
Diverse environmental perturbations reveal the evolution and context-dependency of genetic effects on gene expression levels
Abstract
There is increasing appreciation that, in addition to being shaped by an individual's genotype and environment, most complex traits are also determined by poorly understood interactions between these two factors. So-called “genotype × environment” (G×E) interactions remain difficult to map at the organismal level but can be uncovered using molecular phenotypes. To do so at large scale, we used TM3′seq to profile transcriptomes across 12 cellular environments in 544 immortalized B cell lines from the 1000 Genomes Project. We mapped the genetic basis of gene expression levels across environments and revealed a context-dependent genetic architecture: The average heritability of gene expression levels increased in treatment relative to control conditions, and on average, each treatment revealed new expression quantitative trait loci (eQTLs) at 11% of genes. Across our experiments, 22% of all identified eQTLs were context-dependent, and this group was enriched for trait- and disease-associated loci. Further, evolutionary analyses suggested that positive selection has shaped G×E loci involved in responding to immune challenges and hormones but not to man-made chemicals. We hypothesize that this reflects a reduced opportunity for selection to act on responses to molecules recently introduced into human environments. Together, our work highlights the importance of considering an exposure's evolutionary history when studying and interpreting G×E interactions, and provides new insight into the evolutionary mechanisms that maintain G×E loci in human populations.
It is now clear that, in addition to being shaped by an individual's genotype and environment, most human complex traits are determined by poorly understood interactions between an individual's genetic background and his or her environment (Hunter 2005; Thomas 2010; McAllister et al. 2017). Consequently, there has been a strong interest in mapping “genotype × environment” (G×E) interactions—in which genotype predicts an individual's response to environmental variation—and in understanding how loci involved in G×E interactions evolve and are maintained in our species. However, scientists have struggled in practice to map G×E interactions in humans, largely because (1) the relevant environmental factors are often unknown, difficult to measure, or minimally variable within the study population and (2) large sample sizes are needed to overcome the power limitations posed by point 1. Current state-of-the-art approaches have focused on leveraging very large cohort studies such as the UK Biobank; however, this body of work has uncovered only a handful of G×E interactions for common diseases and complex traits (Tyrrell et al. 2017; Arnau-Soler et al. 2019; Nag et al. 2019; Wang et al. 2021).
An alternative approach is to use in vitro manipulations of the cellular environment paired with transcriptomics to map “context-dependent” expression quantitative trait loci (eQTLs), defined as variants that do not affect gene expression levels under baseline conditions but become associated with transcriptional variation following an in vitro exposure (or vice versa). This approach focuses on the cellular level as a proxy for the organismal level in a tractable, experimental framework. Using this methodology, thousands of G×E interactions, in the form of context-dependent eQTLs, have been identified following cell treatment with pathogens, other molecules that provoke an immune response, drugs, hormones, chemicals, and additional stimuli (Barreiro et al. 2012; Lee et al. 2014; Çalışkan et al. 2015; Moyerbrailean et al. 2016; Nédélec et al. 2016; Quach et al. 2016; Kim-Hellmuth et al. 2017; Piasecka et al. 2018; Findley et al. 2021). The representative set of studies referenced above, as well as many others using similar designs, have consistently shown that context-dependent eQTLs overlap genome-wide association (GWAS) hits for complex traits and diseases; in some cases, this overlap is stronger than for eQTLs that are constant or “ubiquitous” across cellular conditions (Kim-Hellmuth et al. 2017; Findley et al. 2021). This body of work has also revealed that context-dependent eQTLs revealed by immune stimuli are often more strongly enriched for signatures of past adaptation than ubiquitous eQTLs, suggesting there has been historical selection for plasticity in immune function.
Taken together, previous studies thus argue that G×E interactions and context-dependent eQTLs make important contributions to the genetic architecture of gene expression levels. However, given the large sample sizes needed to robustly map context-dependent eQTLs, only a handful of cellular conditions are usually explored in any single study. This leaves us with a poor understanding of (1) the full catalog of G×E interactions, as new cellular perturbations typically reveal new context-dependent eQTLs (Barreiro et al. 2012; Lee et al. 2014; Çalışkan et al. 2015; Moyerbrailean et al. 2016; Nédélec et al. 2016; Quach et al. 2016; Kim-Hellmuth et al. 2017; Piasecka et al. 2018; Findley et al. 2021), and (2) the evolutionary forces that maintain context-dependent eQTLs. In particular, although previous work has focused on the action of positive selection in maintaining context-dependent eQTLs for pathogens and immune stimuli, which have coevolved with humans through evolutionary time, context-dependent eQTLs for “evolutionarily novel” stimuli (e.g., man-made chemicals that have only recently been introduced into our environments) may be maintained by different forces. For example, inefficient purifying selection may maintain G×E loci that are in fact deleterious but are only revealed under rare or newly introduced environmental conditions; in other words, these loci may be sufficiently “hidden” from purifying selection such that they persist in human populations despite their negative effects. However, empirical investigations of the varied evolutionary forces that maintain context-dependent eQTLs, especially in high-powered studies exploring many types of cellular environments, remain limited.
Here, we report a large-scale study in which we profiled genome-wide gene expression levels across 12 different cellular environments using 544 immortalized B cell (lymphoblastoid) lines from the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012). We focused on unrelated European and African individuals for whom whole-genome sequence data were publicly available (Byrska-Bishop et al. 2022), and we used these data to map both ubiquitous and context-dependent eQTLs across all 12 cellular conditions. Our experimental treatments included stimuli familiar to B cells such as immune signaling molecules and hormones but also man-made chemicals and other novel cell stressors that have not coevolved with human B cells through evolutionary time. Our design thus allowed us to address three fundamental questions about the genetic architecture of gene expression levels: (1) To what degree is gene expression determined by ancestry (as has been shown previously) (Nédélec et al. 2016; Quach et al. 2016), and how environmentally robust are these effects? (2) How prevalent are context-dependent versus ubiquitous eQTLs, and what are their respective properties and relevance to human complex traits? (3) What are the evolutionary forces (e.g., genetic drift, inefficient purifying selection, positive selection) that maintain context-dependent versus ubiquitous eQTLs, and do these forces differ depending on the evolutionary history of the cell treatment? Together, our work emphasizes the importance of G×E interactions in shaping complex traits and provides new insight into the evolutionary mechanisms that maintain G×E loci in humans.
Results
Diverse cell exposures induce diverse changes in gene expression levels
We exposed 544 lymphoblastoid cell lines (LCLs) derived from individuals included in the 1000 Genomes study (The 1000 Genomes Project Consortium 2012) to 12 cellular environments, including 10 treatment and two control conditions (Fig. 1A; Supplemental Table S1). We chose treatments that have been previously shown to induce moderate to strong responses in LCLs, focusing on a range of treatment types including immune- and non-immune-related stimuli (Table 1). Specifically, we exposed cells to the following: (1) FSL-1 and (2) gardiquimod, two synthetic molecules that activate the TLR2/TLR6 and TLR7 signaling pathways, respectively; (3) TNF superfamily member 13b (also known as B-cell-activating factor [BAFF]), a cytokine that is a member of the tumor necrosis factor family and a potent B cell activator; (4) interferon gamma, a cytokine that is critical for coordinating innate and adaptive immune responses to viral infections; (5) dexamethasone, a synthetic glucocorticoid hormone and anti-inflammatory drug; (6) insulin like growth factor 1, a hormone that plays a key role in growth-related processes; (7) tunicamycin, an antibiotic that induces endoplasmic reticulum stress and is used as a model for cell stressors that impact protein folding; (8) perfluorooctanoic acid, (9) acrylamide, and (10) bisphenol A, three man-made chemicals and environmental contaminants; (11) water, a vehicle control for all treatments except tunicamycin and dexamethasone; and (12) ethanol, a vehicle control for tunicamycin and dexamethasone but also considered a treatment with water as a control (see Supplemental Table S2). Note that we describe our experiment as including 12 “environments” and 11 “treatments” (i.e., all environments except water because ethanol is both a treatment and a control).
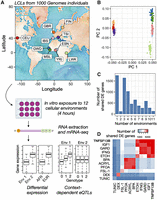
Study overview and environmental effects on gene expression. (A) Lymphoblastoid cell lines (LCLs) derived from individuals included in the 1000 Genomes Project were obtained from Coriell Institute. Specifically, we obtained lines derived from individuals of European and African ancestry as noted on the map (abbreviations for included populations are as follows: [CEU] Utah residents [CEPH], [GWD] Gambian Mandinka, [GBR] British, [IBS] Iberian, [MSL] Mende, [FIN] Finnish, [TSI] Toscani, [ESN] Esan, [YRI] Yoruba, and [LWK] Luhya) (see also Supplemental Table S1). Each cell line was exposed to 12 cellular environments for 4 h, after which we harvested the RNA and performed mRNA-seq. These data were used to understand differential expression as a function of environmental context and ancestry ([AFR] African, [EUR] European), as well as to map ubiquitous and context-dependent eQTLs. (B) Principal components analysis of genotype data for individuals included in this study (colors are as in A). Individual used in this analysis are those for which paired RNA-seq and genotype data were available (Supplemental Table S4). (C) Number of differentially expressed (DE) genes shared between N environments using a mashR, joint analysis approach. N is plotted on the x-axis and ranges from one (i.e., the gene is DE in response to only one environmental treatment) to 11 (i.e., the gene is DE in response to all 11 environmental treatments) (see also Supplemental Table S5). The low number of genes when N = 2 is driven by a large number of dexamethasone (DEX)-specific genes, such that 93.7% of the N = 1 genes are DEX-specific; when DEX is excluded from the data set, most genes are shared between three or more environments (Supplemental Fig. S8). (D) Number of DE genes shared between a given pair of environmental treatments using a mashR, joint analysis approach. The diagonal represents the number of DE genes in response to the focal environmental treatment. Abbreviations are as in Table 1.
Cellular environments
Following 4 h of exposure to each cellular environment, we extracted RNA and used TM3′seq (Pallares et al. 2020) to collect mRNA-seq data from 5223 samples (mean reads per sample ± SD = 2.199 ± 2.731 million). We paired this mRNA-seq data with publicly available genotype data for the same individuals, derived from whole-genome sequencing to at least 30× coverage (Byrska-Bishop et al. 2022). All individuals included in our study were unrelated and showed ancestry of European or African origin (admixed populations were not included in our study design) (Fig. 1B). After filtering, we retained a total of 3886 mRNA-seq profiles from 500 unique individuals (Supplemental Table S3). Genotype data derived from high-coverage whole-genome sequencing were also available for 454 of these individuals (Supplemental Table S4).
We found significant differences in transcriptome dynamics between treated and control condition cells (when analyzing each of the 11 treatment–control pairs separately): On average, 6.39% ± 8.98% (SD) of all 10,157 tested genes responded to a given treatment, with a maximum of 31.64% genes differentially expressed in response to dexamethasone (limma FDR < 10%) (Supplemental Table S5). Gene set enrichment analyses (GSEA) revealed overrepresentation of differentially expressed genes in expected biological pathways. For example, gardiquimod induced differential expression of genes involved in immune system processes such as “cytokine production” (enrichment score = 0.536, q = 0.055), “toll-like receptor signaling pathway” (enrichment score = 0.507, q = 1.03 × 10−2), and “activation of immune response (enrichment score = 0.457, q = 1.03 × 10−2). Similarly, IFNG treatment activated expression of genes related to “response to virus” (enrichment score = 0.583, q = 0.066), “type I interferon production” (enrichment score = 0.540, q = 0.066), and “regulation of innate immune response” (enrichment score = 0.462, q = 0.066). Finally, the strongest pathways induced by tunicamycin were related to endoplasmic reticulum stress, such as “endoplasmic reticulum unfolded protein response” (enrichment score = 0.581, q = 0.189) and “ER overload response” (enrichment score = 0.844, q = 0.189) (Supplemental Fig. S1; Supplemental Table S6). These results suggest that our experimental cell treatments induced appropriate biological responses.
When we used an empirical Bayes approach to perform joint analyses across our entire data set (Urbut et al. 2019), we found that across all 11 treatments only 136 genes were generally environmentally responsive and differentially expressed (mashR LFSR < 10% and all posterior effect size estimates within a factor of two) (see Fig. 1C; Supplemental Fig. S2; Supplemental Table S7). Gene Ontology analysis (Eden et al. 2009) of these genes did not reveal any significant (FDR < 10%) biological pathways or functions; however, the Gene Ontology term with the strongest enrichment was “multicellular organismal response to stress” (fold enrichment = 19.6, P = 4 × 10−4). In contrast, most genes showed some degree of context-dependency: 23.93% of differentially expressed genes were unique to a single treatment (mashR LFSR < 10% and no posterior effect size estimates within a factor of two), and 42.44% were shared between two and 10 treatments (Fig. 1D). Together, these results emphasize the environmental dependency of gene expression, as well as the fact that diverse experimental treatments generally provoke nonoverlapping cellular responses.
Genetic ancestry controls the expression of immune-related genes across cellular conditions
We next explored transcriptional variation as a function of ancestry, comparing individuals of African versus European descent. Of the 10,157 genes we tested, an average of 3.59% ± 2.80% (SD) genes were differentially expressed between ancestry groups (limma FDR < 10%) (Supplemental Table S8). Joint analyses (Urbut et al. 2019) revealed that these ancestry-associated genes were generally shared across conditions, with 69.20% of ancestry-associated genes displaying similar effect sizes across all 12 conditions and 98.92% displaying similar effect sizes across more than two out of three conditions (Fig. 2A,B; Supplemental Fig. S2; Supplemental Table S7). In agreement with previous work (Nédélec et al. 2016), we found that the set of genes controlled by ancestry across conditions were most strongly enriched for immune system processes such as “cytokine-mediated signaling pathway” (enrichment score = 0.311, q = 0.160), “response to virus” (enrichment score = 0.419, q = 0.160), and “inflammatory response” (enrichment score = 0.350, q = 0.160) (Fig. 2C; Supplemental Table S9). Further, we downloaded publicly available sets of genes that are thought to lie along the causal pathway linking genetic variation, gene expression, and human complex traits (inferred through probabilistic transcriptome-wide association studies [PTWAS]) (Zhang et al. 2020) and tested for overlap with our set of ancestry-associated genes. Here, we found that ancestry-associated genes were enriched for complex traits and diseases with immune involvement, such as lymphocyte counts (fold enrichment = 1.10, q = 0.098), platelet counts (fold enrichment = 1.08, q = 0.098), inflammatory bowel disease (fold enrichment = 5.59, q = 0), and Crohn's disease (fold enrichment = 5.59, q = 0) (Supplemental Table S10). Many of these phenotypes are known to differ in prevalence and/or etiology between individuals of African versus European ancestry (Reddy and Burakoff 2003; Coates et al. 2020).
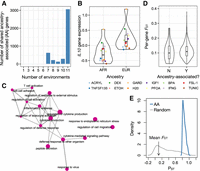
Ancestry effects on gene expression. (A) Number of ancestry-associated (AA) genes shared between N environments using a mashR, a joint analysis approach. N is plotted on the x-axis and ranges from one (i.e., the gene is AA in only one cellular environment) to 12 (i.e., the gene is AA in all 12 cellular environments). (B) Example of an AA gene. The y-axis shows the mean, normalized IL10 gene expression levels estimated in each environment, after regressing out three surrogate variables. (C) Results from gene set enrichment analyses testing for overrepresentation of particular Gene Ontology categories among AA genes (note that genes were sorted by average AA effect size across all 12 cellular environments, and only the top 15 most significant categories are shown). Enrichment map was created with the emapplot function in the R package enrichplot. (D) Distribution of average per-gene FST values for genes that (1) were found to be AA in most (more than two of three) cellular environments or (2) had no effects on ancestry in any cellular environment (results are from a mashR, a joint analysis approach). (E) Phenotypic differentiation (in gene expression; PST) versus genetic differentiation (FST) for AFR versus EUR samples. Plots show the distribution of PST values for (1) AA genes identified in the H2O cellular environment (blue) and (2) a same-sized set of randomly selected genes (gray). The mean genome-wide FST value comparing genetic divergence between AFR and EUR samples is noted on the x-axis with an arrow. We find that all AA genes show PST > FST, indicative of diversifying selection (Lamy et al. 2012; Leinonen et al. 2013). In this panel, we show the H2O environment as a representative example: The results are similar across all 12 environments, and these results are shown in Supplemental Figure S3.
We hypothesized that the observed transcriptional differentiation between African and European individuals was controlled by genetic variation (Nédélec et al. 2016; Quach et al. 2016), and performed two sets of analyses to address this possibility. First, we followed the approach of Nédélec et al. (2016) and asked whether genes with ancestry effects showed higher FST values between African and European populations relative to non-ancestry-associated genes. We found that this was indeed the case for genes with shared effects across more than two-thirds of conditions (Wilcoxon signed-rank test, P = 1.91 × 10−4), as well as for the total set of genes with effects in any condition (Wilcoxon signed-rank test, P = 2.79 × 10−4; Fig. 2D); we note the effect size here is small, but very similar to previous work (Nédélec et al. 2016). Further, when analyzing genes with ancestry effects in one or more conditions, we found that the degree of genetic differentiation was positively correlated with the number of conditions in which an ancestry effect was found (linear model, beta = 3.69 × 10−4, P = 7.85 × 10−5), suggesting that genes with ancestry effects that are unmodified by treatment may be under the strongest genetic control. Second, we compared PST values for ancestry-associated genes identified in one or more conditions versus genome-wide FST values to understand whether ancestry-related variation in the transcriptome (i.e., putatively genetically controlled) showed evidence for being driven by genetic drift (PST = FST), diversifying selection (PST > FST), or stabilizing selection (PST < FST) (Lamy et al. 2012; Leinonen et al. 2013). Our analyses point toward diversifying selection at ancestry-associated genes (all PST > FST), suggesting that there has been selection for different local optima in African versus European populations (Fig. 2E; Supplemental Fig. S3).
Consistent with the high degree of effect size similarity we observed for ancestry effects analyzed across cellular environments, we did not find any compelling evidence for ancestry × treatment effects on gene expression levels using several analysis approaches (see Methods, “Testing the degree to which ancestry and genotype affect the response to treatment”). Although previous work has found that ancestry predicts the magnitude of the response to experimental infection in macrophages and peripheral blood mononuclear cells (PBMCs) (Nédélec et al. 2016; Quach et al. 2016; Harrison et al. 2019), we note that our treatments induced much smaller overall transcriptional shifts in LCLs relative to these previous studies in primary cells, which likely affects our power to detect interaction effects at genome-scale. Further, LCLs are derived from B cells, which are a key component of the adaptive immune system; in contrast, previous work has found that ancestry effects on the response to infection largely involve innate immune system cell types and processes (Nédélec et al. 2016; Quach et al. 2016; Harrison et al. 2019).
Cellular perturbations reveal many context-dependent eQTLs
The primary goal of this study was to understand the genetic architecture of gene expression levels, including the degree to which genetic effects are context-dependent versus unperturbed by environmental challenges. Consistent with previous studies (Grundberg et al. 2012; Wright et al. 2014; Wheeler et al. 2016), we found that there is a substantial genetic component to gene expression levels in LCLs, with an average heritability of 0.111 ± 0.250 (SD) in unstimulated cells. However, we found that this genetic component changed following cell treatments, such that mean per-gene heritability estimates were higher in almost all treatment conditions relative to their respective controls. Specifically, mean heritability estimates increased in nine of 11 treatments, with an average fold increase of 1.736 ± 0.955 (SD) across all 11 treatments (Fig. 3A; Supplemental Table S5). Further, the difference in mean per-gene heritability estimates between treatment and control conditions remained after accounting for the sample size used to estimate heritability in each condition (linear model: β = 2.94 × 10−2, P = 2.90 × 10−13) and after subsampling all environments to an identical sample size (Fig. 3B; Supplemental Table S5). Together, these results are consistent with an increase in additive genetic variation for gene expression levels when cells are perturbed. This result parallels observations of increased heritability for human complex traits in recent decades, despite minimal changes in the genetic makeup of populations (Jelenkovic et al. 2016; Athanasiadis et al. 2022); such environmentally induced increases in heritability are consistent with the idea that cryptic alleles are revealed by environmental perturbations and lead to an increase in additive genetic variance.
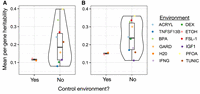
Environmental perturbations increase the heritability of gene expression levels. The y-axis shows the mean per-gene heritability estimated in each environment, using (A) the total available sample size for each environment or (B) a subsample of n = 100 from each environment (plot shows the average of five subsamples).
We confirmed that the genetic architecture of gene expression levels changes following cellular perturbations by mapping cis eQTLs in each of the 12 control or treatments conditions and comparing their effect sizes. These analyses revealed extensive genetic control of gene expression: 11.07% ± 7.64% (SD) of genes contained at least one eQTL (matrixeQTL FDR < 10%), with this number increasing to 15.72% ± 3.69% (SD) when considering only the conditions with the largest sample sizes (Supplemental Table S8). Further, when we pooled our data and performed one analysis using samples from all conditions, we found that almost all genes (92% of those tested) had at least one eQTL (matrixeQTL FDR < 10%). The observation that most genes have an eQTL is consistent with analyses of the Genotype-Tissue Expression (GTEx) data set and other large-scale eQTL studies (GTEx Consortium 2017; Urbut et al. 2019). Further, this result is robust to different statistical thresholds: 91% and 80% of genes have one or more eQTLs when using a 5% and 1% FDR threshold, respectively.
When we performed joint analyses (Urbut et al. 2019) to identify ubiquitous and context-dependent eQTLs, we found that 77.53% of significant eQTLs were shared across all 12 conditions, whereas 6.70% were condition specific and the remaining 15.78% were shared between two and 11 conditions (mean = 10.40 ± 3.42 [SD] conditions) (for general patterns, see Fig. 4A–C; for example eQTLs, see Fig. 4D,E). We also found substantial evidence for a subset of context-dependent eQTLs known as “response eQTLs,” which we define as SNPs that do not affect gene expression levels in the control condition but for which genetic effects are revealed by experimental treatment (or vice versa) (Supplemental Table S5). On average, we found that 11.28% ± 2.91% (SD) of all tested genes contained at least one response eQTL for a given treatment–control pair, with the strongest evidence for response eQTLs observed for the treatment that induced the largest overall shift in transcriptome dynamics (i.e., dexamethasone, which revealed response eQTLs at 14.98% of genes).
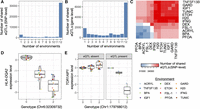
Environmental variation reveals context-dependent eQTLs. (A) Number of eQTLs shared between N environments using a mashR, a joint analysis approach. N is plotted on the x-axis and ranges from one (i.e., the eQTL is present in only one cellular environment) to 12 (i.e., the eQTL is present in all 12 cellular environments). (B) Same plot as in A, but sharing is defined at the gene rather than the SNP level. (C) Number of eQTLs shared between a given pair of cellular environments using a mashR, a joint analysis approach. The diagonal represents the number of eQTLs in the focal cellular environment. Note that ACRYL, BPA, PFOA, and FSL-1 had lower sample sizes than the other eight cellular environments, and their clustering thus reflects differences in power. (D,E) Examples of ubiquitous and context-dependent eQTLs, identified using a mashR, a joint analysis approach. The y-axis shows the mean, normalized expression levels for a given gene estimated in each environment, after regressing out three surrogate variables.
Both ubiquitous and context-dependent eQTLs were identified for many phenotypically and clinically relevant genes. For example, we identified four eQTLs that regulate the expression of NFKB1; three of these eQTLs are ubiquitous (Chr 4: 102692410, 102700807, 102711391), whereas one is condition specific and only observed in GARD-stimulated cells (Chr 4: 103062159; mashR LFSR = 0.042 and posterior effect size = −0.165). Key immune genes were also found to harbor context-dependent eQTLs. For example, we identified two SNPs associated with TLR10 expression only in FSL1-stimulated cells (Chr 4: 38601324 and 38687410; LFSR = 0.029 and 0.023; posterior effect size = −0.128 and −0.173). Similarly, several interferon regulatory factors harbor both ubiquitous eQTLs (IRF2: Chr 4: 184215264, 184515101, and 184747221; IRF3: Chr 19: 49960725 and 49225171; IRF7: Chr 11: 1078619, 354014, 377139, 537130, 636702, 658618, and 840034), as well as genetic effects that are only found following biologically relevant treatments such as IFNG and dexamethasone (IRF2: Chr 4: 184883814 and 184877973; IRF3: Chr 19: 49723947; IRF7: Chr 11: 755659, 793588, and 786452).
To validate and contextualize the eQTLs we identified, we performed two follow-up analyses: (1) we tested for an expected mechanistic pattern, namely, enrichment of eQTLs within accessible chromatin regions and active regulatory regions, and (2) we tested for overlap between our eQTL-containing genes (“eGenes”) and eGenes identified by the GTEx Project in untreated LCLs (GTEx Consortium 2017). First, as others have observed (Nédélec et al. 2016; GTEx Consortium 2017), we found that SNPs within accessible chromatin regions were more likely to be identified as ubiquitous (P = 0.042, hypergeometric test) and context-dependent eQTLs (P = 4.01 × 10−4; enrichment analyses performed using publicly available ATAC-seq data [Buenrostro et al. 2013] from untreated LCLs [Banovich et al. 2018]). We also found that both ubiquitous and context-dependent eQTLs were mostly enriched within ENCODE-defined chromatin states associated with active gene regulation and transcription, such as “active promoter” and “strong enhancer” (Supplemental Fig. S4; The ENCODE Project Consortium 2012). Second, we found that genes containing ubiquitous eQTLs overlapped significantly with GTEx LCL eGenes (P = 6.22 × 10−3, hypergeometric test), suggesting there is a core set of eQTLs identified across study designs and cell states. However, genes containing context-dependent eQTLs did not overlap with GTEx LCL eGenes (P = 0.465), suggesting that our cellular perturbations revealed previously uncharacterized genetic effects on gene expression. In support of this idea, genes with evidence for eQTLs in our data set, but not in GTEx, were enriched for genes that responded to at least one experimental treatment (P = 4.66 × 10−4).
Phenotypic relevance of ubiquitous and context-dependent eQTLs
Our next goal was to understand the phenotypic relevance of context-dependent versus ubiquitous eQTLs and to test the hypothesis that context-dependent eQTLs are especially important for human diseases and adaptively relevant traits. To do so, we asked whether our context-dependent and ubiquitous eQTL SNP sets (or their associated genes) were enriched within three publicly available data sets: (1) GWAS hits for human traits and diseases (Hindorff et al. 2009); (2) sets of genes that are thought to lie along the causal pathway linking genetic variation, gene expression, and 114 complex traits and diseases (inferred through PTWAS) (Zhang et al. 2020); and (3) loss-of-function, mutation-intolerant genes (as curated by ExAC) (Lek et al. 2016).
First, we found that both context-dependent and ubiquitous eQTLs were enriched for loci previously implicated in GWAS of human complex traits and diseases, although contrary to our expectations, these results were slightly stronger for ubiquitous compared with context-dependent eQTLs (P-value = 5.74 × 10−15 and <10−16 for context-dependent and ubiquitous eQTLs, respectively, hypergeometric test) (Fig. 5C). Second, we found that context-dependent eGenes were enriched for 11 complex traits, largely focused on immune system traits and disorders such as platelet counts (q = 2.47 × 10−4, hypergeometric test), neutrophil counts (q = 0.086), and rheumatoid arthritis (q = 0.086) (Supplemental Table S10). In contrast, ubiquitous eGenes were enriched for approximately half the number of complex traits (n = 6), again including many immune phenotypes such as inflammatory bowel disease (q < 10−16) and lymphocyte counts (q = 0.053) (Fig. 5A; Supplemental Table S10).
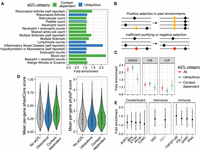
Phenotypic relevance and evolution of context-dependent eQTLs. (A) Diseases and heath conditions for which associated genes (identified via PTWAS) (Zhang et al. 2020) are enriched among genes with one or more context-dependent or ubiquitous eQTLs. The x-axis represents the fold enrichment estimate from a Fisher's exact test. (B) Evolutionary forces that potentially maintain context-dependent eQTLs: (1) positive selection on beneficial mutations (yellow) or (2) inefficient purifying or negative selection that fails to remove deleterious mutations (red). (C) Overlap of ubiquitous, context-dependent, and all (ubiquitous and context-dependent) eQTLs with (1) the full catalog of GWAS-associated loci (Hindorff et al. 2009), (2) iHS outlier loci (Johnson and Voight 2018), and (3) genes annotated as loss of function and mutation intolerant (Lek et al. 2016). The y-axis represents fold enrichment from a Fisher's exact test, with error bars denoting the 95% confidence interval for each estimate. (D) Distribution of mean per-gene phastCons and phyloP scores for genes with no eQTLs, one or more ubiquitous eQTLs, and one or more context-dependent eQTLs. (E) Overlap of response eQTLs, identified in a given condition, with iHS outlier loci (Johnson and Voight 2018). The y-axis represents fold enrichment from a Fisher's exact test, with error bars denoting the 95% confidence interval for each estimate.
Finally, we tested whether ubiquitous or context-dependent eGenes were enriched for mutation-intolerant genes, which are thought to be phenotypically relevant and essential for life given that no adult humans (in a large population sample known as ExAC) contain mutations that would result in a truncated or loss-of-function protein (Lek et al. 2016). Here, we found that context-specific eGenes were enriched among mutation-intolerant genes (odds = 1.306, P = 5.45 × 10−8, Fisher's exact test), but ubiquitous eGenes were not and even trended toward being under enriched (odds = 0.982, P = 0.8102) (Fig. 5C). These results parallel those obtained by the GTEx project: eGenes that were shared across many tissues were significantly depleted for loss-of-function, mutation-intolerant genes, whereas tissue-specific eGenes were significantly enriched (GTEx Consortium 2017). These results were interpreted as evidence for purifying selection on regulatory variants that involve many tissues and are thus more likely to have deleterious, pleiotropic effects. In contrast, tissue-specific eQTLs, and in our case, context-dependent eQTLs, appear to be more likely to “escape” the effects of purifying selection and to persist in loss-of-function, mutation-intolerant genes.
Evolutionary forces maintaining ubiquitous and context-dependent eQTLs
The analyses described in the previous section point toward negative selection as a key evolutionary force that patterns genetic effects on gene expression. However, several previous studies of context-dependent eQTLs uncovered by immune stimulation have instead emphasized a role for positive selection (Nédélec et al. 2016; Quach et al. 2016; Kim-Hellmuth et al. 2017; Harrison et al. 2019). For example, Kim-Helmuth and colleagues (2017) found that monocyte eQTLs with differential effect sizes in baseline versus immune-stimulated conditions were enriched among genomic regions with recent signatures of positive selection; a similar result was found for ubiquitous eQTLs, although the enrichment was weaker. Drawing on this work and our results thus far, we were motivated to further explore the role of both positive and negative selection in generating context-dependent and ubiquitous eQTLs.
To do so, we first drew on publicly available measures of sequence conservation across mammals to understand whether context-dependent and ubiquitous eQTLs fall in rapidly evolving versus conserved regions of the human genome. We found that ubiquitous eGenes showed both lower phastCons (P = 1.765 × 10−5, Wilcoxon signed-rank test) and phyloP scores (P = 1.749 × 10−4) than background expectations, indicating that ubiquitous eGenes are underrepresented in evolutionarily conserved genes. In contrast, context-dependent eGenes showed higher phastCons (P = 5.01 × 10−3, Wilcoxon signed-rank test) and phyloP scores (P = 1.35 × 10−4) (Fig. 5D) than background expectations, indicating that they are overrepresented in conserved genes. Similar to our results using mutation-intolerant gene annotations, these analyses suggest that, in highly conserved and putatively essential genes, ubiquitous eQTLs are selected against and depleted, whereas context-dependent eQTLs are “hidden” from selection and persist (Fig. 5B).
Next, we explicitly investigated a role for positive selection by obtaining per-site estimates of the integrated haplotype score (iHS), a commonly used measure of within-population recent positive selection (Voight et al. 2006; Johnson and Voight 2018). We obtained genome-wide iHS estimates for each of the populations included in our study and identified putative selection candidates as loci that fell in the more than 99th percentile of |iHS| values in two or more populations (as in Nédélec et al. 2016; Kim-Hellmuth et al. 2017). When we overlapped these selection outliers with our eQTLs, we found that loci that functioned as eQTLs (in all or fewer than 12 cellular environments) were more likely to show signatures of positive selection (odds = 1.332, P = 3.702 × 10−13, Fisher's exact test). This result was especially strong for ubiquitous eQTLs (odds = 1.389, P = 1.688 × 10−13) but less so for context-dependent eQTLs (odds = 1.152, P = 0.086) (Fig. 5C), in contrast to patterns observed in previous work focusing on immune stimulations (Nédélec et al. 2016; Kim-Hellmuth et al. 2017). Given that our study focuses on a more diverse set of environmental perturbations, we wondered whether heterogeneity between treatments could explain the lack of enrichment of iHS outliers in the total set of context-dependent eQTLs. When we analyzed each condition separately, we found statistically significant enrichment of iHS outliers in response eQTLs for two immune stimuli, namely, FSL-1 (odds = 1.362, P = 0.036) and TNFSF13B (odds = 1.512, P = 0.031), although we note these effect sizes were small. Further, we found that these enrichment effect sizes were similar for immune versus hormone treatments (β = 0.065, P = 0.297) (Fig. 5E) but that immune response eQTLs were more strongly enriched for iHS outliers than environmental contaminant and cell stressor response eQTLs (β = 0.246, P = 5.82 × 10−4, linear model). Thus, our data are consistent with the hypothesis that positive selection has played a greater role in shaping loci involved in the response to immune and hormone molecules relative to loci involved in the response to man-made chemicals and novel cell stressors.
Discussion
Despite major advances in genomic technologies, understanding the genetic basis of human complex traits remains a challenge. Although decades of GWAS have uncovered many loci for common diseases and health-related traits, there is a general consensus that mapping additive effects will not allow us to account for the total estimated genetic component of most phenotypes. This problem is known as the “missing heritability” problem and has been used to argue for a critical role for G×E interactions (Manolio et al. 2009). More specific support that G×E interactions contribute to human complex traits comes from analyses of variance and observations that the heritability of key traits has increased in recent decades, despite minimal changes in the genetic makeup of populations (Jelenkovic et al. 2016; Czamara et al. 2019; Dahl et al. 2020; Sulc et al. 2020). However, despite multiple lines of evidence that G×E interactions are likely important, we have made little progress in mapping the specific loci involved in G×E interactions. This reality limits our ability to understand their distribution, effect sizes, mechanisms of action, and evolution.
Here, we use a cell culture model to overcome issues faced by observational studies and to maximize our power to detect G×E interactions in the form of context-dependent eQTLs. Specifically, we exposed cells from genetically well-characterized individuals to 12 controlled, in vitro environments and asked whether genetic variation predicted individual responses at the transcriptional level. We found that diverse environmental perturbations induced diverse gene regulatory programs, whereas the genetic control of gene expression levels was more environmentally robust: at least 70% of ancestry effects on the transcriptome were consistent in effect size across environments, as were 78% of eQTLs. These results agree with previous work using smaller sample sizes, fewer environments, as well as cross-tissue comparisons (Moyerbrailean et al. 2016; Nédélec et al. 2016; Quach et al. 2016; GTEx Consortium 2017).
Nevertheless, our experiments do show that a nonnegligible portion of the transcriptome's genetic architecture is environmentally sensitive: 16% of eQTLs were only shared between two and 11 conditions, whereas 6% were specific to a single condition. Further, we found that our observed patterns of cis eQTLs sharing generally mimicked the bimodal distribution observed by GTEx for cross-tissue comparisons (GTEx Consortium 2017). In other words, the largest categories of cis eQTLs were those shared across all 12 environments, followed by those that were specific to a single environment (Fig. 4A–C). Many of the eQTLs revealed by our environmental treatments were both uncharacterized (e.g., unannotated in GTEx LCLs) (GTEx Consortium 2017) and putatively phenotypically relevant (Fig. 5A,C), arguing that environmental diversity should be incorporated into eQTL mapping studies whenever possible (e.g., Moyerbrailean et al. 2016). In fact, although an estimated 88% of all genetic variants associated with complex human traits and diseases lie outside of protein-coding regions (Edwards et al. 2013; MacArthur et al. 2017), the most comprehensive eQTL studies to date (e.g., GTEx) (GTEx Consortium 2017) have only accounted for about one-half of known regulatory GWAS hits. This state of affairs has motivated recent calls to expand the set of cellular conditions under which we study links between genotype, gene regulation, and disease (Umans et al. 2021); our study design provides one feasible avenue for doing so.
Although previous work has applied in vitro environmental manipulations to study G×E interactions (Barreiro et al. 2012; Lee et al. 2014; Çalışkan et al. 2015; Moyerbrailean et al. 2016; Nédélec et al. 2016; Quach et al. 2016; Kim-Hellmuth et al. 2017; Piasecka et al. 2018; Findley et al. 2021), the number of environments we assay here using genome-wide data sets and appreciable sample sizes is unprecedented. Further, previous experiments have focused heavily on environmental manipulations involving pathogens and other immune stimuli, and we speculate that context-specific eQTLs in the immune system may manifest and evolve in different ways than context-specific eQTLs for other stimuli, especially stimuli that have not been common throughout human evolutionary history. According to evolutionary theory, most new mutations are overall deleterious (e.g., because of their pleiotropic effects) and will be selected against; in contrast, context-dependent regulatory mutations can provide more targeted fitness benefits and are thus thought to play a key role in the evolution of adaptively relevant trait (Carroll 2000; Wray 2007; Umans et al. 2021). In support of this argument, several studies have shown that eQTLs that control the gene regulatory response to infection are under positive selection (Nédélec et al. 2016; Quach et al. 2016; Kim-Hellmuth et al. 2017; Harrison et al. 2019). However, it is unlikely that natural selection has similarly shaped eQTLs that control the response to stimuli recently introduced into human environments (e.g., man-made chemicals). In line with this thinking, we find little evidence that positive selection has shaped G×E loci revealed by novel cell stressors or chemicals, but we do find evidence for this pattern for immune-responsive loci (Fig. 5). Our results point toward a model in which cellular perturbations with distinct evolutionary histories may produce consistently divergent patterns (although this model needs further confirmation). Moving forward, we argue for greater consideration of the evolutionary history of a given environmental exposure when studying and interpreting G×E interactions.
There are several limitations to the present study, as well as open directions for future work. First, our sample sizes were more than reasonable for eQTL mapping in all 12 cellular environments, however, they were not identical. Nevertheless, our use of a Bayesian joint analysis framework (i.e., mashR) to pool information across conditions should circumvent many potential issues associated with variable sample sizes; this is because the original sample size for a given environment does not reflect the “effective” sample size used by the program, which provides power gains from sharing information (Urbut et al. 2019). In support of this point, previous analyses of GTEx data with mashR found that genetic effects on gene expression estimated across dozens of tissues in the body clustered by organ system (e.g., brain regions all clustered together) rather than sample size (Urbut et al. 2019). Second, although many SNPs in the human species have minor allele frequencies <5%, our sample sizes were not sufficient to assess the contribution of these rare variants. Emerging evidence suggests these rare variants explain ∼25% of the heritability of gene expression levels (Hernandez et al. 2019), and as future studies with larger sample sizes are performed, it will be exciting to assess their context-dependency. Third, our study design relied on Epstein–Barr virus transformed LCLs, because they are a replenishable and shareable resource and are commercially available for genetically well-characterized individuals (The 1000 Genomes Project Consortium 2012). They have also been used extensively for functional genomic work (Stranger et al. 2007; Veyrieras et al. 2008; Pickrell et al. 2010; Bell et al. 2011; Mangravite et al. 2013), and previous studies have shown that (1) genomic results from LCLs replicate in primary tissues (Stranger et al. 2007; Veyrieras et al. 2008; Pickrell et al. 2010; Bell et al. 2011; Mangravite et al. 2013) and (2) gene expression levels in newly established LCLs maintain a strong individual signature (Çalışkan et al. 2011). However, gene regulation in LCLs is not identical to their progenitor B cells, and the transformation process is known to induce certain artifacts (Redon et al. 2006; Akey et al. 2007). Thus, although we believe LCLs represent a powerful model for high-throughput and large-scale G×E mapping, future work in primary tissues or induced pluripotent stem cells will be important for corroborating the patterns we see here. Finally, we did not generate environment-specific data on additional gene regulatory mechanisms such as DNA methylation, chromatin accessibility, or chromatin state. Although this work was beyond the scope of the present study, it is a key avenue for future research and for understanding the molecular mechanisms that generate context-dependent eQTLs (e.g., environmentally dependent transcription factor binding). Although functional fine-mapping of individual G×E interactions can be laborious and difficult, several recent studies offer promising new avenues for using gene regulatory assays to understand the path from genotype to phenotype (Johnson et al. 2018; Garske et al. 2019), including under diverse environmental conditions.
Technological advances have fueled the ascent of personal genomics and the promise of precision medicine. However, to unlock this potential, we must first understand how the environmental and genetic interactions unique to each individual contribute to variation in complex traits. Our study provides a comprehensive window into the environmental dependency of the human transcriptome, including genetic effects that are only revealed by environmental change—a class of variants known as “cryptic genetic variation” (Gibson and Dworkin 2004; Paaby and Rockman 2014). Cryptic alleles likely drive the significant increase in heritability we observe following cellular treatments, and could play a major role in explaining the “missing heritability” problem; again, these results argue for a critical role for G×E interactions in driving variation in complex traits.
Methods
Cell culture, experimental cell treatments, and mRNA-seq
LCLs were obtained for 544 unrelated individuals from 10 different African and European populations included in the 1000 Genomes study (The 1000 Genomes Project Consortium 2012). Population-specific panels were immortalized by different 1000 Genomes investigators at different times, and thus, batch effects could confound population-level analyses of differential expression; however, we instead focus here on differences between broad ancestry groups (African vs. European) in an effort to circumvent issues with analyzing individual populations. Further, all cell lines were cultured in a controlled setting before our experiments. All cell lines were ordered from Coriell Institute, and live cultures were shipped overnight to Princeton University in randomized batches of 25 (Supplemental Table S1). Cells were cultured in parallel for 5–11 d until 12 million cells were available to seed (at a density of 1 million cells/2.5 mL of media in a 12-well plate). After an overnight incubation period, 12 environmental treatments (Table 1; Supplemental Table S2) were added to each of the 12 wells (for treatment concentrations, see Supplemental Table S2). We note that, following previous in vitro exposure studies (Barreiro et al. 2010; Nédélec et al. 2016; Snyder-Mackler et al. 2016; Harrison et al. 2019), we did not choose our treatment concentrations to be physiologically realistic; instead, we chose treatments concentrations that induced robust gene expression responses across all environments in order to reveal G×E interactions (Table 1). After 4 h, cells were washed, harvested, and preserved in lysis buffer for downstream RNA work. We chose a 4-h exposure to capture the early stages of gene expression responses to perturbation and to ensure that cellular processes that occur on longer timescales (e.g., cell divisions, replication, and death) would minimally impact our results. This timescale has also been used successfully in previous studies of response eQTLs in immune cells (Barreiro et al. 2010; Pai et al. 2016; Khan et al. 2020).
Total RNA was extracted from each sample using Zymo's Quick-RNA 96 kit, following the manufacturer's instructions. mRNA-seq libraries were prepared using the published TM3′seq protocol (Pallares et al. 2020) and a CyBio FeliX liquid handling robot (Analitik Jena). The total data set (n = 5223 libraries) was sequenced across four runs of the Illumina NovaSeq platform. Each sample was sequenced to a mean depth of 2.199 ± 2.731 (SD) million reads using 100-bp single-end sequencing. We note that, because we are using a 3′ rather than a full transcript approach, this amount of sequencing is adequate. For example, previous work has shown that ∼1 million reads provide accurate and reproducible estimates of gene expression levels using TM3′seq (Pallares et al. 2020). This is because the 3′ approach targets only the ends of transcripts (near the poly(A) tail), and consequently, fewer reads are needed to estimate the expression of a given gene because reads are concentrated in a small amount of sequence (rather than being distributed across the entire transcript).
Additional information relating to cell culture, experimental cell treatments, and mRNA-seq data generation is available in the Supplemental Materials.
Low-level processing of mRNA-seq and genotype data
Following sequencing, we trimmed FASTQ files (Martin 2011), mapped the filtered reads to the human reference genome (hg38) (Dobin et al. 2013), extracted counts of mapped reads (Anders et al. 2015), and filtered out low-coverage samples and genes (Supplemental Table S3). Next, we normalized the count data (Law et al. 2014), conducted surrogate variable (SV) analysis, and regressed out three SVs to remove batch and technical effects (Leek and Storey 2007). Phased genotype calls derived from about 30× whole-genome sequence data were downloaded for 454 individuals included in our study (Supplemental Table S4; Byrska-Bishop et al. 2022). We used PLINK (Purcell et al. 2007) to remove low-quality and low-MAF SNP and to perform LD filtering (Purcell et al. 2007). We used a set of 7,205,828 filtered SNPs to generate a PCA in PLINK (Purcell et al. 2007), as well as a genetic relatedness matrix (GRM) in GCTA (Yang et al. 2011). Finally, to prepare for cis eQTL mapping, we extracted the 1,950,183 SNPs that fell within 500 kb of the transcription start or end site of protein-coding genes. Additional information relating to low-level processing of mRNA-seq and genotype data is available in the Supplemental Materials (see also Supplemental Figs. S5, S6).
Testing for ancestry and treatment effects on gene expression levels
To identify genes for which gene expression was predicted by ancestry within a given condition, we used linear models implemented
in limma (Law et al. 2014). Specifically, for each of the 12 cellular environments, we ran the following model on the SV-corrected residuals for each
gene:
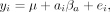 (1)
where yi is the gene expression level estimate for sample i, μ is the intercept, ai represents ancestry of the focal sample (AFR or EUR), βa is the corresponding estimate of the ancestry effect, and ei represents environmental noise.
(1)
where yi is the gene expression level estimate for sample i, μ is the intercept, ai represents ancestry of the focal sample (AFR or EUR), βa is the corresponding estimate of the ancestry effect, and ei represents environmental noise.
To identify genes for which gene expression was significantly affected by a given treatment, we used a similar modeling approach
again implemented in limma (Law et al. 2014). Specifically, for each of the 11 treatments, we ran the following model on the SV-corrected residuals for each gene:
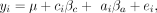 (2)
where all variables are as described above, with the addition of ci, which denotes the condition (treatment or control), and βc, which is the corresponding estimate of the treatment effect. After running both models 1 and 2, we extracted the P-value associated with the effect of interest (ancestry or treatment, respectively) and corrected for multiple hypothesis
testing using a Storey–Tibshirani FDR approach (Storey and Tibshirani 2003). A summary of the results of these analyses is provided in Supplemental Tables S5 and S8. We also extracted the effect size and standard error estimates associated with the effects of interest for downstream analyses.
(2)
where all variables are as described above, with the addition of ci, which denotes the condition (treatment or control), and βc, which is the corresponding estimate of the treatment effect. After running both models 1 and 2, we extracted the P-value associated with the effect of interest (ancestry or treatment, respectively) and corrected for multiple hypothesis
testing using a Storey–Tibshirani FDR approach (Storey and Tibshirani 2003). A summary of the results of these analyses is provided in Supplemental Tables S5 and S8. We also extracted the effect size and standard error estimates associated with the effects of interest for downstream analyses.
Testing for cis eQTL effects on gene expression levels
We used the R package matrixeQTL (Shabalin 2012) to test for cis eQTLs (within 500 kb) that affected gene expression variation in each cellular environment separately. Specifically, for
each of the 12 environments, we ran the following model on the SV-corrected residuals for each gene:
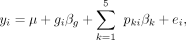 (3)
where gi denotes the genotype of individual i in terms of number of copies of the minor allele (zero, one, or two), and βg is the corresponding estimate of the genotype effect. pki is the loading for principal component k for individual i (from a PCA on the filtered genotype matrix, as described above), and βk is the estimate of the principal component effect. For each gene–SNP combination, we extracted the P-values, effect sizes, and standard error estimates associated with the genotype effect. We then used a Storey–Tibshirani
FDR (Storey and Tibshirani 2003) to correct for multiple hypothesis testing. Results are summarized in Supplemental Table S8.
(3)
where gi denotes the genotype of individual i in terms of number of copies of the minor allele (zero, one, or two), and βg is the corresponding estimate of the genotype effect. pki is the loading for principal component k for individual i (from a PCA on the filtered genotype matrix, as described above), and βk is the estimate of the principal component effect. For each gene–SNP combination, we extracted the P-values, effect sizes, and standard error estimates associated with the genotype effect. We then used a Storey–Tibshirani
FDR (Storey and Tibshirani 2003) to correct for multiple hypothesis testing. Results are summarized in Supplemental Table S8.
We also ran a second analysis, using the same approach described above, but pooling the data across all 12 conditions. For
this analysis, condition was also included as a covariate in the following model:
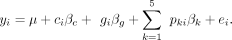 (4)
Finally, we note that we explored an alternative analysis strategy, in which we regressed out one to 20 principal components
from the normalized (but not SV-corrected) gene expression matrix before fitting the models described in Equation 3 (as in Nédélec et al. 2016; GTEx Consortium 2017). We did not find that this approach consistently increased our power to detect eQTLs across the 12 cellular environments
and therefore opted for using the SV-corrected data in which a consistent pipeline could be applied to the full data set.
(4)
Finally, we note that we explored an alternative analysis strategy, in which we regressed out one to 20 principal components
from the normalized (but not SV-corrected) gene expression matrix before fitting the models described in Equation 3 (as in Nédélec et al. 2016; GTEx Consortium 2017). We did not find that this approach consistently increased our power to detect eQTLs across the 12 cellular environments
and therefore opted for using the SV-corrected data in which a consistent pipeline could be applied to the full data set.
Estimating sharing of ancestry, treatment, and genotype effects
To understand the degree to which treatment, ancestry, or genotype effects mapped across different conditions have shared versus context-dependent effects, we used the empirical Bayes approach implemented in the R package mashR (Urbut et al. 2019). Although previous studies have instead used linear models to (1) test for interaction effects between condition and genotype or ancestry or (2) test for ancestry or genotype effects on the fold change in gene expression levels estimated between treatment and control conditions (Nédélec et al. 2016; Kim-Hellmuth et al. 2017; Harrison et al. 2019), joint analysis via mashR is more appropriate to our design and provides many advantages. mashR is explicitly designed for quantitative assessment of effect size heterogeneity across conditions, increases power via joint analysis, and exploits patterns of similarity to provide improved estimates of effect size in each condition. It also provides a common framework for comparing effect sizes across many conditions rather than relying on the comparison of many different “significant” lists derived from arbitrary P-value or FDR cutoffs. Similar meta-analytic approaches have been successfully applied to other high-dimensional data sets, such as GTEx (GTEx Consortium 2017). We used similar pipelines to estimate sharing of treatment, ancestry, and genotype effects using mashR (Urbut et al. 2019), with small modifications appropriate to each predictor variable.
First, using our multitreatment (n = 11) estimates of environmental effect sizes, we evaluated effect size concordance between all pairwise combinations of treatment–control pairs. Here, we followed the pipeline provided by the mashR investigators for data sets that use the same reference or control condition samples across multiple comparisons (https://stephenslab.github.io/mashr/articles/intro_correlations.html). Specifically, we corrected for correlations among conditions in our data and then used a combination of canonical and data-driven covariance matrices to fit the mashR model. From the mashR output, we extracted the posterior mean effect size and local false sign rate (LFSR) estimates. Following the investigators’ recommendations, we considered a gene to have “shared” treatment effects across an arbitrary number of treatment if it has a LFSR < 10% for at least one treatment and posterior effect sizes of similar magnitude (within a factor of two) for the other treatments. We always used the treatment with the lowest LFSR as the reference for the effect size comparison (we note that similar results were obtained when we used the median effect size across all treatments with LFSR < 10%). In cases in which the treatment effect was shared across all 11 treatment–control pairs (aka “ubiquitous”), we considered the treatment effect to be “context-dependent.” Finally, we considered a gene to show a special case of context-dependency, namely, “condition-specific” effects, if it had evidence for treatment effects at a LFSR < 10% and the posterior effect size estimate was not within a factor of two of any other treatment.
Second, using our multicondition (n = 12) estimates of ancestry effect sizes, we evaluated effect size concordance between all pairwise combinations of conditions. To do so, we followed the standard pipeline provided by the mashR investigators (https://stephenslab.github.io/mashr/articles/intro_mash_dd.html), which uses a combination of canonical and data-driven covariance matrices to fit the mashR model. From the mashR output, we again extracted the posterior mean effect size and LFSR estimates and used the same approach described above to identify ubiquitous, context-dependent, and condition-specific effects.
Finally, we assessed effect size sharing for cis eQTLs mapped across all 12 cellular environments. In this case, there were too many tested gene–SNP pairs to evaluate all of them in the mashR framework (n = 8,109,941), so we followed the investigators’ recommendations and focused on 66,614 gene–SNP pairs with some evidence for cis eQTLs from matrixeQTL (Shabalin 2012) (FDR < 10% in at least one condition). We followed the pipeline suggested for eQTLs (https://stephenslab.github.io/mashr/articles/eQTL_outline.html) and used a combination of canonical and data-driven covariance matrices derived from 50,000 randomly chosen gene–SNP pairs to fit the mashR model. We then computed posterior summaries for the 66,614 gene–SNP pairs of interest using the model fit to randomly selected data. Finally, we extracted the posterior mean effect size and LFSR estimates and used the same definitions described above to identify ubiquitous, context-dependent, and condition-specific treatment effects. In some cases, we also summarized our eQTL results at the gene rather than the SNP level; in these cases, we report the number of unique genes that have at least one eQTL that is shared between a given number of conditions (12 = ubiquitous and one to 11 = context-dependent). We note that with these definitions a given gene can be reported in more than one category.
A summary of the mashR treatment, ancestry, and cis eQTL results are provided in Supplemental Tables S5 and S8. An overview of our analysis pipeline is provided in Supplemental Figure S7. All estimates of LFSR and posterior mean effect sizes are available on GitHub and as Supplemental Data. To understand the robustness of our conclusions, we also (1) repeated our treatment and ancestry analyses after randomly down-sampling our data to n = 120 per environment (Supplemental Fig. S8), (2) compared our treatment effect size estimates to previous work (Supplemental Fig. S9), and (3) performed follow-up analyses to understand which treatments were driving the sharing patterns we observed (Supplemental Fig. S10).
Testing the degree to which ancestry and genotype affect the response to treatment
We also used mashR to understand whether (1) individuals of African versus European ancestry responded differently to a given
treatment and (2) genotype affects the response to a given treatment, as evidence for both phenomena has been shown in previous
work (Nédélec et al. 2016). To test point 1, we asked whether the posterior mean estimates of the ancestry effect were different between the treatment
and control conditions for all 11 treatments; if so, this would indicate an effect of ancestry on the response to a given
treatment. We used the same pipeline described for estimating sharing of treatment effects, and we considered a gene to show
differential responses to treatment as a function of ancestry if the LFSR was <10% in either the treatment or control condition
(or both) and the posterior mean effect size estimates were not within a factor of two of one another. Using this approach,
we found no evidence for ancestry effects on the response to treatment (except for one gene in the IGF1 data set). To validate
this result, we also ran models in limma (Law et al. 2014) that included an explicit interaction effect (βcxa) between treatment and ancestry. Specifically, for each of the 11 treatments, we ran the following model on the SV-corrected
residuals for each gene:
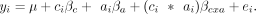 (5)
These results revealed no significant ancestry × treatment (βcxa) effects at a 10% FDR. We note that if we apply mashR to the interaction effect itself (βcxa), we do find a modest result (mean ± SD of 210.18 ± 106.73 genes with a LFSR < 10% per treatment). However, it is not recommended
to perform joint analysis when there is no initial evidence for the effect of interest, and we therefore caution that the
mashR analysis of βcxa is exploratory.
(5)
These results revealed no significant ancestry × treatment (βcxa) effects at a 10% FDR. We note that if we apply mashR to the interaction effect itself (βcxa), we do find a modest result (mean ± SD of 210.18 ± 106.73 genes with a LFSR < 10% per treatment). However, it is not recommended
to perform joint analysis when there is no initial evidence for the effect of interest, and we therefore caution that the
mashR analysis of βcxa is exploratory.
To test point 2, we asked whether the posterior mean estimates of the genotype effect were different between the treatment and control condition for all 11 treatments; if so, this would indicate an effect of genotype on the response to a given treatment. We used the same pipeline described for estimating sharing of cis eQTL effects, and we considered a gene to show differential responses to treatment as a function of genotype if the LFSR was <10% in either the treatment or control condition (or both) and the posterior effect size estimates were not within a factor of two of one another. We found several thousand response eQTLs for each treatment, and these results are summarized in Supplemental Table S5.
Relationship between FST and ancestry-associated gene expression variation
We were interested in testing the hypothesis that genetic variation contributes to the observed differentiation in gene expression between African and European individuals. To do so, we followed the approach of Nédélec et al. (2016) and asked whether genes with ancestry effects show higher FST values between African and European populations relative to non-ancestry-associated genes. To do so, we first calculate the FST for all 7,205,850 variants in our pre-LD filtered genotype data set. We then generated gene-specific estimates by averaging FST values for variants within 5 kb (upstream or downstream) of the transcription start site of a given gene. Next, we used a Wilcoxon signed-rank test to ask whether FST values differed between genes with no evidence for an ancestry effect (LFSR > 10% in all conditions) and (1) genes with any evidence for an ancestry effect in any condition or (2) genes with evidence for an ancestry effect in at least two-thirds of conditions. In a second approach, we also used linear models to test whether the number of cellular environments a gene showed significant ancestry effects in was predictive of the gene's average FST value.
To further understand the contribution of genetic variation and selection to population differences in gene expression, we compared PST values for ancestry-associated genes to FST estimates between African and European individuals. PST is a phenotypic analog of FST (and a proxy for QST), and comparisons between the two measures can thus provide evolutionary insight (Lamy et al. 2012; Leinonen et al. 2013). Specifically, PST > FST is interpreted as evidence for diversifying selection, indicating different local optima for different populations. In contrast, PST < FST signifies uniform selection (also known as homogeneous, convergent, or stabilizing selection) and PST = FST suggests that phenotypic divergence between populations mimics neutral genetic divergence and is thus largely controlled by genetic drift. We used the R package Pstat (Da Silva and Da Silva 2018) to calculate PST for ancestry-associated genes identified in each condition (FDR < 10%), as well as a random sample of 500 genes. We compared these values to the genome-wide average of FST estimates between African and European individuals included in our data set (for whom we also had whole-genome sequencing data, n = 454) (see Fig. 2E; Supplemental Fig. S3). To estimate 95% confidence intervals for the mean genome-wide FST estimate, we performed 1000 replicates of bootstrap resampling.
Evolutionary analysis of ubiquitous and context-dependent eQTLs and eGenes
We performed two sets of analyses to address the roles of positive and negative selection in maintaining ubiquitous and context-dependent eQTLs. First, we obtained two publicly available estimates of sequence conservation: phyloP scores (Pollard et al. 2010) and phastCons scores (Spieth et al. 2005). The phyloP score measures the evolutionary conservation at each individual alignment site, with a positive sign indicating conservation and slower evolution than chance expectations, whereas a negative sign indicates relaxed constraint or positive selection and faster evolution than expected by chance. The phastCons score measures the probability that each nucleotide belongs to a conserved element, with a higher phastCons score representing greater sequence conservation. We obtained the per-site phyloP and phastCons scores from the 100-way vertebrate comparison available via the UCSC Genome Browser (Karolchik et al. 2014). Following the methods of Shang et al. (2020), we averaged the per-site measures across all exons in each protein-coding gene to obtain per-gene phyloP and phastCons scores. Finally, we compared the mean per-gene conservation scores for ubiquitous eGenes and context-dependent eQTL genes to non eGenes using a Wilcoxon signed-rank test.
Second, we investigated a role for positive selection by obtaining per-site estimates of the iHS, a commonly used measure of within-population recent positive selection (Voight et al. 2006; Johnson and Voight 2018). We obtained genome-wide iHS estimates for each of the 10 populations included in our study from Johnson and Voight (2018) and identified putative selection candidates as loci that fell in the more than 99th percentile of |iHS| values in two or more populations (as in Nédélec et al. 2016; Kim-Hellmuth et al. 2017). We then used Fisher's exact tests to test for enrichment of iHS outliers in our ubiquitous and context-dependent eQTL sets, as well as the sets of response eQTLs identified in each condition separately. Finally, we grouped the results of the response eQTL enrichment analyses into three treatment categories (immune stimuli, hormones, and environmental contaminants/novel cell stressors) (see Table 1) and used a linear model to ask whether the enrichment effect sizes consistently differed between the immune stimuli treatments and the other two treatment categories.
All statistical analyses were performed in R version 4.1.2.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE207049. Code is available at GitHub (https://github.com/AmandaJLea/LCLs_gene_exp) and as Supplemental Code. Most supporting data files are available on GitHub, but larger files are posted to Zenodo (https://zenodo.org/record/6595427#.YrnVKJPMJuU). The supporting data files from GitHub and Zenodo are also available as Supplemental Data.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank all members of the Ayroles laboratory for their feedback and support during the completion of this project. We also thank Andrea Graham and members of the Graham laboratory for thoughtful input and discussion. We thank Aaron Wolf, Christina Hansen, and Edward Schrom for assistance with cell culture protocols and facilities. This study was supported by the Lewis-Sigler Institute for Integrative Genomics at Princeton University, as well as a grant from the National Institutes of Health/National Institute of Environmental Health Sciences to J.F.A. (R01-ES029929). A.J.L. was supported by a postdoctoral fellowship from the Helen Hay Whitney Foundation.
Author contributions: A.J.L. and J.F.A. designed the study. A.J.L. performed the experiments and analyzed the data. J.P. generated the mRNA-seq data. A.J.L. wrote the paper, with input from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276430.121.
-
Freely available online through the Genome Research Open Access option.
- Received November 23, 2021.
- Accepted September 7, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








