specific tissues (testis, prostate, placenta, ovary, breast, cervix, endometrium, genital tract, and penis). We only analyzed mutations from canonical transcripts to avoid duplication in the data. Samples with an outlier distribution of mutations, including a relatively low number of mutations on the X Chromosome compared with the autosomes, and samples with a substantially low ratio between the number of unique mutated genes and the total mutations were excluded. The mutations were labeled nonsynonymous (missense, nonsense, and frameshift) and synonymous. After applying all the filtrations, the data set consisted of 1,336,600 nonsynonymous and 3,356,129 synonymous mutations from 7489 males and 4755 females.
To compare somatic mutation rates between the sexes, we performed a randomization test across the different tumor types, similar to the method used in a previous study (Dunford et al. 2017). Separate tests were performed for synonymous and nonsynonymous mutations, the X Chromosome, and autosomes (including the PAR). The status of the genes was treated as binary, with or without a mutation. We only analyzed mutations from the 12 most common tissues in the database, including ∼97% of the mutations. For each tissue and gene, the mutation probability in males was the number of male mutations divided by the total number of mutations across all genes. The male probability for a mutation in each tissue was used to generate random numbers from a binomial distribution, summed across tissues, to have the expected number of male mutations under the null hypothesis. This simulation was repeated 1 million times. The distribution was used to calculate a one-sided P-value based on the number of simulations in which the number of male mutations was higher than observed, divided by the number of simulations. The P-values were transformed into two-sided P-values and corrected for multiple testing using the Benjamini–Hochberg FDR procedure. Genes with FDR-corrected P < 0.05 were considered significant.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Eyal Ben-David for valuable comments on the manuscript. This research was supported by the Israel Science Foundation (grant no. 466/21).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276488.121.
-
Freely available online through the Genome Research Open Access option.
- Received December 14, 2021.
- Accepted November 16, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








