
Alignment of single-cell RNA-seq samples without overcorrection using kernel density matching
- Mengjie Chen1,2,
- Qi Zhan3,
- Zepeng Mu3,
- Lili Wang3,
- Zhaohui Zheng4,5,
- Jinlin Miao4,5,
- Ping Zhu4,5 and
- Yang I. Li1,2
- 1Section of Genetic Medicine, Department of Medicine, University of Chicago, Chicago, Illinois 60637, USA;
- 2Department of Human Genetics, University of Chicago, Chicago, Illinois 60637, USA;
- 3Committee on Genetics, Genomics and Systems Biology, University of Chicago, Chicago, Illinois 60637, USA;
- 4Department of Clinical Immunology, Xijing Hospital, Xi'an 710032, China;
- 5National Translational Science Center for Molecular Medicine, Xi'an 710032, China
Abstract
Single-cell RNA sequencing (scRNA-seq) technology is poised to replace bulk cell RNA sequencing for many biological and medical applications as it allows users to measure gene expression levels in a cell type–specific manner. However, data produced by scRNA-seq often exhibit batch effects that can be specific to a cell type, to a sample, or to an experiment, which prevent integration or comparisons across multiple experiments. Here, we present Dmatch, a method that leverages an external expression atlas of human primary cells and kernel density matching to align multiple scRNA-seq experiments for downstream biological analysis. Dmatch facilitates alignment of scRNA-seq data sets with cell types that may overlap only partially and thus allows integration of multiple distinct scRNA-seq experiments to extract biological insights. In simulation, Dmatch compares favorably to other alignment methods, both in terms of reducing sample-specific clustering and in terms of avoiding overcorrection. When applied to scRNA-seq data collected from clinical samples in a healthy individual and five autoimmune disease patients, Dmatch enabled cell type–specific differential gene expression comparisons across biopsy sites and disease conditions and uncovered a shared population of pro-inflammatory monocytes across biopsy sites in RA patients. We further show that Dmatch increases the number of eQTLs mapped from population scRNA-seq data. Dmatch is fast, scalable, and improves the utility of scRNA-seq for several important applications. Dmatch is freely available online.
Single cell RNA-sequencing (scRNA-seq) technology is transforming the study of cellular heterogeneity (Kumar et al. 2014; Villani et al. 2017), differentiation (Bendall et al. 2014; Durruthy-Durruthy et al. 2014), and cellular response to stress and stimulation (Shalek et al. 2014; Keren-Shaul et al. 2017; Golumbeanu et al. 2018). Gene expression levels in tens of thousands of single cells are now routinely measured in a single scRNA-seq experiment (Macosko et al. 2015), and more scRNA-seq data sets are becoming available each day. However, there remain considerable challenges in scRNA-seq data preprocessing as gene expression measurements from scRNA-seq are much noisier than compared to bulk RNA sequencing (Brennecke et al. 2013; Yuan et al. 2017). This makes integration and comparisons across multiple scRNA-seq experiments particularly difficult because each experiment can vary in capture efficiency, PCR efficiency, and dropout rates, and these technical effects can be cell type– or experiment-specific (Butler et al. 2018; Haghverdi et al. 2018).
Without the ability to integrate multiple scRNA-seq experiments, scRNA-seq studies are generally limited to two general applications: (1) to characterize cell type heterogeneity in a population of cells from one experiment (Kumar et al. 2014; Macosko et al. 2015; Villani et al. 2017); or (2) to infer cellular trajectory during development or response to stimuli from one sample. Although we have learned tremendously about cellular heterogeneity and cellular state transitions from these studies and a number of other applications that use data from a single scRNA-seq experiment, there are important applications in both basic and clinical science that require integration and comparisons across multiple scRNA-seq experiments (Butler et al. 2018; Haghverdi et al. 2018). Important applications that use scRNA-seq data include the identification of differentially expressed genes between two biological conditions and the mapping of expression quantitative trait loci (eQTLs).
Here, we describe Dmatch, a method that enables integration and comparisons across multiple scRNA-seq experiments. Dmatch uses an external panel of primary cells (Mabbott et al. 2013) to identify shared pseudo cell types across scRNA-seq samples and then finds a set of common alignment parameters that minimize gene expression level differences between cells that are determined to be the same pseudo cell types (Fig. 1A). Finally, Dmatch applies an affine transformation in dimensionality reduced space to all gene expression measurements to remove batch or nonbiological effects (Fig. 1A). The affine transformation used by Dmatch preserves cell-to-cell relationships and overall structure among cells and retains the cell densities of the original data sets. Thus, aligned data produced by Dmatch is well suited for downstream analyses and prevents reduction of cellular variation, which can lead to inflated differential gene expression tests, false positives, and false negatives.
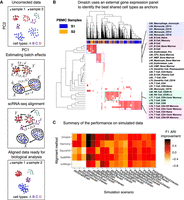
Overview of Dmatch and simulations. (A) Data processing pipeline. First, the uncorrected data are projected onto principal components (PC). Next, an external gene expression panel is used to identify anchor cells to estimate linear batch effects in the form of a rotation and a translation in PC space. Last, the data are corrected by rotating and translating the data points in PC space. The PC loadings are used to recover the aligned data to allow downstream analyses. (B) Dmatch uses a large reference transcriptomes from the Primary Cell Atlas to identify subpopulations from the observed cells based on the projection. These subpopulations are used as anchors to guide the alignment. We show an example applied on real data, which demonstrates the identification of cell clusters corresponding to monocytes, B cells, and two different subclasses of T cells. (C) Heat map showing the ARI F1 scores (Methods) improvements for stimulated data corrected using different alignment methods over unaligned simulated data. Simulations were based on real PBMC data that were split into two batches (see Methods) such that: (1) all cell types were shared or partially shared (All or Partial); (2) noise was added to simulate small, medium, and large batch effect sizes (Small, Medium, or Big); and (3) data were split into two batches such that the cells from each cell type were distributed evenly, unevenly, or very unevenly across the two batches (ratios of 1:1, 1:2, or 1:5, for Even, Uneven, and VeryUneven, respectively). The overall performance of Dmatch as measured by ARI F1 scores was the best overall, followed by Harmony, and fastMNN.
We showcase Dmatch on two major applications, including differential gene expression analysis of cell clusters from biopsies from healthy and disease individuals and cell type–specific eQTL mapping. Dmatch compares similarly or favorably to existing methods on evaluation metrics. Most notably, Dmatch is designed to integrate scRNA-seq data without removing biological variation, that is, without overcorrection. This is an important feature of Dmatch, as we found that overcorrection is often present in data aligned using other existing methods.
Results
Identification of shared cell types across scRNA-seq experiments as anchors
To identify shared cell types across a pair of scRNA-seq data sets, Dmatch computes the Pearson's correlation between gene expression levels of all cells and gene expression quantifications of primary cells from the Primary Cell Atlas (Mabbott et al. 2013). More specifically, the Primary Cell Atlas is a meta-analysis of publicly available microarray data sets compiled from 95 human primary cells from over 100 separate studies (Mabbott et al. 2013; Wu et al. 2016) and was previously used in reference component analysis (Li et al. 2017). Using gene expression measurements from the Cell Atlas, Dmatch computes, for each cell in the scRNA-seq experiments to be aligned a 95-dimensional vector of Pearson's correlations—one correlation per primary cell. Because cells with similar Pearson's correlation vectors are more likely to correspond to the same biological cell types, we reasoned that cells from different scRNA-seq experiments with similar correlation vectors likely belong to the same biological clusters. Dmatch thus clusters cells based on their Pearson's correlation vector and uses these clusters as anchors to determine alignment parameters.
To improve the consistency of the cell clustering, we implemented various strategies to reduce noise in cell type assignment and to detect outlier cells in scRNA-seq data sets. For example, based on empirical tests, we found that the consistency of cell type assignment was generally increased when all Pearson's correlations of the 95-dimensional vectors were set to zero except for those between the cell and the top five reference atlas cell types with the highest Pearson's correlation coefficient (the results were qualitatively similar when the top five to 10 reference cell types were used). To minimize the chance that a rare cell type from the reference panel is chosen as a reference cell type, we required that all reference cell types considered must be ranked as among the top five highest correlated cell types with more than 20 cells from the scRNA-seq data sets. Altogether, this procedure results in a sparse Pearson's correlation matrix, which is then biclustered (e.g., Fig. 1B; Methods). We found that inducing sparsity in this matrix reduced the noise in clustering, which became immediately visible (see Supplemental Fig. S1 for comparison). Dmatch also uses two criteria to rank cell clusters and select two or more clusters as anchors. First, Dmatch favors clusters with at least 100 cells in each scRNA-seq sample. Second, Dmatch performs a Shapiro-Wilk test to rank cell clusters in terms of their likelihood to be drawn from a normal distribution in PC space, an assumption that is required in our optimization (described below). The normality assumption also helps detect cell clusters that visibly consist of two or more distinct cell subclusters or clusters with a large proportion of outlier cells. Clusters that fail the Shapiro-Wilk test are removed from the pool of possible anchors.
Alignment of scRNA-seq data using anchor cell types
Dmatch uses two or more shared cell types as anchors to estimate alignment parameters to reduce cell type–specific batch effects. Specifically, Dmatch first estimates the probability density distribution of anchor cells in each sample separately using Gaussian distributions to model each probability density. Then, Dmatch uses gradient descent to find the best linear transformation (translation d and rotation A) to minimize the Kullback-Leibler divergence (KL divergence) between cells from anchor cell types in one “source” scRNA-seq data set and the matched cells from another “target” data set (Fig. 1A; Methods). Once the parameters that minimize the KL divergence are obtained, the linear transformation is applied to all cells, including nonanchor cell types, from the “source” scRNA-seq data set. The transformed values from the “source” data set, combined with the original values from the “target” data set, form a pairwise alignment between the two scRNA-seq samples. This alignment process can be repeated iteratively to align additional scRNA-seq samples to the “target” data set. Of note, because the KL divergence is not a symmetric distance measure, the choice of which sample to use as a “source” or “target” data set may result in a different alignment. In our work, we consistently used the sample with the largest number of cells as the “target” data set. Despite likely differences in the gene expression values when a different “target” data set is chosen, all downstream analyses are expected to result in findings that are highly consistent.
Although our approach is applicable when only a single anchor cell type is shared across two scRNA-seq experiments, we recommend using at least two anchors to reduce the potential for overcorrection and introducing cell type–specific batch effects. Indeed, batch effects have been observed to depend on mRNA expression level, length, and nucleotide composition (Li et al. 2014; Haghverdi et al. 2018). Thus, batch effects can also be cell type–specific because the expression levels of genes and the amount of total mRNA molecules vary across cell types. Estimating alignment parameters using two or more anchors drastically reduces overcorrecting cell type–specific batch effects, which prevents artificial signals from being introduced in the aligned data. To evaluate the consistency of our alignment when different cell clusters are selected as anchors, we computed the mean squared error (MSE) between data sets produced using different anchors (Methods). We found that the MSE between data sets aligned using different anchors were significantly smaller (average 0.59) than compared to the MSE between unaligned data and aligned data (average 9.74), suggesting that choosing different anchors results in similar alignments.
Evaluation of alignment methods on simulated data
We evaluated the performance of Dmatch and four recently proposed alignment methods, including Seurat V3 (Stuart et al. 2019), fastMNN (scran 1.9.39) (Haghverdi et al. 2018), scMerge (Lin et al. 2019) (0.1.9.1), and Harmony (Korsunsky et al. 2019) (0.0.0.9000). We first applied all methods on simulated scRNA-seq data. To best reflect cellular proportions and number of cell types observed in real data sets, we generated simulated data by manipulating data from a real PBMC sample with 3222 cells from nine cell types (Methods). We generated two batches from the data set by random splitting using different ratios. We introduced batch effects of different sizes by adding Gaussian noise with different means. Furthermore, two scenarios with different levels of difficulty were considered. In the simple case, all nine cell types are shared. In the difficult case, batches are partially overlapping. Specifically, batch 1 and 2 have one and two batch-specific cell types, respectively, and share the remaining six cell types.
Visual inspection of the UMAP plots summarizing data from these simulated batches revealed that Dmatch, Seurat V3, Harmony, and fastMNN showed significant improvement compared to data without alignment (Supplemental Figs. S2–S6). To compare the performance of the alignment methods, we used the Adjusted Rand Index (ARI) (Santos and Embrechts 2009) to evaluate how well cells of the same type clustered together and how well cells from the two batches mixed together (Methods). Specifically, we computed an ARI F1 score (Methods) to summarize the performance of the five methods on each simulation scenario (higher ARI F1 indicates better performance).
In the easy case (9/9 cell types shared), we found that Dmatch achieved the highest ARI F1 score, an average of 0.747, across all scenarios (Fig. 1C). Harmony and fastMNN achieved an average ARI F1 of 0.608 and 0.589, respectively, whereas Seurat V3 and scMerge achieved lower ARI F1 scores. In the difficult case (6/9 cell types shared), all methods had lower ARI F1. Seurat V3, fastMNN, and Dmatch performed better than others. It is important to note here that the ARI F1 score does not assess overcorrection, that is, the removal of true biological variation. Indeed, although Seurat V3 appears to perform better than other methods for some difficult cases in terms of ARI F1 scores, visual inspection of the UMAP plots revealed that Seurat V3- and scMerge-aligned data showed evidence of overcorrection. For example, CD4+ T cells largely overlap with cytotoxic T cells in Seurat V3- and scMerge-aligned data but show separation in data aligned using other methods (Supplemental Figs. S4, S5). In addition, we observed a large reduction of heterogeneity in expression levels for megakaryocytes in Seurat V3-aligned data, again indicating overcorrection (Supplemental Fig. S6).
In summary, Dmatch demonstrates the best overall performance regardless of cell type sharing patterns, imbalance of sample sizes, or size of batch effects.
Alignment of clinical scRNA-seq data from patient biopsies
To evaluate the five alignment methods on real data, we chose to focus on immune cells, as they have been characterized extensively, and many cell types have well-documented markers. To obtain a realistic data set that uses the same scRNA-seq technology to measure gene expression levels at different biopsy sites in multiple individuals, we collected scRNA-seq data using 10x Genomics (Methods) from PBMC (average 4444 cells) from six individuals, including three rheumatoid arthritis (RA) patients, one ankylosing spondylitis (AS) patient, one systemic lupus erythematosus (SLE) patient, and one healthy individual (HC). For each individual except for the AS patient, we also collected scRNA-seq data from bone marrow (average 1013 cells) (Supplemental Table S1). In two of the three RA patients, we further collected scRNA-seq data from synovial fluid (2961 and 2254 cells) at the active site of inflammation. Although these samples were collected and processed on the same day, they were processed on different experimental runs and were not multiplexed nor pooled together. Thus, our data collection reflects a realistic collection procedure for clinical samples that we predict will be widespread.
We first aligned the six PBMC samples we collected to survey the general landscape of immune cells in healthy and disease peripheral blood. Using UMAP to visualize the unaligned data, we observed subtle, but clear, separation between clusters of cells from different individual samples, indicative of the presence of batch effects (Fig. 2A). All alignment methods produced a noticeable reduction in the separation of cells from different samples. The mixing of sample cells after alignment using Harmony, and in particular scMerge, was particularly homogeneous (Fig. 2A).
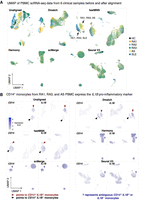
Application of alignment tools on PBMC clinical samples. (A) UMAP dimensionality reduction of PBMC scRNA-seq data from six clinical samples before and after alignment comparing Dmatch, fastMNN, Harmony, scMerge, and Seurat V3. UMAP plots suggest possible overcorrection from scMerge. (B) We observed that CD14+ monocytes express IL1B in a subset of individuals with autoimmune disease (RA1, RA3, AS). This signal was preserved after alignment using Dmatch and, to some extent, fastMNN, whereas it disappeared using Harmony, Seurat V3, and scMerge. Shades of purple represent marker expression in each cell relative to other cells in the same UMAP.
To characterize more systematically the accuracy of the alignment, we sought to determine whether the same cell types from different samples were aligned together while distinct cell types remained separated. To this end, we applied a standard pipeline to identify cell clusters using Seurat on each sample separately (Method). This resulted in 8–15 clusters for the six samples, wherein each cluster expressed informative markers that allowed us to assign a likely immune cell type (e.g., CD4+ T cell, or CD14+ monocyte) (see Methods). Visualizing these markers on the UMAP revealed that, indeed, the alignment procedures were able to align cells expressing the same markers together (Fig. 2A; Supplemental Fig. S7). However, in addition to aligning similar cell types together, scMerge also aligned distinct cell types together, resulting in three major homogeneous cell clusters representing B cells, monocytes, and a large cluster of T cells (Supplemental Fig. S7).
When the samples were analyzed separately using the standard Seurat pipeline (Methods), we observed that CD14+ monocytes from RA1, RA3, and AS (but not RA2, SLE, or HC) expressed the pro-inflammatory marker IL1B, indicating that monocytes in these individuals exhibit a pro-inflammatory state. CD14+ monocytes that express IL1B have been observed in RA patients (Kay and Calabrese 2004), but the expression of IL1B in monocytes from the ankylosing spondylitis patient supports reports that IL1B may be involved in the pathogenesis of a wide number of autoimmune diseases (Dinarello 2011; Wan et al. 2016).
We also observed a cluster in SLE PBMC consisting of cells that expressed high levels of kidney-expressed genes (e.g., ECHS1, MIOX, FXYD2, and ALDOB). The presence of this “kidney” cell cluster is consistent with circulating kidney cells in the peripheral blood of the SLE patient, as the patient exhibits kidney inflammation in the form of lupus nephritis.
Because both pro-inflammatory monocyte clusters and the “kidney” cell cluster were identified in the patient samples when analyzed individually, we reasoned that their presence could not be explained by technical effects. However, investigating data aligned using Seurat v3, Harmony, and scMerge revealed that the clusters representing pro-inflammatory monocytes disappeared subsequent to alignment (Fig. 2B) and the cluster corresponding to SLE-specific circulating kidney cells disappeared after Seurat V3, Harmony, and to some extent, scMerge alignment (Supplemental Fig. S8). In contrast, both clusters are visibly separated in the UMAP representation of data aligned using Dmatch and, to some extent, fastMNN (Fig. 2B; Supplemental Fig. S8).
Overcorrection of batch effect masks biological signal
To better understand the extent to which data aligned using different methods vary, we performed a series of differential gene expression analyses between cell type clusters that were determined using Seurat on each sample separately, henceforth referred to as Seurat clusters (see Methods). We thus set to identify differences in the list of genes identified as differentially expressed across Seurat clusters when different methods were used to align the data. We reasoned that if the lists of differentially expressed genes (DEGs) across Seurat clusters are the same for all methods, then the alignments must be very similar across methods. However, if the lists of DEGs differ, then the alignments must be highly variable.
We began by evaluating the differences between unaligned data and data aligned using Dmatch. For each pair of Seurat clusters, we counted the number of DEGs that were identified using limma-trend (Law et al. 2014; see Methods) using unaligned data as input but not using data aligned from Dmatch as input, and vice versa. We first identified DEGs across pairs of Seurat clusters from different samples that were determined to represent the same cell type from our manual annotation (Methods). In the ideal scenario, the number of DEGs between clusters representing the same cell type should be reduced when using aligned data compared to unaligned data, because DEGs identified when comparing the same cell type across different samples are expected to be due to batch effects. Indeed, when we compared DEGs obtained from unaligned and Dmatch-aligned data, we found that the number of DEGs between same cell types in different samples was drastically reduced (generally 5–25 DEGs from unaligned data vs. 0–3 DEGs from Dmatch-aligned data) (Fig. 3A; Supplemental Fig. S9). The reduction in the number of DEGs therefore suggests that Dmatch was able to efficiently remove the effects of batch.
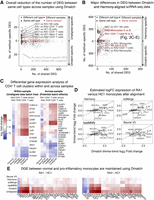
Evidence for overcorrection in aligned scRNA-seq data. (A) Scatterplot showing the number of differentially expressed genes (DEGs) that are identified using unaligned data (positive y-axis) or using data aligned using Dmatch (negative y-axis), versus the number of DEGs that are identified in both data sets (x-axis). Each point represents a comparison between cell type clusters from the same or different cell type, or from the same or different sample. Successful removal of batch effects is supported by smaller numbers of DEGs resulting from Dmatch-aligned data in same cell type comparisons, compared to unaligned data. DEGs inferred from within-sample cluster comparisons are identical between unaligned and Dmatch-aligned data. (B) Scatterplot similar to A shows a large difference between Dmatch and Harmony-aligned data. (C) Heat maps of estimated DEG fold changes between CD4+ T cells versus CD4+ T cells expressing FOS within the same sample (RA3, left heat map), and across two samples (RA1 vs. RA3, right heat map). Dmatch and fastMNN estimates of DEG fold changes within samples are consistent with unaligned data, as we should expect. However, the estimates from scMerge and Harmony are shrunken and inconsistent, respectively, suggesting overcorrection. Across-sample comparison between the two cell types shows an increase in JUN, FOS, and CD69 expression in activated CD4+ T cells, consistent with within-sample comparisons for Dmatch and fastMNN. The signal is reduced or reversed for scMerge and Harmony, respectively. (D) Scatterplot of DEG log fold change comparison between CD14+ monocytes from healthy individual HC and RA patient (RA1). Fold change estimates are reduced or zero for inflammatory markers (IL1B, CCL3, CCL4) using data obtained from fastMNN, Harmony, and scMerge. (E) Heat maps of estimated DEG fold change comparison between RA1-HC1 and RA3-HC1 are consistent and support the presence of overcorrection in data aligned using Harmony, scMerge, and fastMNN, masking real biological signal.
We also considered comparisons across pairs of Seurat clusters determined to be different cell types but from the same sample (Fig. 3A,B, red points). Ideally in this case, DEGs identified across two clusters from the same sample after alignment should be largely the same as those identified before alignment because much of the confounding effects are shared among measurements in a single experiment. In fact, within-sample DEGs identified from unaligned data that are not identified from aligned data suggest overcorrection and a reduction of true biological variation. We found that DEGs from within-sample comparisons were identical between unaligned data and data aligned using Dmatch. Altogether, these results suggest that Dmatch is able to preserve within-sample biological variation while correcting across-sample batch effects.
We next compared DEGs identified from unaligned data to DEGs identified from Harmony, fastMNN, scMerge, and Seurat V3. We found that, although all methods showed reduction of DEGs identified across Seurat clusters determined to be the same cell types, there were also significant differences in the DEGs found between Seurat clusters within a sample (Supplemental Fig. S9). These observations raise the possibility that existing alignment methods are prone to overcorrection.
We next compared DEG results between Dmatch and other methods. We highlight in Figure 3B the differences between Dmatch and Harmony, which showed the largest number of differences (for other comparisons, see Supplemental Fig. S10). For example, many comparisons shared no DEGs, whereas hundreds of genes were identified as differentially expressed in either Dmatch-aligned data only or Harmony-aligned data only. Although the number of DEGs in comparisons between the same cell types was smaller using Harmony and some of the other methods, the large number of method-specific DEGs when comparing clusters within samples suggests overcorrection.
To further investigate possible overcorrection, we focused on identifying cell clusters that show biologically relevant or known differences. We found that samples from RA1 and RA3 both harbored two clusters of cells that expressed classical markers of CD4+ T cells (e.g., CD3D, IL7R, IL32), with one cluster showing high expression levels of activation markers including FOS and JUN (Supplemental Fig. S11). Because both clusters were found in RA1 and RA3 patients, they likely represent real biological variation between naive and activated CD4+ T cells circulating in patient blood. To verify this, we identified DEGs between the naive and activated CD3D+ IL7R+ cell clusters (CD4+ T cells). Within the top 15 most significantly DEGs, we observed overexpression of activation makers such as CD69, JUN, FOS, and DUSP1 in activated T cells when using unaligned data, Dmatch-aligned, and fastMNN-aligned data (Fig. 3C). However, we found that the estimated fold changes were reduced or even opposite when using Seurat V3-, scMerge-, or Harmony-aligned data (Fig. 3C). We also observed that comparing naive CD4+ T cells from RA1 to activated CD4+ T cells from RA3 resulted in a highly consistent list of DEGs to within-sample comparison, further suggesting that cross-sample comparison is likely accurate for fastMNN and Dmatch and inaccurate for Seurat V3, scMerge, and Harmony.
We also found that the list of DEGs between the CD14+ Seurat cluster (CD14+ monocytes) from RA1 and the CD14+ Seurat cluster from HC were different between Dmatch-aligned data and data aligned using other methods. As described earlier, we found that the pro-inflammatory marker IL1B was overexpressed in RA1 CD14+ monocytes compared to monocytes from the healthy individual. We also observed overexpression of additional consistent markers such as CCL3, CCL4, and NFKBIA (Fig. 3D,E). In contrast, starting from data aligned using fastMNN, scMerge, Seurat V3, and Harmony, the differences in gene expression levels between RA1 and HC monocytes were drastically reduced in effect sizes (MNN) or disappeared completely (Harmony, Seurat V3, and scMerge) (Fig. 3D,E). IL1B and cytokines (e.g., CCL3 and CCL4) have long been recognized to be overexpressed in samples from rheumatoid arthritis patients (Koch 2005; Dinarello 2011). Thus, our observations suggest that heterogeneity between the two monocyte clusters are not a result of uncorrected batch effects. Instead, the lack of DEGs detected across the two monocyte clusters using Harmony, scMerge, Seurat V3—and to some extent fastMNN—indicates that the two biologically variable clusters are aligned together erroneously.
Altogether, these results suggest that Dmatch allows correction of batch effects from scRNA-seq data, while avoiding overcorrection. In contrast, although Harmony, scMerge, Seurat V3, and fastMNN are all able to correct batch effects by homogenizing scRNA-seq data from different experiments, they do so at the cost of removing real biological signals.
Variation in gene expression levels in peripheral and tissue-resident immune cells
We next evaluated scRNA-seq alignment across biopsy sites. To this end, we used all methods to align scRNA-seq data from PBMC and bone marrow (BMMC) from all individuals (11 total samples) and again evaluated the number of DEGs across Seurat clusters. We then conducted a hierarchical clustering on all samples based on the number of DEGs detected, using the Euclidean distance as the distance metric. We found that, compared to the clustering from unaligned data, the clustering using aligned data from all methods resulted in more clearly defined clusters (Fig. 4A; Supplemental Fig. S12). The samples clustered according to inferred cell type, rather than sample provenance, and samples within the same cell type clusters showed fewer DEGs within cluster than compared to unaligned data, suggesting a reduction of batch and technical effects.
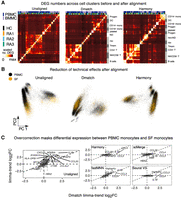
Cross-biopsy alignments between PBMC and SF immune cells. (A) Heat maps showing the number of DEGs identified (Methods) across Seurat clusters in PBMC and BMMC samples using unaligned data, Dmatch-aligned data, and Harmony-aligned data (others in Supplemental Fig. S12). Improvement of clustering by cell type using aligned data indicates removal of batch effects from unaligned data. (ND) Not determined. (B) PCA of scRNA-seq samples from PBMC and SF before alignment and after alignment using Dmatch and Harmony. Although unaligned data show likely batch effects specific to biopsy site, this effect is reduced after alignment. (C) Scatterplots of estimated gene expression log fold changes between CD14+ monocytes from RA2 PBMC and RA2 synovial fluid (SF). We found that only Dmatch- and fastMNN-aligned data reflected overexpression of pro-inflammatory genes in RA2 SF versus PBMC monocytes.
The study of differences between peripheral immune cells and resident immune cells in the pathological sites requires integration of peripheral samples and samples from the pathological sites. Because cell type composition can differ across these sites, we predicted that Dmatch would outperform other methods as, based on our simulations, existing methods struggled to align samples in the presence of cell types that were not shared or a high variability in cell type proportions. To test our prediction, we aligned our scRNA-seq data from synovial fluid (SF) (from RA1 and RA2) to scRNA-seq data from PBMC (all individuals). PC analysis on uncorrected data suggests that SF and PBMC scRNA-seq showed clustering within biopsy site, indicative of the presence of batch effects (Fig. 4B). Alignment of the data using Dmatch showed clear improvement in mixing of cells from SF and PBMC samples (Fig. 4B). Furthermore, we observed that alignment using other tools such as Harmony also showed improved mixing and a clear reduction of batch effects (Fig. 4B; Supplemental Fig. S13).
To better understand the differences in alignments produced by the five methods, we again compared the list of DEGs across Seurat clusters in PBMC and the synovial fluid scRNA-seq data set without alignment and after alignment using the five methods. We found that using unaligned data generally led to a larger number of DEGs across samples from different biopsy sites (Supplemental Fig. S14), consistent with the large batch effects observed from the PCA (Fig. 4B).
However, we found that many pairs of cell clusters had DEGs that were only identified in Dmatch and fastMNN, but not Harmony, scMerge, or Seurat V3. Among the pairs of clusters with DEGs specific to Dmatch and fastMNN are two clusters of monocytes from RA2 PBMC and RA2 SF. Upon examination, we found that, whereas monocytes from RA2 PBMC did not express IL1B or other pro-inflammatory markers, IL1B and other pro-inflammatory markers were highly overexpressed in RA2 SF monocytes. This is in contrast to RA1 samples, in which pro-inflammatory monocytes were found in both PBMC and SF. Dmatch-aligned data revealed that monocytes in RA1 SF expressed higher levels of CCL3 compared to RA1 PBMC monocytes, suggesting a stronger or more robust activation of monocytes in SF than in PBMC. Our finding suggests that the presence of IL1B+CCL3+CCL4+ monocytes may be a potent biomarker of rheumatoid arthritis, even though not always detectable in patient PBMC.
We were able to identify IL1B overexpression on monocytes only using data aligned by Dmatch and fastMNN—and the extent of the overexpression estimated using fastMNN alignments was reduced compared to that estimated by Dmatch. IL1B overexpression and thus this entire population of pro-inflammatory monocytes could not be detected in scRNA-seq data aligned using Harmony, scMerge, and Seurat V3.
Thus, we conclude that although all methods were able to integrate scRNA-seq samples from multiple biopsy sites with modest differences in cell type compositions, nearly all existing methods also removed true biological variation between cells from the different biopsy sites.
Alignment improves power of eQTL mapping from population-level single-cell RNA-seq data
PCA is often used to estimate and correct batch effects to increase mapping power in eQTL studies from bulk RNA-seq data (Li et al. 2016). However, because PCA-based methods often fail to correct batch effects across scRNA-seq samples, we hypothesized that Dmatch and other alignment methods can increase eQTL mapping power beyond standard PCA-based batch correction methods. To test this, we re-analyzed population scRNA-seq data from peripheral blood mononuclear cells collected in a previous study (van der Wijst et al. 2018). UMAP of the unaligned data shows minimal batch effects (Fig. 5A), as has been generally observed from high-quality, droplet-based scRNA-seq from peripheral blood.
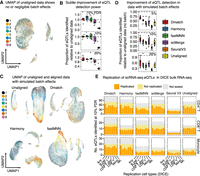
Evaluation of alignment methods on eQTL mapping. (A) UMAP of unaligned PBMC scRNA-seq data from 45 donors (van der Wijst et al. 2018). The cells from the 45 donors were pooled into eight samples for sequencing. (B) Alignment slightly improves mapping power. (C) UMAP of unaligned PBMC data with spiked-in batch effects (see Methods). (D) Alignment improves eQTL mapping power substantially when batch effects are simulated to reflect a realistic and practical scRNA-seq collection strategy. (E) Replication of eQTLs identified in a bulk RNA-seq study of immune cell type eQTLs from the DICE consortium. The eQTLs that were not tested correspond to SNPs whose genotype could not be determined or to genes filtered out due to low expression.
We mapped eQTLs in eight cell types using the same labels identified by the original authors (van der Wijst et al. 2018). To this end, we computed the mean count of all genes in each cell type separately. We then performed eQTL mapping, as we have done extensively in the past (Li et al. 2016, 2018), by appropriate normalization and standardization of the data and false discovery test correction (Methods). We found that alignment generally improved the number of eQTLs detected but only slightly (2%–10%) (Fig. 5B).
Owing to the pooled design employed by the authors, the scRNA-seq data showed little to no batch effects, which can explain the subtle improvement of eQTL detection power after alignment. To test this possibility, we next added batch effects (Methods) to each of the batches to simulate a more practical data collection pipeline, wherein clinical samples are collected continuously and immediately processed for scRNA-seq. UMAP of the unaligned data clearly shows the introduced batch effect (Fig. 5C), which is consistent with scRNA-seq data from clinical samples that were immediately processed after collection (Fig. 2A). When using unaligned data with simulated batch effects to identify eQTLs, the number of eQTLs identified in each cell type was reduced by an average of 40%–60%. These observations demonstrate the necessity to account for potential batch effects in eQTL mapping studies that use scRNA-seq data. Thus, we next identified eQTLs starting from data aligned using different methods. We found that eQTL mapping starting with data aligned using Dmatch resulted in the most eQTLs identified (1.3- to 1.5-fold as many as starting with unaligned data) compared to data aligned using other methods (0.9- to 1.1-fold as many) at 10% FDR (Fig. 5D). To visualize the differences in eQTLs recovered from data aligned using the different methods, we compared the significance of the association for the QTLs. We found that the linear regression P-values were generally highly correlated but in many cases were more significant for Dmatch-aligned data compared to data aligned using other methods (Supplemental Fig. S15). This suggests that data aligned using Dmatch improves QTL mapping power. We also validated the eQTLs mapped using data from the DICE consortium, which collected population-level bulk RNA-seq data for a large number of sorted cell types. We found that, although the rates at which eQTLs identified from data aligned using the different methods were largely similar, Dmatch identified more eQTLs, which generally led to a higher number of eQTLs that are replicated overall (Fig. 5E; Supplemental Fig. S16).
Thus, we conclude that alignment of scRNA-seq data increases eQTL mapping power. However, the improvement in mapping power varies across alignment methods and is likely to be reduced when biological variation is removed due to overcorrection.
Discussion
There has been a great interest in using scRNA-seq to discover gene regulatory signatures that underlie various biological processes and phenomena. One major obstacle to fully exploit the power of scRNA-seq is the extensive batch and technical effects in scRNA-seq experiments. These unwanted effects not only limit the re-usability of scRNA-seq data produced by different groups but make it difficult to compare two scRNA-seq experiments generated by the same group and individual. To overcome this obstacle, a growing number of methods were proposed to “align” scRNA-seq experiments to facilitate comparison and allow downstream analysis of the merged data sets. Many of these alignment strategies have been designed to align data sets that have been generated using different scRNA-seq platforms or even to align scRNA-seq data from different species. However, there is a paucity of studies that evaluate the performance of these methods on aligning data sets that were generated using the same methods in perhaps different contexts, for example, healthy versus disease condition. Indeed, although most methods appear to perform well on the task of aligning conspicuous cell clusters together (e.g., the monocytes from one data set to monocytes of another data set), it is unclear whether more subtle biological variation is lost during the alignment process.
We developed Dmatch to align scRNA-seq experimental data that are obtained using the same scRNA-seq platform to allow downstream analyses including differential gene expression analysis. Dmatch uses an external panel of reference transcriptomes (Primary Cell Atlas [Mabbott et al. 2013]) that was compiled from over 100 separate studies to identify anchor cell types that can be used for identifying batch or technical effects. Although the cell type annotation in the Primary Cell Atlas may be relatively coarse (95 distinct cell type annotations), we found that Dmatch was able to find appropriate anchors to align samples consisting of immune cell types. Although we expect this strategy to work less well for more specialized scRNA-seq samples that contain cell types that are not represented in the Primary Cell Atlas, we envisage that ongoing efforts, such as that of the Human Cell Atlas (Regev et al. 2017), will establish better cell type references that can be used by Dmatch to identify more appropriate anchor cell types. In particular, we found that, in some cases, a new reference panel must be created with finer grained data, for example, brain cell types when analyzing scRNA-seq data from brain tissues, to identify appropriate anchor cell types for alignment. In order for Dmatch-type methodologies to work in these specific cases, the user can provide a reference panel with relevant cell lines or cell types. Specifically, if there are no significant correlations and biclustering pattern between the scRNA-seq data and the panel, the users must find an alternative reference panel with relevant cell lines for Dmatch to properly remove batch effects. The lack of significant correlation can be diagnosed from the visualization tools implemented in Dmatch as well as inspection of the correlations output by Dmatch.
In this paper, we showcased the application of Dmatch on scRNA-seq data from immune cell types. Several of the parameters used to identify anchor cell types were chosen empirically. For example, we found that using a 0.1–0.4 cutoff threshold for the Pearson's correlation matrix and keeping the top five primary cell lines that are highly correlated with at least 20 cells worked well for all data sets in the study. However, to ensure successful batch effect removal using Dmatch, users are encouraged to inspect the distribution of Pearson's correlation coefficients between cells in the data sets and primary cell lines in the reference panel (Fig. 1A). This will allow the user to adjust the cutoff thresholds or other parameters as needed. These parameters allow aberrant signals due to technical artifacts to be removed in order to identify external expression data that can be used to find appropriate anchors for alignment. The higher the Pearson's correlation cutoff threshold is set, the more confident the results of biclustering will be and thus the more likely the identified anchors will be suitable for Dmatch alignment. However, setting the cutoff threshold too high may result in no external expression data to help identify suitable anchors. Thus, the parameters should depend on the reference panel used and how representative the reference panel is of the data sets under study. The cutoff should therefore be a parameter to examine case by case.
Dmatch uses kernel density matching to estimate two parameters, a translation parameter and a rotation parameter, to align two scRNA-seq data sets. Because Dmatch transforms data linearly and this transformation depends only on two parameters, we reasoned that the resulting transformed data should be less prone to overcorrection. The resistance to overcorrection, however, comes at the cost of not being able to detect, and therefore correct for, batch or technical effects that are entirely nonlinear. Indeed, we found that alignments from Dmatch resemble uncorrected data more so than alignments from other methods, suggesting that the corrections made by Dmatch are relatively conservative. Nevertheless, Dmatch performed similarly or favorably to all evaluated methods in terms of reduction of batch effects in simulated data and in terms of producing correctly aligned cell type clusters across samples by visual inspection of PCA or UMAP plots.
A major difference we observed between the evaluated alignment methods in this study was that Dmatch appeared to be less, or even not, prone to overcorrection, whereas Harmony, scMerge, Seurat V3, and to some extent, fastMNN, all showed signs of overcorrection in real data analysis. For example, we found several cell clusters that likely represented distinct cell states (e.g., pro-inflammatory and normal monocyte) but were made indistinguishable in alignments obtained using Harmony, scMerge, and Seurat V3. Even in the case of fastMNN, the differences in gene expression that distinguished these clusters were often reduced substantially. Thus, although all methods appear to work relatively well in terms of correcting batch or technical variation across scRNA-seq samples, methods vary in terms of their propensity to remove true biological variation. Indeed, we found that data aligned using scMerge showed the strongest signature of overcorrection in data that we collected. In this study, we also found that overcorrection can impact eQTL mapping. Indeed, although eQTL analysis is generally tolerant to undercorrection, as covariates can be added to the linear regressions that test for associations, overcorrection of true biological—in this case, inter-individual—variation will strictly reduce eQTL mapping power.
In summary, we present Dmatch, a novel method for alignment of scRNA-seq data, which allows users to integrate multiple scRNA-seq experiments for downstream analyses. Although we found that all evaluated methods performed well in terms of their ability to remove batch and technical effects, only Dmatch alignments appeared to be resistant to overcorrection. Thus, Dmatch unlocks several important applications, such as differential gene expression analysis and eQTL mapping using scRNA-seq data. These applications are becoming staple tools in genomic studies as scRNA-seq continues to increase in popularity and maturity.
In addition to aligning scRNA-seq data, Dmatch can be extended to align scATAC-seq data or perform integrative analyses that use both scRNA-seq and scATAC-seq. There are two ways that Dmatch can be used in this setting: (1) to use the RNA-seq reference panel to identify anchor cells by correlating gene expression to ATAC-seq counts in corresponding promoter regions; or (2) to use a new reference panel established using bulk ATAC-seq data from primary cell lines. Additional work is required to explore these possibilities.
Methods
Identifying anchor cells by projecting scRNA-seq to a reference panel
We propose to use reference transcriptomes from the Primary Cell Atlas, a meta-analysis of a large number of publicly available microarray data sets compiled from human primary cells (745 samples, from over 100 separate studies). After preprocessing using quantile normalization and feature selection, a panel of 5209 gene measurements for 95 annotated cell types (e.g., epithelial cells, fibroblasts and endothelial cells, etc.) were used for projection. This processed reference panel is downloaded directly from R package RCA (Li et al. 2017). To identify subpopulations from the observed single cells, we projected all cells from our scRNA-seq samples to this reference panel by quantifying the Pearson's correlation (the Spearman's correlation can also be used) between measurements for each single cell and the measurements for the 95 annotated cell types. We then enforced sparsity to the original Pearson’s correlation matrix by (1) keeping only the top five primary cell lines in the reference panel which were highly correlated with any cell in the experimental data sets, and (2) keeping only the primary cell lines in the reference panel which were among the top five primary cell lines with more than 20 cells in the experimental data sets. The correlations of cell lines that do not meet these two criteria were set to zero. The cutoffs were selected empirically for 10x Genomics data and could be adjusted based on the quality of signals in the data sets. Of note, the identities of the single cells were described by a 95-dimensional projected vector on the reference panel. We further performed a biclustering which allowed simultaneous clustering of the rows and columns on the sparsity-enforced Pearson's correlation matrix. We used hlcust() implemented in R for the hierarchical clustering with Euclidean distance as the distance metric. In our experiments, complete linkage, Ward's minimum variance method, and Ward's minimum variance square linkage method achieved the best performance. Finally, we determined clustering membership based on the biclustering results. The cell clusters shared across samples were used as anchor cell types for alignment. If two samples share more than two cell clusters, the default is to select two clusters based on (1) the number of cells in each cluster; a recommended cluster has at least 5% of the total number of cells in each sample, and (2) the results from a Shapiro test on the normality.
Removal of batch effects
We assume that the batch effects can be represented as linear transformations on reduced dimensions. We used PCA for dimension reduction in the analysis, which could be replaced by other nonlinear alternatives. We assume that the density of each selected anchor cell type on a pair of PCs follows a 2D Gaussian distribution. Taking advantage of orthogonality, we can perform the correction independently on every consecutive pair of top PCs. In the default setting, Dmatch accounts for variation loaded on the top 30 PCs. Overall, we seek to correct for batch effects through applying linear transformations estimated from density matching.
To illustrate the general ideas, we describe the alignment procedure for measurements from two cell types, type A and type
B cells in individual 1 and 2, respectively. At population level, the density for measurements in individual 1 is q and the density for measurements in the individual 2 is p without any perturbation. Let Y′ represent measurements of type A cells from individual 1, X′ represent measurements of type B cells from individual 1, Y represent measurements of type A cells from individual 2, X represent measurements of type B cells from individual 2 after some unknown affine perturbation on the original density.
Let D(q, p) represent some distance measure for two densities, for example, Hellinger distance, total variation distance, χ2 distance, or Kullback-Leibler divergence. We seek an affine transformation on {Y, X}, including a translation d and a linear map A, which can minimize
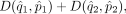 where
where 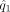 and
and 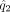 are the estimated density functions based on Y′ and X′, respectively, and
are the estimated density functions based on Y′ and X′, respectively, and 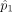 and
and 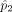 are the estimated density functions based on YA − d and XA − d, respectively. The solution of this problem depends on the distance measure as well as the density estimation method. We
use Kullback-Leibler (KL) divergence, which possesses good properties for our application and results in a closed-form objective.
The KL divergence from p to q is defined as
are the estimated density functions based on YA − d and XA − d, respectively. The solution of this problem depends on the distance measure as well as the density estimation method. We
use Kullback-Leibler (KL) divergence, which possesses good properties for our application and results in a closed-form objective.
The KL divergence from p to q is defined as
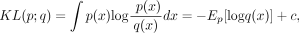 where q is a reference density that we want to align to. It is a measure of how much “evidence” each sample will, on average, bring
when we are trying to distinguish p(x) from q(x) and when we are sampling from p(x). Accordingly, our problem can be formulated as
where q is a reference density that we want to align to. It is a measure of how much “evidence” each sample will, on average, bring
when we are trying to distinguish p(x) from q(x) and when we are sampling from p(x). Accordingly, our problem can be formulated as
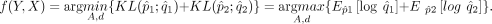 This is equivalent to calculating maximum likelihood estimators for A, d, given empirical density of q1 and q2. We use Gaussian distributions to estimate the densities for X, Y, X′, and Y′, respectively. Let µy, D1 and µx, D2 represent the estimated mean vector and covariance matrix for Y′ and X′, respectively. The objective becomes the following:
This is equivalent to calculating maximum likelihood estimators for A, d, given empirical density of q1 and q2. We use Gaussian distributions to estimate the densities for X, Y, X′, and Y′, respectively. Let µy, D1 and µx, D2 represent the estimated mean vector and covariance matrix for Y′ and X′, respectively. The objective becomes the following:
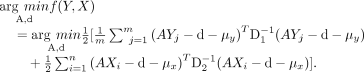 The gradients with respect to A and d equal the following:
The gradients with respect to A and d equal the following:
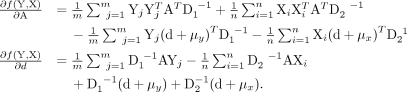
Further simplification leads to the following matrix form representation:
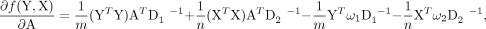 where ω1 = 1m ⊗ (d + μy)T and ω2 = 1n ⊗ (d + μx)T.
where ω1 = 1m ⊗ (d + μy)T and ω2 = 1n ⊗ (d + μx)T.
We use coordinate descent to solve the above estimation problem. At each step, d will be updated by
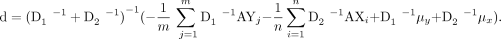 Theoretically, A is allowed to take many forms of linear transformation. In the default implementation, we constrained the linear map in the
affine transformation to a single rotation, reducing the search space of parameters, that is,
Theoretically, A is allowed to take many forms of linear transformation. In the default implementation, we constrained the linear map in the
affine transformation to a single rotation, reducing the search space of parameters, that is,
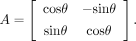 As such, we replace the gradient search by an angle search in the algorithm
As such, we replace the gradient search by an angle search in the algorithm
 where
where
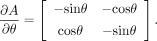
Simulation study
We created the simulated data by manipulating a real PBMC data set (standard filtering and log-normalized), which contained a total of 3222 cells from nine cell types (Ding et al. 2020). We added normally distributed noise to the data set to model batch effects, through the mean parameter of which we controlled the effect sizes. Specifically, we used a normal distribution with a mean of 0.28 to model large batch effects, a mean of 0.18 to model medium effects, and a mean of 0.08 to model small effects. All standard deviations are set to 1. We split the data into two sets, and the ratios of numbers of cells in the two sets are set to 1:1, 1:2, or 1:5 to represent scenarios with an equal number of cells, a moderately unequal number of cells, and a very unbalanced number of cells, respectively.
We tested two patterns for cell type sharing. In the first case, two splits share all the cell types; in the second case, they share six out of nine cell types. For each pattern, we simulated batch effects with different sizes: big, medium, and small. The evaluation of the size of the batch effects is done by inspecting the cells in the PC space. For each effect size, we generated 10 data replicates. In total, we evaluated three ratios of numbers of cells divided into the two batches, two different sharing patterns, and three batch effect sizes. The cellular proportions in the original real data set are listed below in Table 1. For scenarios in which all cell types are shared, we kept the ratio in two splits. For scenarios with partial overlapping, one sample is set to have one batch-specific cell type and the other batch to have two specific cell types. Three batch-specific cell types are randomly sampled from the nine possible cell types. Thus, the cellular proportions will vary for different replicates. We included cellular proportions from one example replicate.
Cellular proportions in the original data set and an example simulation with partial overlapping cell types
Performance evaluation
We use the ARI F1 score to evaluate the performance of the batch correction methods. The ARI F1 score combines two ARI scores that aim to capture (1) the purity of cell type, and (2) the mixing of batches.
The ARI measures the similarity between two clusterings. To assess cell type purity and mixing of the two batches, we first performed a k-means clustering on the first 20 PCs of 80% down-sampled data, which include all cell types and batches, using the kmeans() function from the stats package in R and by setting k = 9, the number of distinct cell types in the simulation. Using this data, the ARI (ARIcell type) was computed by comparing the cell type labels against the k-means clustering labels using the adjustedRandIndex() function in the R package mclust (Scrucca et al. 2016). A high ARIcell_type indicates that the k-means clusters correspond well to the cell type clusters, reflecting separation by cell types. To assess mixing of the batches, we similarly computed ARI (ARIbatch) by comparing the batch labels against the k-means clustering labels. A high ARIbatch indicates separation of cells by batch. Thus, the ideal alignment method should result in a high ARIcell_type and a low ARIbatch.
To produce a representative ARI score for each method, both batch and cell type assessments were repeated 100 times with subsampling.
The median ARI scores were then normalized to a range between 0 and 1, and a combined F1 score was obtained for each method
by computing the harmonic mean of the ARI scores defined as
 From the equation above, it can be seen that a higher ARI F1 corresponds to a better alignment.
From the equation above, it can be seen that a higher ARI F1 corresponds to a better alignment.
Data collection
Samples of peripheral blood (PB) and bone marrow (BM) were obtained from three RA patients, one SLE patient, and one HC. In addition, a sample of PB was obtained for one AS patient. Synovial fluid samples were obtained from the knee joints of two RA patients. RA, AS, or SLE patients fulfilled the 2010 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) RA classification criteria (Aletaha et al. 2010), the SpondyloArthritis International Society classification criteria for axial spondylarthritis (Rudwaleit et al. 2009), or the ACR Revised Criteria for Classification of SLE (Hochberg 1997), respectively. Donors were enrolled at the Department of Clinical Immunology of Xijing Hospital in Xi'an, and their clinical characteristics are summarized in Supplemental Table S1. Donors with chronic disease, cancer, and chronic infections such as Hepatitis B, C, and HIV were excluded. PB, BM, and SF were collected by EDTA-treated Vacutainer tubes in a sterile manner (BD Biosciences). SF was diluted with two volumes of phosphate-buffered saline (PBS) and treated with bovine testicular hyaluronidase (10 mg/mL; Sigma-Aldrich) for 30 min at 37°C. Mononuclear cells were isolated using Ficoll-Paque gradient separation (GE Healthcare Bio-Sciences). This research was approved by the ethical standards committee of Xijing Hospital (KY20192006-C-1). All donors provided written informed consent in accordance with the Declaration of Helsinki.
Data processing
We used standard QC to filter our data such that all retained genes are expressed in more than 5% of cells and all cells express at least 200 genes. For feature selection, we normalized gene expression measurements for each cell by the total expression, multiplied by a scale factor, and calculated log variance versus mean ratio (logVMR) for each gene across all cells. Next, we selected the top 50%–75% genes (after inspecting the logVMR) as the features to calculate the Pearson's correlation between cells in the data sets under study and primary cell lines in the reference panel. Because genes must be shared between the reference panel and the data sets under study, we retained a high percentage of total genes based on logVMR.
Identification of cell clusters using Seurat and markers
We used Seurat (version 2.3) with default recommended settings on each of our scRNA-seq samples separately to identify clusters (referred to as Seurat clusters). To annotate Seurat clusters, we inspected the top 10 marker genes that were the most overexpressed in each cluster compared to the remaining cells as identified using the “FindAllMarkers” function from Seurat. More specifically, we used 2–3 representative markers to annotate the following cell types: CD4+ T cells (CD3D, IL7R); CD8+ T cells (CD3D, CD8A, CD8B); NK cells (NKG7,GNLY); B cells (MS4A1, CD79A); CD14+ monocyte (LYZ, CD14); CD16+ monocyte (LYZ, FCGR3A); red blood cells (HBB, HBA1, HBA2); and dendritic cells (FCER1A, CST3).
Alignment of scRNA-seq data from patient and healthy biopsies
To align the scRNA-seq data from patient and healthy individual biopsies (PBMC, PBMC + BMMC, and PBMC + SF), we used the default parameters for Seurat V3 (Stuart et al. 2019), fastMNN (scran 1.9.39) (Haghverdi et al. 2018), scMerge (0.1.9.1) (Lin et al. 2019), and Harmony (0.0.0.9000) (Korsunsky et al. 2019) and used the 30 first PCs as input data (except for scMerge, for which the full data set was used as input, as required).
Dmatch aligns scRNA-seq samples by finding the alignment parameters that minimize the difference in the densities between anchor cell clusters from the “target” and “source” scRNA-seq samples and by applying the transformation to all cells including cells in the “source” data set. Thus, the alignment process can fail if batch effects are highly inconsistent among cell clusters (and thus anchor cell clusters). To detect such inconsistencies, Dmatch computes an alignment vector for each anchor cell cluster as the linear transposition in a 2D PC space from the center of an anchor cell cluster from the “source” sample to the same anchor cluster of the “target” sample. If the linear transpositions of two anchor cell clusters are very different, then the batch effects are likely to be highly inconsistent and result in the incorrect estimation of alignment parameters. In other words, differences in the linear transposition are indicative that different anchor cell clusters provide conflicting information regarding how batch effects should be corrected.
We found no inconsistent batch effects for our PBMC and PBMC + BMMC alignments and therefore used the default parameters for Dmatch to align PBMC and PBMC + BMMC samples with 30 PCs as input. However, when we computed the angle between the alignment vectors for the alignment of RA1 SF onto HC PBMC, we found that the angles were small (consistent) for pairs of anchor clusters in PCs 1–6 but large (inconsistent batch effects) for many pairs of anchor clusters in PCs 7 and onwards (cf. Table 2 and Table 3). Thus, we used the six first PCs as input for our PBMC + SF alignment. We found no qualitative difference when we used the first six PCs or all 30 PCs for the other methods.
Angles between the alignment vectors for pairs of clusters for the SLE PBMC to HC PBMC alignment
Angles between the alignment vectors for pairs of clusters for the RA1 SF to HC PBMC alignment
Functions for computing alignment vectors and their angles are included in the Dmatch R package (R Core Team 2019) and can be used to select the appropriate number of PCs to input when the alignment vectors are inconsistent.
Differential expression analysis
To estimate differential expression across Seurat clusters, we used limma-trend as implemented in Soneson and Robinson (2018) and in which limma-trend was shown to perform well in nearly all major evaluation criteria including true-positive rates and maximum false-positive rates. To evaluate the differences between differential expressed genes identified using unaligned data and data aligned using the various methods (Fig. 3A,B), limma-trend was applied to identify DEGs across all pairs of Seurat clusters from all samples combined. To be considered a DEG, we required a P-value <10−10 and an absolute log2 fold change difference to be greater than 1. Although these cutoffs were chosen arbitrarily, we found that varying the P-value cutoff to 10−5 or 10−20, or the absolute log2 fold change difference cutoff to 0.5 or 1.5 did not qualitatively change our interpretations.
To identify DEGs that are shared or specific to a method (Fig. 3A,B), we simply counted the number of DEGs that were identified at the aforementioned P-value and log2 fold change cutoffs for each of the different tools. We then plotted the number of DEGs that were identified specifically using method A (positive y-axis), specifically using method B (negative y-axis), and DEGs that were identified using both methods (positive x-axis) for all pairwise comparisons across Seurat clusters. Comparisons across Seurat clusters from the same scRNA-seq sample are colored in red, and comparisons across Seurat clusters with the same cell type assignment are marked as crosses (again, see Fig. 3A,B).
To produce the heat maps in Figure 4A, we again called DEGs across Seurat clusters across PBMC and BMMC scRNA-seq samples. We counted the number of DEGs for each pairwise comparison and used the heatmap.2 command from the R package gplots (R Core Team 2019) to produce the heat maps using hierarchical clustering with the average linkage clustering.
Population PBMC scRNA-seq data set and simulated batch effects
We analyzed processed data from van der Wijst et al. (2018) and used their cell type annotations. These data are also available through the single-cell eQTLGen Consortium (https://eqtlgen.org/single-cell.html) and at the European Genome-phenome Archive (EGA; https://ega-archive.org/) under accession number EGAS00001002560. When performing UMAP on processed data, we observed that samples mixed well, indicative of limited batch effects. To add batch effects to the scRNA-seq data in order to simulate a simpler collection procedure without the need to pool samples together, we once again used the simulation model from fastMNN (Haghverdi et al. 2018). Briefly, perturbation effects were incorporated by generating a Gaussian random vector for each data set and adding it to the expression profiles for all cells in that data set. More specifically, gene expression profiles were log-normalized, and eight perturbation parameters were generated for each of the eight samples from van der Wijst et al. (2018) (mean, standard deviation of the Gaussian random vector): list(c[1,0.28],c[1.6,0.4], c[1.8,0.35], c[2.5,0.45], c[2.2,0.38], c[1.8,0.22], c[1.95,0.4], c[2.0,0.3]). These eight Gaussian random vectors (each with length: n cells × n genes) were then added to each of the eight samples. The above parameters were chosen based on the PCA and t-SNE plots.
eQTL mapping
To map eQTLs in scRNA-seq data, we computed, for all individuals separately, the mean expression levels of all cells in each annotated cell types: B cells, CD4+ T cells, CD8+ T cells, dendritic cells, NK cells, monocytes, and combined cells (pseudobulk, or bulk). We then mapped eQTLs in each cell type separately using a standard eQTL pipeline by quantile-normalizing expression levels across genes within individuals and standardizing the expression across individuals (Li et al. 2016). We then used FastQTL (Ongen et al. 2016) to identify associations between genetic variants and gene expression levels and the Benjamini-Hochberg procedure to control false discovery rates at the 0.05, 0.10, and 0.20 levels. No PCs were used as covariates.
Replication using DICE eQTLs
To replicate the eQTLs identified using the population scRNA-seq data, we quantified gene expression levels from DICE RNA-seq data for each different cell type separately using kallisto v0.46.1 (Bray et al. 2016). We then used a standard eQTL pipeline (Li et al. 2016) and FastQTL (Ongen et al. 2016) with three genotype PCs and a variable number of phenotypic PCs. More specifically, PCA on genotype data was performed using the smartpca program from EIGENSOFT (version 6.1.4), after pruning SNPs using PLINK (v1.9) with parameters ‐‐indep-pairwise 50 2 0.2. Individual outliers reported by smartpca were excluded from eQTL mapping. Phenotypic PCs were calculated in R using the prcomp function. We empirically chose the number of phenotypic PCs that maximized the number of significant eQTLs (5% FDR) in each cell type (Supplemental Table S2).
To determine the replication of an eQTL from aligned scRNA-seq data, we asked whether the linear regression between the gene expression and the most significantly associated SNP in the aligned scRNA-seq data was also significant in each of the cell types in the DICE data set (P <0.05). To determine the replication in “any” cell type, we asked whether the linear regression was significant, correcting for the number of cell types (P < 0.05/6).
Data access
Processed scRNA-seq data generated in this study are available as Supplemental Data File S1. The source code for Dmatch can be found at GitHub (https://github.com/qzhan321/dmatch) and as Supplemental Code S1. Scripts to reproduce simulations and evaluation of the alignment methods on simulated data are available at GitHub (https://github.com/qzhan321/Dmatch-data-code-rep) and as Supplemental Code S2.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Sebastian Pott for comments on the manuscript. This work was supported by the U.S. National Institutes of Health (R01GM130738 to Y.I.L. and R01GM126553 to M.C.) and by the National Defense Basic Scientific Research Program of China (NO.2015CB553704) to P.Z. M.C. also acknowledges support from the Sloan Research Foundation Fellowship and an HCA Seed Network grant from the Chan Zuckerberg Initiative. This work was completed in part with resources provided by the University of Chicago Research Computing Center.
Author contributions: Y.I.L. and M.C. conceived of the project. M.C. and Q.Z. performed analyses and implemented the software. Y.I.L., Z.M., and L.W. also performed analyses. Y.I.L. and M.C. wrote the manuscript with contributions from Q.Z. P.Z., Z.Z., and J.M. collected the experimental data.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.261115.120.
-
Freely available online through the Genome Research Open Access option.
- Received January 13, 2020.
- Accepted January 19, 2021.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








