
Evidence of widespread, independent sequence signature for transcription factor cobinding
- Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, USA
-
↵1 These authors contributed equally to this work.
Abstract
Transcription factors (TFs) are the vocabulary that genomes use to regulate gene expression and phenotypes. The interactions among TFs enrich this vocabulary and orchestrate diverse biological processes. Although simple models identify open chromatin and the presence of TF motifs as the two major contributors to TF binding patterns, it remains elusive what contributes to the in vivo TF cobinding landscape. In this study, we developed a machine learning algorithm to explore the contributors of the cobinding patterns. The algorithm substantially outperforms the state-of-the-field models for TF cobinding prediction. Game theory–based feature importance analysis reveals that, for most of the TF pairs we studied, independent motif sequences contribute one or more of the two TFs under investigation to their cobinding patterns. Such independent motif sequences include, but are not limited to, transcription initiation–related proteins and known TF complexes. We found the motif sequence signatures and the TFs are rarely mutual, corroborating a hierarchical and directional organization of the regulatory network and refuting the possibility of artifacts caused by shared sequence similarity with the TFs under investigation. We modeled such regulatory language with directed graphs, which reveal shared, global factors that are related to many binding and cobinding patterns.
Regulatory biomolecules cooperatively decode the human genome and ultimately orchestrate a variety of tissue-specific cellular processes. One of the key challenges for understanding the mechanisms underlying gene expression and human diseases is how to analyze and delineate the genome-wide landscape of multiple characteristic biochemical signatures. An important aspect is the regulation of transcription factors (TFs), which bind to DNAs and drive the expression or suppression of genes. TFs do not work alone; they cooperate and interact with each other and thus create complex transcription patterns of the genome (Lemon and Tjian 2000; Perez-Pinera et al. 2013; Li et al. 2014; Ang et al. 2016).
Studying the collaborative patterns of TFs is a much more complicated problem than characterizing the contributors of a single TF binding event, for which open chromatin and the presence of the TF motifs have been recognized as the key determinants (Pique-Regi et al. 2011; Neph et al. 2012; Tsompana and Buck 2014). Naïvely, one might deduce that the main contributors to the cobinding patterns of a TF pair should be the presence of the two TF motifs and open chromatin. Yet, empirically many open questions remain to be investigated: are there other independent sequence signatures contributing to TF cobinding? If such sequence signatures do exist, are they generic for a TF when it is paired with any other TFs, or are they unique to a specific TF pair? Are there global, important sequence signatures affecting many TF cobinding patterns? We attempt to answer these questions with machine learning and game theoretical approach–based feature analysis.
The Encyclopedia of DNA Elements (ENCODE) and the NIH Roadmap Projects have provided invaluable resources of the in vivo biochemical signatures, including chromatin accessibility and TF binding. Many exciting computational approaches have been developed to predict transcription factor binding sites (TFBSs) and to study what controls the binding patterns (Bulyk 2003; Blanchette et al. 2006; Kumar and Bucher 2016; Chen et al. 2017; Quach and Furey 2017). However, only a few models are developed for the prediction of TF cobinding patterns, and most of them focus on the pairing with a single motif (e.g., CTCF [Liu et al. 2016]). Although pioneering works have made substantial contributions, there is an urgent need to develop a generalizable model for the prediction of TF-cobinding and identify key contributors for this process.
This study aims at addressing these questions from a machine learning perspective. Towards this goal, we developed an algorithm that focuses on dissecting the predictive elements for genome-wide TF cobinding across diverse cell types based on chromatin accessibility, TF binding motifs, and gene locations. What is unique to this model is that not only the motifs of the TF pairs under investigation but also the motifs of other seemingly irrelevant TFs are considered in building the model. This allows us to further adopt game theory–based approaches to identify independent contributors to the binding patterns of a TF pair. Additionally, we created regulatory graphs based on this feature importance analysis.
Results
Designing a machine learning model to dissect individual contributors that predict TF cobinding
Our goal is to dissect the individual contributors to TF cobinding, which include motif locations of the TF pair, other TF sequence signatures, chromatin accessibility, and gene locations. Our approach is to first establish a predictive model to predict TF cobinding using features extracted from the above data types and then use game theoretical feature analysis to identify independent contributions of each feature.
To this end, we first created the gold standard cobinding data set for training the model. Following one of the mainstream conventions in the TF binding field (Levy and Hannenhalli 2002; Valouev et al. 2008; Yu et al. 2016), chromosomes are segmented every 200 bp, with a moving step of 50 bp (Fig. 1A). If a 200-bp region is bound by two transcription factors at the same time in a specific cell line as examined by ChIP-seq experiments, it is defined as a positive example. Otherwise, the region is considered as a negative example (Fig. 1A). We carried out the experiments on 13 commonly seen and well-studied TFs (ATF3, CTCF, E2F1, EGR1, FOXA1, FOXA2, GABPA, HNF4A, JUND, MAX, NANOG, REST, and TAF) using the ENCODE data set (Supplemental Tables S1, S2). The TFs were chosen by two criteria: belonging to different families grouped by binding site similarity as defined by sequence homology to a previously characterized DNA-binding domain (Supplemental Table S1; Lambert et al. 2018); and having sufficient ChIP-seq data across ENCODE cell lines for subsequent analysis of TF-TF interactions. A total of 228 TF cobinding profiles involving 56 different TF-TF pairs were prepared to train and evaluate the model.
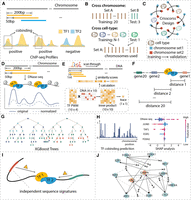
Machine learning–based method for identifying independent sequence signatures for TF binding and cobinding. (A) Preparing gold standard co-occupancy profiles from the ChIP-seq data of single TF binding profiles. For each 200-bp genomic interval of interest, it was labeled as ‘cobound’ if the labels of both TFs at this interval were ‘bound.’ (B) Train-test data partition for cross-chromosome and cross-cell type cases. (C) Crisscross design of model training and validating. Chromosomes were split into two sets. The blue and red squares represent the chromosome set 1 and set 2, respectively. The starting point of the arrow represents the training data set, and the endpoint of the arrow represents the validation data set. (D) DNase-seq-based features. Quantile normalization was applied to the original reads to eliminate experimental biases. In the plot, the dashed line is the values after quantile normalization and the solid line is the original reads. (E) DNA sequence and motif-based features. (F) Distance-to-gene features. Top 20 closest distances to proximal genes are calculated according to GENCODE annotation. (G) Illustration of tree-based machine learning models. (H) Predictions are continuous values between 0 and 1, which were generated by averaging results from all built models, and SHAP analysis (figure is illustrative only) was used to calculate feature importance in predicting TF cobinding. (I) Independent sequence signatures affect TF cobinding. (J) Illustration of TF cobinding effector network. Dashed lines connect TF pairs, and solid arrows connect TF-specific effectors with TF- or TF pair–specific effectors with TF pairs.
We sought to evaluate the model performance from two aspects: (1) cross-cell type predictions, that is, when the binding pattern is known in some cell lines and we have corresponding open chromatin status in other cell lines sharing the same genomic sequence; (2) predictions on new chromosomes, that is, when we move the model learned from one set of chromosomes to others. The former is useful for imputing a large amount of cobinding patterns across cell lines. The latter would be useful for studying binding patterns associated with SNPs or moving to other species. Toward this goal, we partitioned the chromosomes (Chr) into the training set (Chr 2, Chr 3, Chr 4, Chr 5, Chr 6, Chr 7, Chr 9, Chr 10, Chr 11, Chr 12, Chr 13, Chr 14, Chr 15, Chr 16, Chr 17, Chr 18, Chr 19, Chr 20, Chr 22, and Chr X), which are used to evaluate model performance across cell types in Case 1, and the test set (Chr 1, Chr 8, and Chr 21), which are used to evaluate model performance across chromosomes in Case 2 (Fig. 1B). This partition was made following several traditions in the TF-binding field (Keilwagen et al. 2019; Li et al. 2019). We randomly selected one cell line as the test cell line for each TF-TF pair and ensured that the number of test pairs in each cell line did not differ too much after the random selection.
TF co-occupancy is a much less explored field than single TF binding. With very limited prior experience in feature engineering, we consider it logical to adapt the feature engineering methods based on the state-of-the-field in the TF binding field. Open chromatin and motifs are two widely accepted factors that affect TF binding (Zhou and Liu 2004; Pique-Regi et al. 2011; Schmidt et al. 2017). Proximity to genes is another important factor, with some disagreement between studies (Chen et al. 2020). For the completion of the study, we considered all three of these. Based on several previous studies (Kelley et al. 2016; Li and Guan 2019a; Li et al. 2019; Kelley 2020), we first used long-range DNase-seq-based features to capture open chromatin long-distance information. Moving windows of 200 bp in size, with a step of 50 bp until 1500 bp, are included as features (Fig. 1D). Additionally, the variability of open chromatin signals was suggested to be informative for TF binding (Pique-Regi et al. 2011; Davie et al. 2015; Schmidt et al. 2017; Li et al. 2019). Thus, we used the maximum, minimum, average DNase signals in each of the 200-bp regions as additional features. To capture and correct for the DNA position biases, we also used the difference between the DNase value and the average value across all cell lines. Then, we scanned TF motifs along DNA sequences and used the top four sequence similarity scores in each 200-bp region as the motif-based features. Again, we took motifs that are even not within 200 bp under consideration (up to −750 to 750 bp) in order to study the effect of other TF signatures in cobinding (Fig. 1E). Finally, we used the closest distances to the 20 proximal genes as the distance-to-gene features, as previous studies suggested that TF binding is closely related to the positioning of open reading frames (Fig. 1F; Clements et al. 2007; Maienschein-Cline et al. 2012; Ezer et al. 2014). This resulted in a total of 526 features, for each TF-TF-cell line combination, which were nested trained with XGBoost (Fig. 1C,G; see Methods), a classical classifier used for TF binding predictions to build models for TF cobinding.
Robust performance in predicting the rare events of TF cobinding across cell types and in previously unseen genome sequences
This algorithm demonstrated strong predictive performance when evaluated on the testing data set covering 56 TF pairs. We first calculated the area under the receiver operating characteristic curve (AUROC) to evaluate the model performance (Fig. 2A,D; Supplemental Table S3). The median AUROC of cross-cell line predictions is 0.998 in 56 TF pairs, and the range of our prediction AUROCs is between 0.986 and 0.999. In addition to AUROC, we further calculated the area under the precision-recall curve (AUPRC) (Fig. 2B,C,E; Supplemental Table S3; Davis and Goadrich 2006). Whereas the AUROC baseline is consistently 0.5, the AUPRC baseline is the proportion of positive examples, which is 0.000504 in our data, because TF co-occupancy is extremely rare in the human genome. The median of AUPRC was 278 times over the baseline across 56 TF pairs.
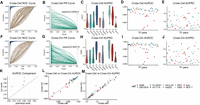
Cross-cell type and cross-chromosome evaluation of TF cobinding predictions. (A) The ROC curves of predicting cross-cell type TF cobinding. The brown lines are ROC curves of the logistic regression model. (B) The PR curves of predicting cross-cell type TF cobinding. The brown lines are PR curves of the logistic regression model. The dashed line with AUPRC of 0.000612 is the average AUPRC of the logistic regression. (C) The AUPRCs of cross-cell type predictions of 56 TF-TF pairs in 10 cell types. (D,E) The cross-cell type AUROCs and AUPRCs of 56 TF-TF pairs. Different colors indicate different cell types. All cell lines represented in the figure are testing cell lines. (F–J) Represent cross-chromosome evaluations. (K) AUROC comparison of our method (y-axis) and a previous work (x-axis) (Liu et al. 2016). The cross-cell type predictions of ATF3-CTCF on cell lines A549, H1-hECS, HepG2, and K562 are compared. (L) The AUROC and (M) the AUPRC comparison of cross-cell type (x-axis) and cross-chromosome (y-axis) predictions.
The model represents a substantial improvement over the previous work, whose test set was limited to CTCF-associated TF pairs but could nevertheless serve as a benchmark for this study (Liu et al. 2016). In this benchmark work, Liu et al. focused on distinguishing between TF-TF cobinding and single TF binding events. Although our model was not directly designed for this purpose, it can still predict such events, as single-binding events were also used as negatives when we constructed the gold standard. In the test set cell line-TF pair combination, we selected the evaluation chunks limited to the single-bound and cobound intervals, for the CTCF-associated pairs shared between our study and their study on four cell lines (A549, H1-hECS, HepG2, and K562). Compared with the AUROC values of 0.80, 0.79, 0.68, and 0.80 in the previous research, our method reached AUROC values of 0.8724, 0.8245, 0.8039, and 0.9211, and AUPRC 0.2367, 0.2365, 0.0955, and 0.2438, respectively, on this evaluation (Fig. 2K). This improvement in performance might come from the extraction of maximal, minimal, and variance of the DNase features and long-range features, which are additional in this study. Of note, differentiating cobinding and single-binding events is a much more challenging task, explaining why these AUROCs are comparably lower than the global AUROCs, and the model presented in this paper was not designed for that purpose.
We also benchmarked with two existing software: TACO and coTRaCTE (Jankowski et al. 2014; van Bömmel et al. 2018). Both are unsupervised methods. Both TACO and coTRaCTE make predictions for a subset of the chromosome and do not consider the regions that are not open. Thus, for a comprehensive comparison, we compared both on their restricted subset, as well as the whole genome by assuming all nonpredicted sites are zero. For the restricted subset of the chromosomes, TACO's AUROC is 0.49867 and AUPRC is 0.01103; for the method presented in this study on the same region, the values are 0.857 and 0.061. For coTRaCTE, its performance was AUROC = 0.68450 and AUPRC = 0.08259. The comparison highlights the importance of supervised learning in cobinding prediction.
Next, we tested the model on its ability to make predictions on the three held-out chromosomes (Chr 1, 8, and 21) on test cell lines. We obtained AUROCs with a range from 0.985 to 0.9995, with an average value of 0.997 (Fig. 2F,I; Supplemental Table S4). For the AUPRCs, the improvement of our predictions over the baseline ranges from 155× to 1759× (Fig. 2G,H,J; Supplemental Table S4). Compared with cross-cell line prediction, the cross-chromosome performance can reach equally high AUROC values (difference less than 0.001) and even better AUPRCs. Compared with single-bound examples, no-bound examples were easier to separate (Supplemental Table S5). For single-bound, the average AUROC and the average AUPRC are 0.817 and 0.336 for cross-cell type evaluation, and 0.820 and 0.348 for cross-chromosome evaluation. For no-bound, the average AUROC and the average AUPRC are 0.998 and 0.411 for cross-cell type evaluation, and 0.998 and 0.403 for cross-chromosome evaluation.
Of note, predicting cobinding in previously unseen genome sequences is often considered a much more difficult task than making predictions for a chunk of sequence that already occurs in the training set (i.e., cross-cell line predictions). We compared the performance of these two situations (Fig. 2L,M) and found a strong correlation, indicating that the performance could depend on intrinsic difficulties in the TF pair rather than the sequence. In fact, the performance is almost indistinguishable compared to cross-cell line predictions.
To evaluate the individual contribution of each type of feature, we separately evaluated model performance by including only DNase, only sequence motif, and only distance to genes, as well as a model with motif and gene distance (but excluding DNase) (Fig. 3). DNase is by far the most predictive feature, as it reflects chromatin status and achieves a cross-cell type AUROC of 0.995 and AUPRC of 0.149. The cross-chromosome AUROC is 0.995 and AUPRC is 0.148 (Fig. 3C). Removing DNase, but retaining the other two, we obtained the cross-cell type AUROC of 0.9450 and AUPRC of 0.0573. The cross-chromosome AUROC is 0.943 and AUPRC is 0.0553 (Fig. 3A). Motif-based features make the second most important contribution: the cross-cell type AUROC is 0.941 and AUPRC is 0.0492. The cross-chromosome AUROC is 0.940 and AUPRC is 0.0470 (Fig. 3D). Gene distance-based features are the least important: the cross-cell type AUROC is 0.773 and AUPRC is 0.00577. The cross-chromosome AUROC is 0.7620 and AUPRC is 0.00572 (Fig. 3B).
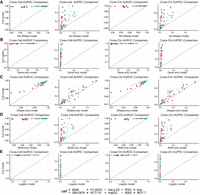
Performance of models with different features. (A) Comparison of performance between model without DNase-based feature and model with full features. (B) Comparison of performance between model with gene distance–based feature and model with full features. (C) Comparison of performance between model with DNase-based feature and model with full features. (D) Comparison of performance between model with sequence motif–based feature and model with full features. (E) Comparison of performance between logistic regression model with full features and XGBoost model with full features.
We also benchmarked with logistic regression and compared the model performance (Fig. 3E). For the logistic regression model with full features, the cross-cell type AUROC is 0.613 and AUPRC is 0.000612. The cross-chromosome AUROC is 0.622 and AUPRC is 0.000716. The adoption of the XGBoost model contributes to the improvement of the performance.
The above-described algorithm has practical application to differentiate TF cobinding status and predict TF cobinding sites, especially given the large amount of missingness in the ENCODE database. The predictions made by this algorithm are capable of differentiating TF cobinding versus single TF binding events and, certainly, versus nonbinding events, as has been demonstrated in the performance analysis in previous paragraphs (AUROCs). Here, we used some intervals from JUND-MAX in cell line K562 in the cross-cell line experiment to illustrate this point (Fig. 4). We were able to distinguish these three cases with the prediction values of 0.9938 for the cobinding case (Fig. 4A), 0.8174 for the single-binding case (Fig. 4B), and 0.1851 for the no-binding case (Fig. 4C). These results indicate that the algorithm leverages both TF motif sequences and chromosome accessibility to predict TF cobinding.
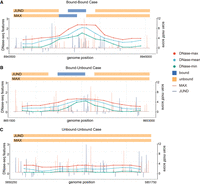
Examples of predicting TF-TF cobinding and un-cobinding events. The comparison of TF binding signals from ChIP-seq (the blue/orange horizontal bar on the top), DNase-seq-based features (the red/blue/green scatter points and lines in the middle), and TF motif hits (the blue/orange vertical bars on the bottom). (A) A cobound (Bound-Bound) case of the JUND-MAX pair in Chromosome 2 between position 8944200 and 8944400 in K562. The red, blue, and green dots represent the maximum, mean, and minimum DNase-seq values, respectively, in a 200-bp interval. The two bars on the top highlight the binding locations MAX and JUND. Our prediction for this interval is ‘cobound’ (0.9938). (B) An un-cobound (Bound-Unbound) case in Chromosome 2 between position 8652400 and 8652600. Specifically, JUND is bound while MAX is unbound in this interval. The prediction for this interval is ‘un-cobound’ (0.8174). (C) An un-cobound (Unbound-Unbound) case in Chromosome 2 between 5850900 and 5851100. Both JUND and MAX are unbound. The prediction for this interval is ‘un-cobound’ (0.1851).
Game theoretical approach–based analysis suggests widespread, strong, and independent sequence signatures related to TF cobinding patterns
The above model is unique in that not only the motif presence of the TF pair under investigation is used as a predictive feature but also the seemingly irrelevant motifs of different TFs. This allows us to investigate the determinants of transcription factor co-occupancy beyond what is already known: open chromatin and the presence of motifs. A technical challenge in finding independent contribution is addressed by a recent advance in game theory application: an improved SHapley Additive exPlanations (SHAP) analysis (Fig. 1H,I; Shapley 1988; Lundberg et al. 2018). Mimicking the process of finding out the contribution of football players in a game, this analysis assigns the independent contribution of each of the features considering the context and the existence of other features. This is more appropriate than direct correlation analysis where correlations do not conclude contribution but could be a consequence of shared patterns with another important feature.
First, we analyzed the 526 features encompassing open chromatin (DNase), TF motif presence, other TF sequence signature, and gene location (Fig. 5A,B; Supplemental Fig. S1). We found that DNase and the presence of TF motifs under investigation are globally the most important factors, and the proximity to gene location plays a small role. Additionally, the DNase and TF features that are closer to the center of the 200-bp chunk would be more important (Fig. 5C–E).
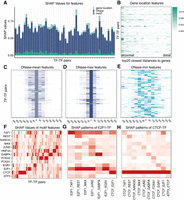
Game theoretical approach–based analysis of determinants of TF cobinding. (A) SHAP values of DNase-seq-based features (blue), TF motif–based features (dark green), and distance-to-gene features (light green). DNase-seq-based features play the most important role in all TF-TF pairs. (B) The spatial distribution of distance-to-gene features. Columns correspond to the top 20 closest distances to proximal genes and rows are the 56 TF-TF pairs. Distances to proximal genes are more important than those to distal genes. (C–E) Spatial distributions of DNase-seq-based features. In the heat map, each row corresponds to a TF-TF pair and each column corresponds to one 200-bp genomic interval. Zero represents the feature calculated from the target interval, and ± represents the upstream/downstream neighboring intervals. (F) The spatial distribution of TF motif–based features. Each column is a TF-TF pair and each row is the motif-based features averaging at one TF. The motifs-based features for TFs that are the component of the TF-TF pair have higher contributions. In addition to the component TF, other TF sequences are also predictive. (G) The SHAP value pattern of TF pairs tied with E2F1. For most pairs, E2F1 motif–based features have the highest SHAP values. For E2F1-TAF1, both E2F1 and TAF1 have higher SHAP values than other TFs. (H) The SHAP value pattern of pairs tied with CTCF. For most pairs, CTCF motif–based features have the highest SHAP values. Other TF motifs like NANOG and TAF1 also play an important role.
To study the contribution of independent sequence signatures, for each TF pair, we calculated the SHAP value for each feature (Fig. 5F; Supplemental Figs. S2, S3). The original SHAP values for all motif features are a 416 × 56 matrix. The 416 rows come from 13 TFs multiplied by 32, because each TF has a total number of 32 features, depending on their distance to the center of the chunk under investigation. We took the mean of those 32 features for 13 TFs, respectively, and scaled the SHAP values to 0−1 for each cell line-TF pair combination. An alternative by taking the maximum values was also investigated, and the patterns are very similar to taking the mean (Supplemental Fig. S4).
The first most prominent observation is that proximity to an open chromosome region and the appearance of motifs of the TF pair are the most important features. For example, for pairs tied with E2F1, E2F1 and TAF1 are the two most important features (Fig. 5G). For the effect of TAF1, overall it is a positive indicator for all E2F1-related pairs (Supplemental Fig. S3). Similarly, for pairs tied with CTCF, CTCF is the most important feature (Figs. 5H, 6E). For the DNase-mean and DNase-max features, the central interval is more important than neighboring intervals when predicting TF co-occupancy events. Additionally, proximity to genes only contributes to about half of the binding patterns of TF pairs. However, we also found other irrelevant TF sequence signatures as significant contributors. In the majority (62.5%) of the TF pairs we studied, independent motif sequences contribute one or more TFs under investigation to the TF cobinding patterns. In seven cases among the 56 TF pairs we studied, there exist independent sequence signatures that contributed more than both TFs under investigation. Excluding cases related to FOXA1-FOXA2, which share great sequence similarity, there remain three cases (Table 2; Supplemental Fig. S5). Moreover, we found that, for seven pairs tied with ATF3, the average contribution of JUND is the highest among those 13 TFs; it is marginally higher than ATF3 itself. This is a strong piece of evidence of the existence of an independent TF-specific effector.
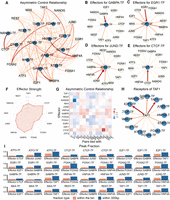
Asymmetric regulatory relationships of TF-TF pairs in controlling cobinding. (A) In this network, each node represents a TF or a TF-TF pair. Each edge that connects a TF and a pair represents the contribution of that TF to the pair when predicting cobinding events. The width and color of an edge represent the strength of the contribution. (B–E) Directed regulatory networks of TF-TF pairs effectors. In each network, the blue node represents a TF-TF pair and the light color node represents a TF motif signature. The arrow direction represents the contribution of a TF to a TF-TF pair. The width and color of an edge represent the strength of the contribution. In B, GABPA, TAF1, and CTCF are the three most effective effectors for pairs tied with GABPA. In C, JUND, TAF1, and EGR1 are the most effective effectors for pairs tied with EGR1. In D, JUND is the most dominant effector for pairs tied with JUND. In E, CTCF is the most dominant effector for pairs tied with CTCF. The four examples show that the contribution of different TF effectors and both pair-relative and pair-irrelative TFs affect cobinding. (F) Radar plot of effectors strength averaging over pairs. Among the 13 TFs, CTCF, JUND, and TAF1 are the three most contributive effectors compared with other TFs. (G) Asymmetric control heat map. Each cell represents the difference between (1) the contribution of TF1 to pairs tied with TF2 and (2) the contribution of TF2 to pairs tied with TF1. Positive and negative values are shown in red and blue, respectively. (H) Contribution of TAF1 is different when predicting the cobinding of pairs tied with different TFs. (I) Peak fractions of TF-specific effectors binding with TF pairs. Red bars: the fraction of the effector binding in the bins under investigation; blue bars: the fraction of the effector binding within 300 bp of the bins under investigation.
As mentioned above, some of these signatures could be a result of shared sequences or complementary sequences with the two TFs under study. For example, FOXA2 is an effector for pairs tied with FOXA1, and FOXA1 and FOXA2 share similar motifs. However, many effectors and their TFs do not directly show sequence similarity. For example, GABPA is an effector for pairs EGR1-TF (Fig. 6C), but the motif sequences of GABPA and EGR1 do not share many similarities, nor do they form a protein complex. Thus, some of these sequence signatures seem to speak a language other than forming complexes to facilitate certain shared processes. To investigate the contribution of cobinding in this feature importance analysis, we calculated two types of peak fractions: fractions in which effectors are bound with TF pairs in the same bin; and fractions in which effectors are bound with TF pairs within 300 bp (Fig. 6I; Supplemental Table S6) because a single binding event will result in consecutive sets of bins annotated to ‘bound.’ The above two criteria are, in fact, quite representative and generous in considering the contribution of cobinding. The average fraction of peaks that the top effector binds in the same bin with the TF-TF pair is 0.248. The average fraction of peaks in which the top effector binds with TF pairs within 300 bp is 0.490. For example, for ATF3-GABPA and effector MAX, the fraction that MAX bound with ATF3-GABPA in the same bin is 0.624, and the fraction that MAX bound with the pair within 300 bp is 0.893. For ATF3-TAF1 and effector CTCF, the fraction that CTCF bound with ATF3-TAF1 is 0.124, and the fraction that CTCF bound with the pair within 300 bp is 0.198. This indicates that combining is an important mechanism, resulting in the observed signatures. However, at the same time, even for the top effectors, the binding only contributes to an estimated 25% to, at most, 50% of the times, indicating that it only partially explains these sequence signatures. As a matter of fact, 27.06% of relationships between a sequence signature and a TF pair are negative, suggesting that there exist other mechanisms beyond binding.
Independent sequence signatures can be categorized into generic, TF-specific, and TF pair–specific
Such independent contributors of sequence signatures reflect, to some extent, known biology and could be roughly grouped into three categories, which are not necessarily mutually exclusive. First, the sequence signature is related to many TFs, for example, GABPA is related to 10 TFs (CTCF-TF, E2F1-TF, EGR1-TF, FOXA1-TF, FOXA2-TF, HNF4A-TF, JUND-TF, MAX-TF, REST-TF, and TAF1-TF) in this study, and JUND is related to four TFs (ATF3-TF, EGR1-TF, CTCF-TF, and HNF4A-TF) (Table 1). Certainly, in this case, JUND has been found to directly interact with ATF3 (Chu et al. 1994). TAF1 is an effector for pairs CTCF-TF, E2F1-TF, GABPA-TF, and MAX-TF (Table 1). Transcription initiation factor TFIID subunit 1, TAF1, appears in this global effector list, as the initiation of transcription by RNA polymerase II requires the coordination of TFIID and the binding of this complex at the promoter region (Bieniossek et al. 2013). Of note, even though TAF1 is a global effector, its strength differs for different TFs, with the strongest in TAF1, E2F1, and GABPA (Fig. 6H). We term such effectors as generic effectors. Potential important ones are GABPA, JUND, TAF1, and HNF4A.
TF-specific effectors, which are related to cobinding patterns involving a specific TF, as identified for all TFs with more than two samples available; the TF under investigation is excluded in the table
Second, for the sequence signature that is related to a particular TF, no matter what other TF we are studying, we term it a TF-specific effector (Table 1). To identify a TF-specific effector, we first calculated the mean of absolute SHAP values of each effector on pairs that tied with the specific TF. Then, we selected the top three effectors, excluded the specific TF itself, and identified the remaining ones as TF-specific effectors. For example, HNF4A is a TF-specific effector for pairs tied with JUND (Fig. 6D), and TAF1 is a TF-specific effector for pairs tied with GABPA (Fig. 6B). TFs sharing strong sequence similarity will come out as an artifact of such TF-specific effector, for example, FOXA2 for FOXA1 and vice versa. Third, the sequence signature that is related to a particular TF pair, in the sense that it is stronger than any of the TFs of the pair under study, is termed as a TF pair–specific effector (Table 2). For example, HNF4A is the TF pair–specific effector for FOXA1-REST. We acknowledge this is an extremely stringent criterion, and if we relax it, many more TF pair–specific effectors may arise, and the full spectrum is provided in Supplemental Figures S2 and S3.
TF pair–specific effectors, defined as TFs whose sum of absolute SHAP values is larger than both of the TFs of the pair; FOXA1 and FOXA2 are mutually excluded in this analysis due to high sequence similarity
We found TF-specific and TF pair–specific effectors tend to be tissue-specific. FOXA2 is a TF that activates liver-specific gene expression (Tuteja et al. 2008). The cobinding of FOXA2-TAF1 is linked to the appearance of another tissue-specific transcription factor, HNF4A, which is critical for liver development (Babeu and Boudreau 2014; Thakur et al. 2019). Such tissue specificity seems to be elegantly orchestrated through sequence combination, in that the HNF4A motif comes out as an important effector for FOXA1, FOXA2, and TAF1. Similarly, FOXA2 does not appear as an effector for transcription factors other than FOXA1, which shares sequence identity with FOXA2.
We acknowledge one potential limitation in the above analysis: regions bound by a large number of transcription factors might confound meaningful signals. To examine this effect, we repeated the above analysis by removing the HOT sites (High-Occupancy-Target sites) from the analysis. The HOT sites are defined by counting the number of binding TFs in each 200-bp bin and cutting off using the 99th percentile threshold to define the HOT sites (Wreczycka et al. 2019), which was deemed in line with previous studies (Gerstein et al. 2010). SHAP patterns excluding HOT sites do not differ substantially from the ones including the HOT sites (Supplemental Figs. S6–S13). The correlations between SHAP values with and without HOT sites range from 0.9939 to 0.9995. For example, for cell line K562, GABPA is the most dominant effector for JUND-TF pairs. Similarly, for cell line HepG2, FOXA2 is the most dominant effector for FOXA1-TF pairs. After removing the HOT sites, GABPA remains the top feature of the JUND-TF pair in K562, and FOXA2 also remains the top feature of FOXA1-TF pairs in HepG2. This result indicates the HOT sites had minimal impact on the feature importance analysis.
Independent sequence signatures for TF binding are rarely mutual but mostly directional
For both TF-specific effector and TF pair–specific effector, we observe striking cases where the independent TF sequence signature weights more than the original TFs in predicting cobinding. This motivated us to create a directed graph that represents such contribution relationships (Figs. 1J, 6). First, from the regulatory graph, we observe that the control relationship is rarely symmetric, with the exception of TFs that share strong sequence similarity, for example, FOXA1 and FOXA2. Although the motif signature of TF-A is important for another pair TF-B and TF-C, it is not necessary that TF-B and TF-C will be a strong signature of pairs involving TF-A (Fig. 6A,G). This refutes the possibility of significant artifacts caused by sequence signatures sharing sequence similarity with the TFs under investigation and supports the directional regulatory relationship.
Second, we observe sequence motifs that appear to be hubs in affecting many TFs (Fig. 6A,F). In the network (Fig. 6A), the width and color of each edge connecting a TF and a TF-TF pair represent the contribution of that TF motif features to the TF-TF pair. The weight value of an edge connecting TF-A and TF-B-TF is calculated by averaging the SHAP feature importance of all pairs tied with TF-B for TF-A's motif features. The strength of each sequence motif is calculated by averaging weights connected to that motif in the network (Fig. 6F). From the plot, CTCF, GABPA, and HNF4A are the three most contributive effectors (Fig. 6F), and GABPA, HNF4A, TAF1, and JUND affect many TF-TF pairs in this network under investigation (Table 1), whereas others control more specific and fewer TFs and TF pairs. Overall, this graph supports a hierarchical organization of the regulatory network that is orchestrated by a few sequence signatures and rewires into more specific functions and specificity through combinations of more specific sequence signatures.
Discussion
Unlike single TF binding, the study of cobinding is a less explored area. Although chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) has been applied to the combination among 10 cell lines and 13 transcription factors in the ENCODE database, experimentally measuring the genome-wide co-occupancy of every TF pair in every cell type is practically infeasible. Thus, characterizing the TF cobinding patterns has been an open scientific question for years. Existing software such as TACO and coTRaCTE also used DNase-seq and TF motifs to predict TF-TF co-occurrence (Jankowski et al. 2014; van Bömmel et al. 2018). TACO modeled TF motif complex enrichment based on cell type–specific open chromatin regions from the ENCODE DNase-seq data set. Similarly, coTRaCTE first defined cell type–specific DNase Hypersensitive Sites (DHSs) through the t-statistic measure. Then, the TRanscription factor Affinity Prediction method (TRAP) (Roider et al. 2007) was used to estimate the TF binding sites among the DHSs based on TF motifs. In this work, we further leveraged the large-scale TF ChIP-seq data to build machine learning models for TF-TF co-occurrence detection. Consistent with previous studies, we also found that open chromatin and TF motifs play central roles.
Specifically, we developed machine learning methods to dissect the contributors to TF cobinding. Our focus is on dissecting the independent sequence contributors. The design of the algorithm thus included features of other TFs irrelevant to the ones under investigation. This experiment resulted in the finding of independent sequence signatures that contribute as strongly as or even more strongly than the TFs under investigation. This analysis also allowed us to construct a directed graph of how these sequence signatures affect each other.
Such sequence signatures can be roughly divided into three categories: TF pair–specific, TF-specific, and TF-generic. For example, TAF1 is an important signature for a range of transcription factor binding patterns. On the other hand, TF-specific and TF pair-specific sequence signatures tend to involve TFs controlling more specific biological processes. For example, the cobinding patterns of FOXA2, a TF that activates liver-specific gene expression, are affected by HNF4A, another liver-specific TF. The result from SHAP analysis and network analysis overall suggests a hierarchy of organization of sequence signatures that coordinates to fulfill global functionality of transcription by global effectors as well as tissue specificity by the interplay of binding sites of tissue-specific TFs. Analysis of feature importance could be affected by a variety of factors such as the frequency of occurrence of a feature and the base learners. Upscaled and more comprehensive studies covering these areas could be carried out in the future.
The human genome encodes for a limited number of 1500 transcription factors (Ignatieva et al. 2015), but they are sufficient to facilitate the formation of cellular diversity and development dynamics. One key is the combinatory interplays of multiple TFs. This study identifies the sequence signatures by using a machine learning approach and statistical analysis and contributes to our understanding of the rules that drive such interplay.
Methods
Data sets for model building and testing
In this study, we used the DNase-seq and ChIP-seq data from the ENCODE Project, covering 13 TFs (ATF3, CTCF, E2F1, EGR1, FOXA1, FOXA2, GABPA, HNF4A, JUND, MAX, NANOG, REST, and TAF1) in 10 cell types (A549, GM12878, H1-hESC, HCT116, HeLa-S3, HepG2, K562, MCF-7, iPSC, liver). The ChIP-seq data were downloaded from https://www.synapse.org/#!Synapse:syn6181337 (conservative peaks) and the ENCODE Project website (https://www.encodeproject.org/). The accession IDs are listed in Supplemental Table S1. The DNase-seq data were downloaded from https://www.synapse.org/#!Synapse:syn6176232. The data were processed following the standard ENCODE analysis pipeline (Data Processing Pipelines 2020, https://www.encodeproject.org/pipelines/, accessed October 5, 2020). The reference human genome is GRCh37. The conclusions in this manuscript would not be significantly affected if using GRCh38 as the reference genome. GRCh38 has improvements over GRCh37 such as annotation of the centromere regions and the addition of alternate loci, but these differences should not greatly impact the patterns of transcription factor cobinding. In particular, the DNase-seq data provided the cell type–specific chromatin accessibility, which was highly associated with TF binding events, and we used the filtered alignment files of DNase-seq. The ChIP-seq data provided the single TF binding sites across various cell types. For each 200-bp interval sliding every 50 bp in the human genome, we defined three types of binding labels—“bound” (B); “unbound” (U); and “ambiguous” (A)—by overlapping 200-bp intervals with peaks generated from SPP peak caller (Kharchenko et al. 2008) at the irreproducible discovery rate (IDR) cutoff of 5% (Li et al. 2011; Bionetworks S. Synapse | Sage Bionetworks, https://www.synapse.org/#!Synapse:syn6131484/wiki/402033, accessed November 1, 2019). We further defined a “cobound” genomic bin of a TF-TF pair when the single TF ChIP-seq labels from both TFs were “bound,” serving as the positive examples. When only one TF ChIP-seq label is “bound,” or none of the TFs is bound, the interval is considered as a negative example.
Data set partition for cross-cell type and cross-chromosome training and testing
In this study, we used a total of 228 TF co-occupancy profiles involving 56 different TF-TF pairs. For each TF-TF pair, we randomly held out one cell type for testing and the other cell types for model building. In this way, the performance of our models was evaluated in a cross-cell type manner (Supplemental Fig. S14; Supplemental Table S2). To build a robust and generalizable model for predicting TF co-occupancy, we designed a “crisscross” strategy to exploit the training data and avoid overfitting (Fig. 1C). In particular, we partitioned the cell types and chromosomes into the training set for model building and the validation set for hyperparameter tuning. This is because we used XGBoost, an iteration-based machine learning model, which required the validation-based early stopping. First, the 20 training chromosomes were randomly split into set1 (Chr 2, Chr 4, Chr 6, Chr 7, Chr 12, Chr 13, Chr 15, Chr 16, Chr 17, Chr 20, Chr X) and set2 (Chr 3, Chr 5, Chr 9, Chr 10, Chr 11, Chr 14, Chr 18, Chr 19, Chr 22). The model was trained on one chromosome set and validated on the other set to avoid overfitting on chromosomes. Second, for a TF-TF pair with N training cell types, 2N XGboost models would be built. The first model was trained on chromosome set1 in cell type 1, and validated on chromosome set2 in cell type 2. Similarly, the kth model was trained on chromosome set1 in cell type k and validated on chromosome set2 in cell type k + 1. The Nth model was trained on chromosome set1 in cell type N, and validated on chromosome set2 in cell type 1. In this way, we trained the model in one cell type but validated it against a different cell type, which improved the generalizability of our method in unseen cell types. After that, we switched chromosome set1 and set2 by training a model on chromosome set2 in cell type k and validating on chromosome set1 in cell type k + 1. In this way, we obtained another N models. Finally, predictions from the 2N models were averaged as the final prediction.
An additional test was carried out for cross-chromosome evaluation. In this test, different from cross-cell type evaluation which tested on the 20 training chromosomes, we tested on the three left out chromosomes (Chr 1, Chr 8, Chr 21). Of note, the two approaches of cross-validation were aimed to assess model transferability in different scenarios. The follow-up feature analysis focused on cross-cell prediction models.
The distributions of single TFBSs and co-occupied TFBSs were investigated (Supplemental Table S7; Supplemental Figs. S15–S17). To validate whether a TF-TF pair was significantly co-occupied, we applied a nonparametric paired Wilcoxon signed-rank (Wilcoxon 1945) test to all 56 TF pairs after pairwise comparison selection. The overall distributions of co-occupancy counts and P-values (Supplemental Table S8; Supplemental Figs. S18, S19) are shown in Supplemental Figure S19, C–E. The results showed that all pairs detected were significantly cooperative (all P-value < 0.0001, with family-wise error rate 0.01), which also corresponds to literature reports. In particular, the ratio of observed co-occupancy between FOXA1 and FOXA2 was significantly higher than other TF pairs, which may be attributed to the similarity in their motifs (Kulakovskiy et al. 2018) and regulatory functions (van der Sluis et al. 2008; Li et al. 2012). We also observed a high ratio of co-occupancy between HNF4A and FOXA2 (Supplemental Fig. S19B,E). In fact, the molecular interaction between them has been reported previously (Wallerman et al. 2009).
Feature extraction
A total of 90 DNase-seq-based features, 416 TF motif–based features, and 20 distance-to-gene features were used in our model. To remove the potential batch effect, the original DNase-seq filtered alignment was quantile-normalized before extracting DNase-seq-based features. For each 200-bp genomic bin, the maximum, minimum, and mean DNase-seq values were calculated as 3M-DNase features, and Δmaximum, Δminimum, and Δmean DNase-seq values were extracted as Δ3M-DNase features (Li et al. 2019). The Δ3M-DNase features were the difference between the DNase-seq signal of a specific cell line and the average signals of all 10 cell lines used in this study. We further considered 14 upstream and downstream neighboring bins to extract the corresponding 3M-DNase and Δ3M-DNase features, resulting in a total of 90 = (3 + 3) × 15 DNase-seq-based features. This neighboring information is also reported to improve predictive performance in recent studies.
In addition to the DNase-seq-based features, we also integrated TF motif features of all 13 TFs in the models. Specifically,
for each TF, the position weight matrix (PWM) (Stormo et al. 1982) can be represented by an a-by-4 matrix M, 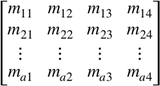 where a is the length of the TF motif. Similarly, DNA sequence can be one-hot encoded and represented by a 4-by-b matrix X,
where a is the length of the TF motif. Similarly, DNA sequence can be one-hot encoded and represented by a 4-by-b matrix X, 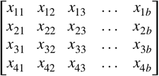 where b is the number of nucleotides in the human genome and four rows represent four types of nucleotides A/C/G/T. Each item in
X was a binary value and for each column, or nucleotide position, the row where x = 1 corresponded to the nucleotide type in that position. By multiplying M and X, we can obtain an a-by-b matrix Y,
where b is the number of nucleotides in the human genome and four rows represent four types of nucleotides A/C/G/T. Each item in
X was a binary value and for each column, or nucleotide position, the row where x = 1 corresponded to the nucleotide type in that position. By multiplying M and X, we can obtain an a-by-b matrix Y, 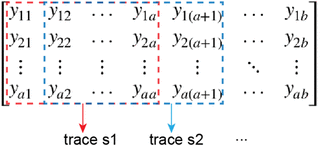
where a is the length of the TF motif and b is the length of a chromosome. In order to obtain the TF motif match score for each nucleotide position, we calculated the trace of the submatrix Y[, i:(i + a−1)] for i = 1, 2, …, b−a + 1. For example, when i = 1, the trace of the submatrix within the red rectangle was calculated as s1. Similarly, when i = 2, the trace of the submatrix within the blue rectangle was calculated as s2. Therefore, for each TF, obtained a 1-by-(b−a + 1) score vector S = [s1 s2 s3 … sb−a+1]. This score vector S was further padded with zeros at both ends to the length a. Then, we extracted bin-level motif-based features from this vector S. For each 200-bp interval, the top three scores among that 200 positions were saved. Similarly, the top three scores of the product between TF PWM and the reverse complement of the genome sequence were calculated. Then, the top four of the two sets of top three scores were considered as motif features. Similar to the DNase-seq-based features, we also considered eight neighboring intervals and generated a total of 32 = 4 × 8 motif features for each TF. To consider the potential interactions of all TFs, we used the 32 motif features from all 13 TFs under consideration in this study. In addition to the DNase-seq-based and motif-based features, we further extracted the distance-to-gene features. Specifically, for each 200-bp interval, we calculated the top 20 closest distances to proximal genes based on GENCODE annotation (Li et al. 2019).
Using XGBoost to model nonlinear interactions between features and to facilitate subsequent game theory–based feature importance analysis
The tree-based XGBoost models (Chen and Guestrin 2016) were trained on a total of 526 (90 + 416 + 20) features mentioned above. The parameters of XGBoost are: (1) the step size shrinkage is 1; (2) the maximum depth of a tree is 7; (3) the minimum sum of weights in a child is 5; and (4) the minimum loss reduction is 0.1. We used cross-entropy loss, as the label is binary, specifically: −[y × log(ypred) + (1−y) × log(1−ypred)], where y represents the true label and ypred represents the prediction value. The tree-based models can learn efficiently the nonlinear interactions between features and are robust to noise and outliers in data (Li et al. 2018; Li and Guan 2019b). Furthermore, we set the maximum number of iterations to 1000 and applied a validation-based early stopping strategy in a crisscross fashion, across both cell types and chromosomes. Final predictions were obtained by averaging the results of all models.
Evaluating predictive performance with AUROC and AUPRC
To evaluate the predictive performance of our model, we used the area under the receiver operating characteristic curve and the area under the precision recall curve as the primary scoring metrics (Davis and Goadrich 2006). For each cutoff in the AUROC and AUPRC curves, true positive (TP) was the number of correctly predicted cobound bins, false positive (FP) was the number of predicted cobound bins whose true labels were un-cobound, true negative (TN) was the number of correctly predicted un-cobound bins, and false negative (FN) was the number of predicted un-cobound bins whose true labels were cobound.
Benchmark comparison
We benchmarked the performance of TF cobinding prediction using methods TACO and coTRaCTE. Briefly, these two methods focus on open chromatin regions and estimate TF cobinding enrichment based on motif information. They assign a P-value for each genomic region under consideration. For TACO, we defined the prediction score by multiplying the P-values of two TFs in the same region. For coTRaCTE, we first transformed the original P-value of a TF pair into −log10(P-value). Then, we rescaled them to the range between zero and one and used one minus the rescaled value as the prediction score.
SHAP analysis to identify independent sequence signatures
We used SHAP (Shapley 1988; Lundberg et al. 2018) to interpret the output of our model and reveal independent sequence signatures’ contributions to cobinding events. The typical feature importance calculation in the gradient boosting model is based on the improvement of each attribute in the performance (Chen and Guestrin 2016; Xia et al. 2017). In contrast, SHAP is based on the magnitude of feature attributions (Molnar 2019). We used SHAP feature importance, which is measured as the mean absolute Shapley values, in our analysis.
For our model, we calculated a 526 × 56 matrix containing SHAP values for 526 features and 56 pairs. For a specific TF or TF-TF pair, we calculated the relative feature importance by analyzing the corresponding submatrix. Additionally, to reveal the control relationship between sequence motif features and TF-TF pairs, we assigned SHAP values to a directed regulatory network, and the edges with different weights were indicated by widths. In the network, each node represents a TF or a TF-TF pair and each edge represents the SHAP feature importance of that sequence feature to the TF-TF pair. For an edge connecting ‘TF1’ and ‘TF2-TF’, the weight was calculated by averaging SHAP values of the TF1 motif feature predicting all pairs tied with TF2. For example, there are seven pairs tied with FOXA2 (FOXA2-TAF1, FOXA2-REST, FOXA2-AMX, FOXA2-JUND, FOXA2-HNF4A, FOXA2-GABPA, and FOXA1-FOXA2) (Supplemental Fig. S2); we only selected a submatrix of the corresponding seven columns from the complete SHAP value matrix and calculated the average absolute SHAP value for each TF as the weights of the edges connecting the nodes TF’ and ‘FOXA2-TF’.
Software availability
The source code of the analysis is available as Supplemental Code and at GitHub (https://github.com/GuanLab/Sequence_Analysis_for_TF-co-binding).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work is supported by the National Science Foundation (NSF#1452656) and the National Institutes of Health (R35-GM133346).
Author contributions: Y.G. directed the study; M.Z. and H.L. performed the experiments; M.Z. and X.W. created the figures; M.Z., Y.G., and H.L. wrote the manuscript. All authors have read and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.267310.120.
- Received June 12, 2020.
- Accepted December 3, 2020.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








