
Mutagenesis of human genomes by endogenous mobile elements on a population scale
- Nelson T. Chuang1,2,3,
- Eugene J. Gardner1,2,6,
- Diane M. Terry1,2,
- Jonathan Crabtree2,
- Anup A. Mahurkar2,
- Guillermo L. Rivell4,
- Charles C. Hong5,
- James A. Perry5 and
- Scott E. Devine1,2,4,5
- 1Graduate Program in Molecular Medicine, University of Maryland, Baltimore, Baltimore, Maryland 21201, USA;
- 2Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, Maryland 21201, USA;
- 3Division of Gastroenterology, Department of Medicine, University of Maryland School of Medicine, Baltimore, Maryland 21201, USA;
- 4Greenebaum Comprehensive Cancer Center, University of Maryland School of Medicine, Baltimore, Maryland 21201, USA;
- 5Department of Medicine, University of Maryland School of Medicine, Baltimore, Maryland 21201, USA
Abstract
Several large-scale Illumina whole-genome sequencing (WGS) and whole-exome sequencing (WES) projects have emerged recently that have provided exceptional opportunities to discover mobile element insertions (MEIs) and study the impact of these MEIs on human genomes. However, these projects also have presented major challenges with respect to the scalability and computational costs associated with performing MEI discovery on tens or even hundreds of thousands of samples. To meet these challenges, we have developed a more efficient and scalable version of our mobile element locator tool (MELT) called CloudMELT. We then used MELT and CloudMELT to perform MEI discovery in 57,919 human genomes and exomes, leading to the discovery of 104,350 nonredundant MEIs. We leveraged this collection (1) to examine potentially active L1 source elements that drive the mobilization of new Alu, L1, and SVA MEIs in humans; (2) to examine the population distributions and subfamilies of these MEIs; and (3) to examine the mutagenesis of GENCODE genes, ENCODE-annotated features, and disease genes by these MEIs. Our study provides new insights on the L1 source elements that drive MEI mutagenesis and brings forth a better understanding of how this mutagenesis impacts human genomes.
Three types of endogenous mobile elements continue to mutagenize human genomes—namely, Alu, LINE-1 (L1), and SVA elements (Mills et al. 2007). When these mobile elements are inserted into genes or other functionally important genomic sites, they can cause human diseases (for review, see Hancks and Kazazian 2016; Kazazian and Moran 2017). Kazazian et al. (1988) discovered the earliest examples of disease-associated mobile element insertions (MEIs) in two cases of Hemophilia A that were caused by independent de novo L1 insertions that disrupted the 14th coding exon of the coagulation factor VIII gene. Four years later, Miki et al. (1992) discovered a somatic L1 insertion that disrupted the 16th coding exon of the APC tumor suppressor gene in a patient with colorectal cancer. Since then, MEIs have been implicated in at least 130 diverse cases of human diseases (for review, see Hancks and Kazazian 2016; Kazazian and Moran 2017).
Human mobile elements initially were thought to be active exclusively in the germline and then repressed in somatic tissues throughout adulthood. However, it has become clear that L1 also is active in at least some somatic tissues, including neuronal tissues of the brain (Coufal et al. 2009; Baillie et al. 2011; Evrony et al. 2012; Upton et al. 2015; for review, see Terry and Devine 2020) and human cancers (Iskow et al. 2010; Lee et al. 2012; Solyom et al. 2012; Shukla et al. 2013; Helman et al. 2014; Tubio et al. 2014; Doucet-O'Hare et al. 2015; Ewing et al. 2015, 2020; Rodić et al. 2015; Scott et al. 2016; Rodriguez-Martin et al. 2020; Yamaguchi et al. 2020). Growing evidence also suggests that L1 can evade somatic repression in other normal cells and tissues, including the esophagus, pancreas, and colon (Doucet-O'Hare et al. 2015; Ewing et al. 2015; Scott et al. 2016; Yamaguchi et al. 2020), as well as the liver, brain, embryonic stem cells, and cells undergoing neuronal differentiation (Sanchez-Luque et al. 2019). Many aspects of L1 regulation and dysregulation remain poorly understood in these diverse settings owing to a limited knowledge of the full-length L1 source elements that drive MEI mutagenesis.
Full-length L1 human-specific (FL-L1Hs) elements are the only active autonomous mobile elements in humans (Kazazian and Moran 2017). FL-L1Hs elements are 6 kb in length and have two open reading frames (ORF1 and ORF2) that encode the protein machinery that is necessary for L1 mobilization. ORF1 encodes a nucleic acid chaperone (Martin et al. 2005), whereas ORF2 encodes an endonuclease (EN) (Feng et al. 1996) and a reverse transcriptase (RT) (Mathias et al. 1991). When the ORF1p and ORF2p proteins are translated from the L1 mRNA, they bind to that same mRNA to form a ribonucleoprotein particle (RNP) (Doucet et al. 2010). The RNP then is imported back into the nucleus, where the L1 mRNA is used as a template to generate a new copy of L1 by a process that is known as target primed reverse transcription (TPRT) (Luan et al. 1993). Both Alu and SVA are nonautonomous mobile elements that do not encode any of the proteins that are required for mobilization. Instead, they hijack the L1 machinery for their own mobilization (Dewannieux et al. 2003; Hancks et al. 2011; Raiz et al. 2012). Because Alu and SVA elements are mobilized by the same process as L1, newly integrated copies of Alu and SVA have L1-like insertion preferences for AT-rich hexamer sequences and are flanked by L1-like target site duplications (TSDs).
Previous studies have examined FL-L1Hs elements in the reference human genome (REF) and in non-reference (non-REF) human genomes (Brouha et al. 2003; Beck et al. 2010). Brouha et al. (2003) amplified and cloned 82 FL-L1Hs copies from BAC clones that were sequenced by the Human Genome Project (International Human Genome Sequencing Consortium 2001) and tested them for activity in a cell culture–based assay for retrotransposition. Each FL-L1Hs was cloned into an episomal plasmid and tested for its ability to generate new “offspring” L1 insertions in HeLa cells (Moran et al. 1996). The study estimated that there are 80–100 active FL-L1Hs elements in the reference human genome, although most of the activity was attributed to eight highly active or “hot” L1s. Beck et al. (2010) later cloned and sequenced 68 non-REF FL-L1Hs elements from eight diverse individuals and tested them in a similar cell culture–based assay. The collection of relatively young, non-REF elements tested by Beck et al. (2010) was enriched for highly active hot L1s compared to the REF elements that were studied by Brouha et al. (2003), suggesting that younger non-REF FL-L1Hs copies are inherently more active than older REF FL-L1Hs elements.
Although these two pioneering studies provide great insights on both REF and non-REF FL-L1Hs elements in humans, the mutagenic potential of FL-L1Hs elements across diverse human populations remains largely unexplored. In both these studies, the FL-L1Hs elements were identified from BAC and fosmid clones that had been fully sequenced with Sanger sequencing; thus, the internal sequences could be examined for the presence of intact ORFs and subfamily status. In contrast, shotgun Illumina WGS does not provide the interior sequences of FL-L1Hs elements because reads that correspond to the interior regions of FL-L1Hs elements cannot be uniquely mapped to individual L1 loci. Thus, despite the massive amount of Illumina WGS and WES data that have been generated over the last decade, our understanding of sequence variation within non-REF FL-L1Hs elements, and the impact of this variation on L1 activity, remains limited.
Nevertheless, several large-scale Illumina WGS and WES projects have emerged recently that have provided exceptional opportunities to discover MEIs and study their impact on human genomes. This includes the TOPMed project, which has sequenced high-coverage whole genomes from human cohorts with heart, lung, and blood phenotypes; the GTEx project, which has sequenced high-coverage whole genomes to study RNA expression in diverse human tissues; the New York University Genome Center, which has sequenced high-coverage genomes from the 1000 Genomes Project (1KGP), and the United Kingdom Biobank (UKBB), which has sequenced whole-exome sequences from participants of the UKBB project. These resources provide unprecedented opportunities and challenges to study endogenous mobile elements on a population scale.
Results
MEI discovery on a population scale
We previously developed the mobile element locator tool (MELT) to perform mobile element discovery in low coverage Illumina genomes (∼7×) that were sequenced by the 1KGP (Sudmant et al. 2015; Gardner et al. 2017). However, as we began to perform MEI discovery on Illumina genome sequences with higher coverages (30–40×), it became evident that we needed to further improve the efficiency and scalability of MELT. Our strategy began by improving the Individual Analysis step of MELTv2.1.5, because this was a particularly inefficient step when processing high-coverage Illumina whole-genome sequences. Our improved code reduced the runtime for that step from 387 min 32 sec to 19 min 33 sec on the NA12878 Illumina high-coverage genome sequence (a 19.8× speedup in runtime). We then created a cloud-based implementation of this improved MELT engine (CloudMELT) using Toil, Docker, and Common Workflow Language tools to parallelize MEI discovery (Methods; Supplemental Fig. S1). This approach allowed us to perform MEI discovery on thousands of high-coverage Illumina genomes in days rather than months.
We then used MELT and CloudMELT to perform MEI discovery on 57,919 human samples and identified 104,350 nonredundant MEIs (including 80,562 Alu, 16,525 L1, 6956 SVA, and 307 HERV-K insertions) in five human populations (Fig. 1A; Supplemental Table S1A). These five highly diverse populations include (1) the TOPMed Amish population (1112 high-coverage WGS samples), (2) the TOPMed Jackson Heart Study population (3331 high-coverage WGS samples), (3) the GTEx population (639 high-coverage WGS samples), (4) the 1KGP population (3202 high-coverage WGS samples), and (5) the United Kingdom Biobank (UKBB) population (49,635 WES samples). A Venn analysis of these data indicates that the majority of the MEIs in our collection (65,192/104,350 or 62.3%) are novel compared to other MEIs that recently have been discovered with earlier versions of MELT (Fig. 1B; Supplemental Table S1B; Sudmant et al. 2015; Cao et al. 2020; Collins et al. 2020). Likewise, only 200 of our L1s were found in dbRIP (version 3) and only 2019 were found in euL1db. These data indicate that MEI discovery is far from complete in humans and that there is an unmet need for highly scalable MEI discovery tools such as CloudMELT.

MEIs discovered in this study. (A) MEIs that we discovered are broken down by population and MEI type. At the top (labeled “Merged”), 104,350 nonredundant MEIs were discovered. (B) A comparison of our study with three other published MEI discovery projects (1KGP LC, GTEx, and gnomAD-SV) (Sudmant et al. 2015; Cao et al. 2020; Collins et al. 2020, respectively). The four-way Venn diagram comparing these studies with our data set indicates that 65,192 (or 62.3%) of our MEIs are novel. All these MEI discovery studies were performed with MELT.
Full-length L1 human-specific (FL-L1Hs) elements
We next examined the size distribution of the L1 elements that we discovered in the five populations. L1 elements often become truncated during the process of retrotransposition, and we wished to determine how many FL-L1Hs (6 kb) elements were discovered among the 14,066 L1 MEIs for which MELT could successfully generate a length estimate. We found a total of 3728/14,066 (26.5%) FL-L1Hs elements and 10,338/14,066 (73.5%) 5′-truncated elements (Fig. 2A; Supplemental Table S2A). We next examined the FL-L1Hs copy number distributions in the four WGS populations in our study and found that the number of non-REF FL-L1Hs elements varies considerably both within and across the populations (Fig. 2B,C; Supplemental Table S2). For example, although the average copy number for non-REF FL-L1Hs elements in the 1KGP populations is 44.3, the copy numbers range from 25 to 63 (Fig. 2B; Supplemental Table S2E). The non-REF FL-L1Hs copy numbers also vary in the Amish, Jackson Heart, and GTEx populations (Fig. 2B; Supplemental Table S2). Non-REF FL-L1Hs copy numbers also vary across the 1KGP superpopulations (Fig. 2C; Supplemental Table S2E). These data suggest that the levels of L1 mutagenesis and L1-mediated disease could vary considerably both within and across diverse populations.
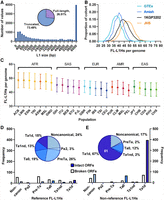
FL-L1Hs elements discovered in this study. (A) We identified 3728 FL-L1Hs elements that are 6 kb or longer within the collection of 16,525 L1 MEIs that were discovered in this study. Because MELT could estimate the lengths of 14,066/16,525 (85.1%) L1s, we calculated the percentages in A using the denominator of 14,066. (B) Comparisons of FL-L1Hs elements per genome in the four WGS studies of our study. The GTEx and Amish populations have fewer FL-L1Hs copy numbers compared to the 1KG population and the Jackson Heart Study. (C) Population distribution of FL-L1Hs elements arranged by superpopulation of the 1KGP 2504 high-coverage genomes. (D,E) REF and non-REF FL-L1Hs elements with their subfamily distributions. Corresponding bar plots indicate the number of FL-L1Hs elements with two intact ORFs (solid bars). (E) The majority of FL-L1Hs elements in the non-REF group belong to the Ta1d subfamily and have two intact ORFs (bottom right). Note also the expansion of Ta1d elements in non-REF populations compared to REF (from 15% to 67%; compare purple sections in D and E). The white 61 numeral in E indicates the number of elements in this group with documented activity in the literature (Supplemental Table S3A).
Although it would be useful to examine the interior sequences of these FL-L1Hs elements to determine whether they encode two intact ORFs and belong to active L1 subfamilies, the interior sequences of these elements are not recovered with shotgun Illumina WGS. Reads that correspond to the interior regions of FL-L1Hs elements cannot be uniquely mapped to individual L1 loci because of the short read lengths and the repetitive nature of FL-L1Hs elements. Thus, we developed a long-read, Pacific Biosciences (PacBio)-based approach to fully sequence a sampling of 698/3728 (18.7%) of the FL-L1Hs elements that we discovered in this project (Methods; Supplemental Fig. S2). We focused on FL-L1Hs elements that were discovered in the 1KGP samples because genomic DNA for these samples can be obtained from the Coriell repository, and these populations also are quite diverse. We used custom primers in conjunction with long-range PCR to amplify and sequence these 698 non-REF insertions from 311 independent 1KGP Coriell samples plus one additional patient sample (Methods; Supplemental Table S3A,B). We sequenced elements across the entire allele frequency spectrum, including both rare and common non-REF FL-L1Hs elements. After sequencing, we identified a total of 647/698 (92.7%) FL-L1Hs elements that are at least 6 kb in length. The remaining 51 elements have small 5′ truncations or internal deletions (Supplemental Table S3A).
Most (519/647 or 80.2%) of our sequenced 6-kb elements have two intact ORFs, and the majority of elements with two intact ORFs (381/519 or 73.4%) belong to the youngest most active Ta1d subfamily (Fig. 2E). Moreover, we found that the Ta1d subfamily has undergone a substantial expansion in non-REF populations compared to other L1 subfamilies (Fig. 2D,E). Note that FL-L1Hs Ta1d elements (purple) expanded from 15% of REF elements (Fig. 2D) to 67% of non-REF elements (Fig. 2E). We also examined published studies to determine whether our non-REF Ta1d FL-L1Hs elements have been active in (1) cell culture assays, (2) the germline, or (3) somatic cancers. We found that 61 of the Ta1d FL-L1Hs source elements were active in one or more of these studies (Fig. 2E, white numeral; Supplemental Table S3A). Overall, these data indicate that the Ta1d subfamily is the most rapidly expanding and active group of FL-L1Hs elements in human populations.
Three novel highly active FL-L1Hs subfamilies in human populations
We leveraged the data from our sequenced FL-L1Hs elements to determine whether we could identify new L1 subfamilies that might be particularly active. In fact, we identified three novel subfamilies that recently evolved from the highly active Ta1d subfamily (Fig. 3A,B). These subfamilies are defined by interior sequence changes at positions 1026, 3337, and 3440 (Fig. 3A). We named these subfamilies Ta1d-CAT, Ta1d-CCA, and Ta1d-TCA based on the sequences that are present at these three positions. The Ta1d-TCA subfamily appears to be the youngest and most active subfamily, because it has undergone the largest recent expansion in non-REF populations (Fig. 3C,D, dark purple). Note that the Ta1d-TCA subfamily went from 9% in REF (Fig. 3C) to 32% in non-REF (Fig. 3D). However, the Ta1d-CAT and Ta1d-CCA subfamilies also are relatively abundant in non-REF populations (Fig. 3D). We next identified collections of FL-L1Hs elements that belong to these three subfamilies and also had been sequenced and tested for activity in cell culture assays previously (Beck et al. 2010). Two of our three new subfamilies (Ta1d-CCA, Ta1d-TCA) had the highest levels of activity in the cell culture assay compared to the other subfamilies tested, with the Ta1d-TCA subfamily having the highest level of activity (Fig. 4A). We conclude that the Ta1d-TCA subfamily is the most active new subfamily in humans, followed by Ta1d-CCA and Ta1d-CAT (Figs. 3B–D, 4A).
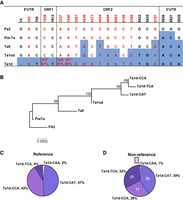
Three novel Ta1d subfamilies of FL-L1Hs elements. (A) Table of canonical positions defining L1 subfamilies building upon those published previously (Boissinot et al. 2000; Brouha et al. 2003). Positions in red are new canonical positions discovered in our sequenced FL-L1Hs elements. Note that positions 1026, 3337, and 3440 define three new subfamilies according to the sequences at those positions. (B) A phylogenetic tree was constructed using subfamily consensus sequences to evaluate the relationship of known subfamilies versus new ones (Supplemental Table S3C). The tree was calculated using the neighbor-joining method and distances were corrected using the Kimura 2-parameter model. The numbers at each node represent the percentage of replicate trees that clustered together in 1000 bootstrap tests. (C,D) Reference and non-reference proportions of Ta1d subfamilies that show expansion of the Ta1d-TCA subfamily in non-reference populations (D). Note the expansion from 9% to 32% (dark purple) when comparing the Ta1d-TCA subfamily in reference (C) versus non-reference (D). The white numerals in D indicate the number of FL-L1Hs elements that were found to be active in the literature for each subfamily (Supplemental Table S3A).
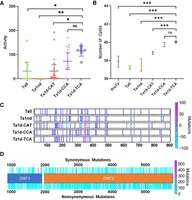
Analysis of new subfamilies, CpGs, and other interior sequence changes in our FL-L1Hs elements. (A) We identified elements from various L1 subfamilies (including our three novel subfamilies) that previously had been sequenced and tested in cell culture retrotransposition assays (Beck et al. 2010). Note that the new Ta1d-TCA subfamily has the highest levels of activity followed by the new Ta1d-CCA subfamily. The Ta1d-CAT and Ta0 subfamilies have similar activities. Significance was calculated by one-way ANOVA corrected for multiple comparisons by the Tukey method. Error bars represent 95% confidence intervals. (B) We also plotted the number of CpGs by subfamily in our sequenced FL-L1Hs elements and found that our three new subfamilies (Ta1d-CAT, Ta1d-CCA, and Ta1d-TCA) have 1–2 additional CpGs in their promoter regions compared to older subfamilies. (C) Mutations in the promoter region causing a gain or loss of CpGs for each L1 subfamily. The color shows the frequency of gain or loss as denoted by the diverging color map. (D) Mutations within the two ORFs. Note the synonymous and nonsynonymous mutations above and below the ORF map, respectively. The frequency of the mutations also is shown with the diverging color map.
Other internal variation in FL-L1Hs elements
We next examined CpGs in the promoter regions of our sequenced FL-L1Hs elements and found an inverse relationship between subfamily age and the number of CpGs in the promoters of these elements. In particular, the older subfamilies (i.e., L1-Pre-Ta and L1-Ta0) had fewer CpGs compared to the younger subfamilies (L1-Ta1nd and our novel Ta1d subfamilies). In fact, our three novel subfamilies had the most CpGs, with an average of one or two additional CpGs compared to the older subfamilies (Fig. 4B). These data suggest that the birth of new, highly active L1 subfamilies is accompanied by an increased number of CpGs in L1 promoters, perhaps to more tightly regulate these elements. These data also are consistent with previous findings that older, less active elements lose CpGs over time owing to the deamination of methylated cytosines (Walser et al. 2008).
We identified additional internal sequence changes in our 647 FL-L1Hs elements that could potentially impact L1 function (Fig. 4C,D; Supplemental Fig. S3; Supplemental Table S4A,B). In addition to CpG changes, mutations that impact transcription factor binding sites in L1 promoters or cis-regulatory RNA binding sites also could be envisioned to modulate L1 activity. Likewise, a large number of synonymous and nonsynonymous changes have occurred within the ORF1 and ORF2 sequences of these elements (Fig. 4D; Supplemental Fig. S3; Supplemental Table S4B). Stop codons, frameshift mutations, and nonsynonymous changes that introduce dissimilar amino acid substitutions would be expected to influence L1 the greatest. Many of the interior sequence changes are unique to a specific L1 locus and can be used to track source/offspring relationships or the expression of a given FL-L1Hs locus (Scott et al. 2016). Together with the Brouha and Beck collections, our collection of sequence-resolved FL-L1Hs elements will serve as an excellent resource to study the impact of internal sequence variation on L1 regulation, activity, and evolution.
We also identified 3′ transductions that are associated with our sequenced FL-L1Hs elements (Supplemental Table S3A; Supplemental Fig. S4). Together with other published 3′ transduction data, we determined that 76/698 (10.9%) of our sequenced FL-L1Hs elements generated 3′ transductions, likely a result of weak poly(A) signals within the FL-L1Hs elements that are bypassed in favor of downstream signals (Supplemental Table S3A). Thirty-one of these elements (40.8%) were active only in the germline, 28 (36.8%) were active only in somatic tissues, and 17 (22.4%) were active in both the germline and somatic tissues (Supplemental Table S3A). We found that 23/698 (3.3%) of our FL-L1Hs elements were active in previously published cell culture mobilization assays (Supplemental Table S3A; Beck et al. 2010). Thus, taken together with the 3′ transduction data, 91/698 (13%) of our sequenced FL-L1Hs elements have one or more forms of evidence for retrotransposition activity. We also compared the germline L1 insertions that we discovered in this study (all sizes) with those discovered in three large studies of somatic L1 insertions in human cancers and found minimal overlap between these data sets (Supplemental Fig. S5). This suggests that there are few, if any, L1 integration hotspots in the human genome that are shared by germline and somatic cells.
Population analysis
We next counted the number of MEIs per individual in the 26 diverse 1KGP populations and found higher numbers of MEIs compared to previous studies with the same 26 populations (Fig. 5A; Supplemental Table S5A; Sudmant et al. 2015). This outcome is not surprising, given the higher WGS coverages that were used in this study (30–40×, compared to ∼7× previously) (Sudmant et al. 2015). The highest counts per individual were observed in the AFR populations, which is consistent with previous studies (Fig. 5A; Supplemental Table S5A; Sudmant et al. 2015). We also observed MEI count variation within each of the 26 populations that is mostly caused by differences in Alu counts (Fig. 5A; Supplemental Table S5A). Sharing analysis of the MEIs across the 26 diverse populations revealed MEIs that are found in all 26 populations (ALL), more than one (SHARED), or that are unique to a specific population (UNIQUE) (Fig. 5B; Supplemental Table S5B). MEIs that are unique to a specific population include singletons (allele count = 1) and non-singletons (allele count > 1) (Fig. 5C; Supplemental Table S5B). We performed the same analysis with the Amish, JHS, and UKBB populations (Supplemental Fig. S6A–C; Supplemental Table S5C) and found that the JHS population had higher MEI counts per individual than the Amish and UKBB populations. The JHS participants are solely African Americans from Jackson, Mississippi. Therefore, a higher number of MEIs per individual in this population is consistent with previous observations of higher levels of genetic diversity in African and African diaspora populations (Fig. 5A; Sudmant et al. 2015). The Amish population, in contrast, is a closed European-derived founder population that underwent a bottleneck upon moving to Lancaster, Pennsylvania, in the mid- to late-1700s (Pollin et al. 2008). Thus, the lower levels of MEI variation in the Amish group compared to the JHS population is consistent with the population histories of these two groups. Finally, the UKBB population had much lower MEI counts because MEI discovery was performed in whole-exome sequences instead of whole-genome sequences.
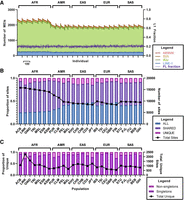
MEI counts per individual and population sharing across the 26 diverse 1KGP populations. (A) The numbers of MEIs per individual are depicted for the 26 diverse 1KGP populations: (light blue) LINE1; (light green) Alu; (light brown) SVA; (red) HERV-K. Dark lines of the same colors represent the boundaries between element classes. The dark purple line indicates the number of non-REF FL-L1Hs elements per individual. Note that the AFR populations have the highest MEI counts, consistent with previous studies (Sudmant et al. 2015). (B) Sharing of MEIs across the 26 diverse populations of the 1KGP. (C) MEIs that are unique to one of the 26 1KGP populations are broken down into singleton and non-singleton categories. We also performed similar analysis comparing the Amish, Jackson Heart Study, and the UKBB populations (Supplemental Fig. S6).
In addition to performing subfamily analysis on FL-L1Hs elements (Fig. 2D,E), we also performed subfamily analysis on the Alu and SVA MEIs that we discovered in this study (Supplemental Figs. S7, S8). In agreement with previous studies, the majority of Alu subfamilies are young AluY subfamilies that are known to be polymorphic and active in humans (e.g., 81% of the elements belong to AluY and the AluYa, AluYb, and AluYc lineages similar to what has been observed previously) (Supplemental Fig. S7; Gardner et al. 2017). Likewise, our data indicate that the SVA-E and SVA-F subfamilies have undergone recent expansions in the populations of our study (Supplemental Fig. S8A,B). Together with the data presented for L1 (Fig. 2D,E), these data indicate that a few dominant lineages of Alu, L1, and SVA elements are responsible for the majority of ongoing MEI mutagenesis in human populations.
Impact of MEI mutagenesis on genes and other important genomic features
We also leveraged our collection of Alu, L1, SVA, and HERV-K insertions to examine the impact of MEI mutagenesis on human genes and other annotated genomic features. We combined our MEIs with the others shown in Figure 1B to create a nonredundant set of 158,873 MEIs and then compared the coordinates of these MEIs with those of GENCODE genes and ENCODE-annotated features (Fig. 6A,B; Supplemental Table S6A,B). Of these MEIs, 97,103 map to GENCODE-annotated transcripts, with 274 MEIs disrupting 5′ UTRs, 2449 disrupting 3′ UTRs, 4639 disrupting exons, and 781 disrupting coding exons (CDS) of annotated genes (Fig. 6A; Supplemental Table S6A). Two MEIs disrupt start codons, 11 disrupt stop codons, and 101 disrupt splice sites (Fig. 6A). MEIs also disrupt 11,586 ENCODE cis-candidate regulatory elements (cCREs) (Fig. 6B; Supplemental Table S6B).
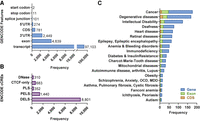
MEI mutagenesis patterns in GENCODE, ENCODE, and various databases. (A) Comparisons of the combined collection of 158,783 MEIs depicted in Figure 1B revealed intersections with GENCODE v35 gene annotations. The total number of GENCODE transcripts intersected by MEIs is 97,103. All features impacted by MEIs (exons, etc.) are found within these transcripts. The UTRs and CDSs also are included in the exon group, because they also are exons. In cases in which multiple transcript models are intersected by MEIs, all transcripts and features are listed in Supplemental Table S6A and are delimited by commas in the same order for each column. We identified insertions that disrupt various subregions of genes including 781 MEIs that disrupt CDS exon sequences. (B) MEIs disrupt ENCODE-annotated cis-candidate regulatory elements (cCREs). (C) MEIs disrupt genes that have been linked to various diseases in the Online Mendelian in Man (OMIM) database. Although all these MEIs disrupt genes and their annotated features, these insertions may or may not have functional consequences. MEIs that disrupt coding exons or ENCODE transcriptional regulators likely produce functional consequences. MEIs that occur within introns, UTRs, and other functionally important sites also can impact gene function (e.g., Watanabe et al. 2005; Lanikova et al. 2013). However, the precise functional consequences of gene-disrupting MEI insertions can be difficult to predict and must be validated experimentally.
We next searched the Online Mendelian Inheritance in Man (OMIM) database to identify MEIs that disrupt genes implicated in human diseases. We identified 16,606 MEIs that disrupt 2335 OMIM-annotated genes. These genes were further grouped by disease, and we identified 20 clusters of disease genes that are disrupted by MEIs (Fig. 6C; Supplemental Table S7A). For example, 177 genes that have been implicated in diverse human cancers are disrupted by MEIs in our collection (Fig. 6C; Supplemental Table S7B). We next expanded this approach for the cancer group to include the COSMIC database, which curates genes that have been implicated in cancers; the Genome Association Database (GAD), which curates loci that have been implicated in human cancers from GWAS studies; and the Candidate Cancer Gene Database (CCGD), which curates genes that have been implicated in mouse studies involving transposon mutagenesis. We identified MEIs that disrupt 7166 genes in these four databases. Among this collection, we found 376 MEIs that disrupt the CDSs of cancer-related genes, including well-defined tumor suppressor genes and oncogenes (e.g., APC, BRCA2, DNMT1, FANCC, FANCI, FANCM, MSH6, and XRCC2) (Supplemental Table S7C). Individuals who harbor such MEIs presumably would be at greater risk for developing cancers, particularly if the second allele of a tumor suppressor gene should be mutated or silenced during their lifetime. We also identified similarly relevant genes and mutations for the remaining disease groups in OMIM (Fig. 6C; Supplemental Table S7).
Discussion
FL-L1Hs source elements
We discovered 3728 non-REF FL-L1Hs elements and sequenced 698 of these elements using a novel PacBio-based approach. This provided us with new opportunities to determine whether these elements (1) have two intact ORFs, (2) belong to active subfamilies, or (3) belong to new subfamilies that are rapidly expanding in human populations. Because the majority of the non-REF FL-L1Hs elements in our collection have two intact ORFs and are very young (mostly belonging to the three novel Ta1d subfamilies that we identified in this study), many of these elements would be expected to be active in humans. This conclusion is supported by Beck et al. (2010), in which the collection of non-REF FL-L1Hs elements likewise was enriched for younger, non-REF elements that were highly active in cell culture assays.
We also learned that the number of non-REF FL-L1Hs copies varies considerably both within and across populations. For example, the number of non-REF FL-L1Hs elements in the 1KGP populations ranged from 25 to 63. The 1KGP individual with the lowest number of non-REF FL-L1Hs elements (i.e., 25) might be expected to have lower levels of MEI mutagenesis than the individual with the highest copy number (63) (Fig. 2). However, the individual with copy number 25 could have a large fraction of very hot L1s, whereas the individual with copy number 63 could have mostly inactive copies. Even a single detrimental mutation can severely diminish or eliminate L1 activity (Feng et al. 1996; Moran et al. 1996). Thus, more work is necessary to understand the mutagenic threat that is posed by the full collection of FL-L1Hs elements in an individual's genome. One way to approach this would be to perform cell culture assays with all the non-REF elements in both individuals’ genomes to measure the combined mutagenic output of each collection (25 vs. 63). These profiles could be further integrated with REF FL-L1Hs element activity (Brouha et al. 2003). Another goal moving forward will be to develop better approaches to predict which elements are highly active versus less active. This might be achieved by testing a large number of elements in cell culture assays and then developing a model that can be used to predict the activity of each new element or profiles of elements. Such experiments could be performed with our collection of FL-L1Hs elements combined with the elements that already have been tested (Brouha et al. 2003; Beck et al. 2010).
We discovered three novel L1 subfamilies that have evolved from the highly active L1Ta1d subfamily—namely, Ta1d-CAT, Ta1d-CCA, and Ta1d-TCA (Figs. 3, 4). We named these subfamilies based on the three positions in L1 that define these elements (1026, 3337, and 3440) and the sequences that are found at those three positions (CAT, CCA, TCA). All three of these positions are located within L1 ORFs (1026 is in ORF1, whereas 3337 and 3440 are in ORF2). However, only one of the changes (at position 3440) creates a nonsynonymous amino acid substitution (lysine to methionine) just upstream of the RT domain of ORF2. The other two (synonymous) changes also could help to boost L1 activity, for example, by eliminating host factor binding sites that mediate L1 suppression. Additional mechanistic studies will be necessary to determine how sequence changes at these positions impact FL-L1Hs activity.
We examined previous studies to determine whether any of the well characterized FL-L1Hs elements that are highly active or have been implicated in human diseases belong to our three novel subfamilies. We found that three of the most active REF elements in Brouha et al. (2003) belong to the Ta1d-TCA and Ta1d-CCA subfamilies (including LRE3, which is a Ta1d-TCA element) (Supplemental Table S3D). We also determined that the following elements all belong to our three novel subfamilies: (1) 39 non-REF elements from Beck et al. (2010), (2) the L1.3 element from Dombroski et al. (1993), and (3) the three FL-L1Hs elements that gave rise to somatic L1 insertions in a case of colorectal cancer (Scott et al. 2016; Supplemental Table S3D). In contrast, the well-studied L1RP element (Schwahn et al. 1998) does not belong to these subfamilies (Supplemental Table S3D). These data suggest that many (but not all) of the highly active FL-L1Hs source elements that have been studied previously belong to our three novel subfamilies.
Several studies indicate that FL-L1Hs loci can have multiple alleles at the same site with internal sequence changes that influence L1 mobilization. For example, Lutz et al. (2003) discovered two alleles of the L1.2 locus (L1.2A and L1.2B) with internal sequence differences that caused a 16-fold change in mobilization in cell culture–based assays. Likewise, Seleme et al. (2006) discovered new alleles of three hot L1 source elements that also impacted L1 mobilization in cell culture assays. Finally, Sanchez-Luque et al. (2019) reported allelic variants of a Chromosome 13 FL-L1Hs source element where two of the three alleles were inactive in cell culture assays because of an internal stop codon and a missense mutation. Thus, additional alleles of our 698 sequenced loci likely exist in human populations, and at least some of these may harbor internal sequence changes that impact retrotransposition activity.
Another factor that can influence FL-L1Hs activity is the genomic context in which the element is located. Lavie et al. (2004) found that the genomic sequences upstream of individual FL-L1Hs loci can influence L1 promoter activity and can enhance or repress FL-L1Hs expression. Likewise, Philippe et al. (2016) found that a relatively small subset of FL-L1Hs loci accounted for the bulk of L1 expression in 12 commonly used cell lines. Moreover, they found that these expressed loci were differentially regulated across the 12 cell lines. An integrated approach was used to study these loci that included RNA-seq analysis of 3′ bypass transcription and 5′ antisense promoter transcripts, along with analysis of active H3K4me3 and H3K27ac chromatin marks (Philippe et al. 2016). Collectively, these data indicate that locus-specific upstream genomic sequences and cell-specific factors contribute to FL-L1Hs regulation.
Our collection of FL-L1Hs elements will be useful for expanding these studies to more broadly examine the impact of diverse genomic contexts on FL-L1Hs activity. As outlined above, the flanking genomic sequences could be examined along with L1 promoter methylation and other chromatin features to better understand how FL-L1Hs element regulation varies across diverse genomic loci (Lavie et al. 2004; Philippe et al. 2016; Sanchez-Luque et al. 2019; Ewing et al. 2020). The expression of each FL-L1Hs locus could be studied using one of several possible approaches (Philippe et al. 2016; for review, see Lanciano and Cristofari 2020). For example, we previously showed that the internal mutation profiles of FL-L1Hs elements can be leveraged to measure the expression of specific FL-L1Hs loci using RNA-seq (Scott et al. 2016). The unique internal mutations that we have identified in our sequenced FL-L1Hs elements now could be leveraged for this purpose (Fig. 4C,D; Supplemental Table S4; Supplemental Fig. S3). Likewise, 76/698 (10.9%) of our sequence-resolved FL-L1Hs elements have produced 3′ transductions (Supplemental Fig. S4; Supplemental Table S3A), which could be leveraged to study the expression, regulation, and mobilization of these elements (Philippe et al. 2016; for review, see Lanciano and Cristofari 2020).
With a comprehensive approach using several of these tools, somatic cancers and neuronal tissues could be examined to determine how FL-L1Hs elements evade somatic repression in these tissues and which elements can achieve this status. Other normal epithelial and neuronal tissues in which FL-L1Hs elements can evade somatic repression also could be examined, including the esophagus, pancreas, colon (Doucet-O'Hare et al. 2015; Ewing et al. 2015; Scott et al. 2016; Yamaguchi et al. 2020) as well as the liver, brain, embryonic stem cells, and cells undergoing neuronal differentiation (Sanchez-Luque et al. 2019). Aberrantly high levels of L1 expression and retrotransposition often are associated with neuronal diseases; in many cases, the underlying mechanisms leading to L1 up-regulation are unknown (Terry and Devine 2020). The regulation and dysregulation of FL-L1Hs elements in these contexts remains an important but relatively unexplored area of research for understanding how collections of FL-L1Hs work together to mutagenize genomes and cause human diseases.
Natural mutagenesis of human genomes by endogenous mobile elements on a population scale
We also examined the population distributions and subfamilies of the MEIs that we discovered in this study. Our analysis indicates that there is substantial variation in the MEI counts per individual in these populations, largely caused by variation in the number of Alu MEIs (Fig. 5A; Supplemental Fig. S6). Likewise, we identified MEIs in each population that are unique to a single population (Fig. 5C; Supplemental Table S5; Supplemental Fig. S6). These population-specific variants might be useful to determine the genetic ancestries of individuals or might help to account for population-specific phenotypes or susceptibility to diseases. We also examined the subfamily status of the Alu, L1, and SVA insertions in our MEI collections, and found that a few dominant lineages of Alu, L1, and SVA elements are responsible for the majority of ongoing MEI mutagenesis in human populations (Supplemental Figs. S7, S8; Supplemental Discussion).
Our collection of 104,350 MEIs combined with the remaining MEI collections in Figure 1B (a total of 158,873 nonredundant MEIs) allowed us to examine MEI mutagenesis on an unprecedented scale in humans. We found 781 MEIs that disrupted the coding sequences (CDS) of GENCODE-annotated genes, and we expect that most of these would disrupt gene function. We also identified MEIs in other annotated features of GENCODE genes, and a subset of these likely affect gene function as well (Fig. 6A). Finally, we identified 11,586 MEIs that disrupt ENCODE cCREs and thus might impact the expression of genes that are regulated by these cCREs (Fig. 6B). Most of these MEIs belong to young subfamilies, are rare, and likely were integrated into human genomes relatively recently. Many of these MEIs undoubtedly were mobilized by FL-L1Hs source elements that belong to the three novel Ta1d subfamilies that we discovered in this study. Such source elements are young and active (Figs. 3D, 4A) and would be responsible for mobilizing not only new L1 insertions but new Alu and SVA insertions as well.
Analysis of the OMIM database revealed 16,606 MEIs that are located within 2335 genes that have been implicated in a wide range of human diseases, including cancers, degenerative diseases, intellectual disability, deafness, heart disease, and others (Fig. 6C). Overall, these data indicate that MEI mutagenesis impacts a wide range of genes and gene features that are relevant to a broad spectrum of human diseases. Because the majority of MEIs that we discovered in this study are rare (81,645/104,350 or 78.2%), MEIs that disrupt CDSs or other functionally important genomic features likely would fit a rare variant model of human disease.
Methods
CloudMELT pipeline
The Individual Analysis step of MELTv2.1.5 (Gardner et al. 2017) was optimized to provide a 19.8× speedup in that step (for additional details, see Supplemental Methods). The resulting MELT engine is termed MELTv2.1.5.fast. CloudMELT version 1.0.1 is a port of MELTv2.1.5.fast to the Amazon Web Services (AWS) cloud computing environment. The pipeline is written with Toil version 3.17 to create a reproducible data analysis workflow (Vivian et al. 2017). Docker was used to create an image of the optimized MELTv2.1.5.fast engine with its required dependencies, and the image was loaded to an AWS container repository (Merkel 2014). CloudMELT requires users to configure initial parameters for their run: define the reference genome version, provide the uniform resource identifiers (URIs) of the genomes for their data set, set MELT flags/arguments, and optionally designate minimum storage and memory requirements. Users then launch a computation cluster on AWS with a “leader” node and a desired number of “worker” nodes (Supplemental Fig. S1). The CloudMELT workflow is created and uploaded to the leader node. The MELT Docker image and the necessary AWS permissions are distributed to the worker nodes.
CloudMELT is then launched as follows. Each worker node has a user-specified number of computational units, or virtual central processing units (vCPU), that can handle each MELT process. For phase 1 of the CloudMELT pipeline, the genome alignment files (i.e., BAM or CRAM) are downloaded to the respective worker nodes for the MELT Preprocess and Individual Analysis steps to discover the user-defined MEIs (Alu, L1, SVA, and HERV-K). Please refer to the original MELT paper for a detailed description of each MELT runtime step (Gardner et al. 2017). Phase 2 is the MELT Group Analysis step in which the phase 1 output is processed on a single worker node. The Group Analysis step was modified to provide deterministic results when running CloudMELT. Phase 3 is the MELT Genotyping step in which the phase 2 output is used by each worker node to determine the genotype of the candidate MEIs. Finally, in phase 4 the Make VCF step will aggregate the phase 3 output into a VCF file (Supplemental Fig. S1).
Previously, we performed whole-genome MEI discovery simulations and extensive PCR validation studies (on 400 independent MEI sites) and achieved an overall false discovery rate (FDR) below 5% with low coverage Illumina WGS data (Sudmant et al. 2015; Gardner et al. 2017). Our analysis of high-coverage Illumina WGS data in this study indicated that the higher coverages yielded more accurate calls and lower FDRs. Therefore, we readjusted our filtering process to achieve an overall FDR of <5%. An initial 1KGP call set of PASS MEIs (39,830) was generated with the original (more stringent) criteria, and an extended call set was generated that includes 14,707 additional high quality MEIs (for a total of 54,537 calls) (Fig. 1A). This adjusted filtering was used for all other data sets. As we developed CloudMELT, we compared the results obtained with MELTv2.1.5 versus the improved MELTv2.1.5.fast and CloudMELT to ensure comparable results. We also examined 50 to 100 MEIs of each MEI type in the Integrative Genomics Viewer (IGV) (Thorvaldsdottir et al. 2013) to ensure that there was strong evidence to support the calls and that our overall FDRs were <5%.
Data sets used in this study
In each case, “reference human genome or REF” refers to the build of the human genome that was used for each cited study or data set (e.g., hg19 or GRCh38). “Non-reference or non-REF” refers to a newly sequenced genome, fosmid, or BAC clone that was aligned to the matching REF build to discover non-REF MEIs. 1KGP, GTEx, and UKBB BAM files aligned to build GRCh38 of the reference human genome were used for initial MEI discovery in these populations. TOPMed BAM files aligned to build hg19 of the reference human genome were used for initial MEI discovery in these populations and the MEI coordinates were converted to build GRCh38 coordinates. The FL-L1Hs elements that were sequenced were obtained from build hg19 coordinates and converted into build GRCh38 coordinates (Supplemental Table S3A). All downstream analysis was performed with the GRCh38 reference genome sequence.
The high-coverage Illumina Amish, Jackson Heart Study, and GTEx WGS data were obtained from the NCBI database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) under Institutional Review Board approvals (accession numbers phs000956.v4.p1, phs000286.v6.p2, and phs000424.v8.p2, respectively). The high-coverage 1KGP WGS data were obtained from AWS (s3://1000genomes/1000G_2504_high_coverage/) (Ebert et al. 2021). The UKBB WES data were obtained from the UK Biobank resource (https://www.ukbiobank.ac.uk/). The data outlined in Figure 1B were obtained from Sudmant et al. (2015), Cao et al. (2020), and Collins et al. (2020). GENCODE and ENCODE v35 data were obtained from the University of California Santa Cruz Genome Browser (https://genome.ucsc.edu/). OMIM and GAD data were downloaded from https://david.ncifcrf.gov/summary.jsp, COSMIC data were downloaded from http://cancer.sanger.ac.uk/cosmic, and CCGD data were downloaded from http://ccgd-starrlab.oit.umn.edu.
Long-read amplification and sequencing
We sequenced the full interior regions of 698 FL-L1Hs elements as follows. Genomic DNA samples were purchased from Coriell (Coriell Institute for Medical Research). PCR primers flanking the FL-L1Hs elements were designed with Primer3 within the 500 bp regions flanking each insertion (Koressaar and Remm 2007; Untergasser et al. 2012) and purchased from Integrated DNA Technologies (IDT). The target size of the primer was 22–26 bases with a melting temperature of 63°C–68°C. If no primers initially were found, the amount of flanking DNA was increased. In some cases, primers were manually designed to target higher melting temperatures (68°C–74°C). PCR amplification was performed using LA Taq DNA polymerase (TaKaRa Bio USA, Inc.) and a PCR protocol for long-range amplification: 90 sec at 94°C, followed by 32 cycles of (1) 30 sec at 94°C, (2) 30 sec at 57°C, (3) 8 min 30 sec at 68°C, with a final elongation step for 10 min at 68°C. This protocol has a relatively low error rate (Supplemental Methods). Although amplification was attempted on 789 FL-L1Hs sites, 49 sites could not be amplified even with repeat attempts. Each filled site was obtained from a single individual. We required FL-L1Hs candidates for sequencing to be at least 6000 bp in length as predicted by MELT. Several dozen amplicons were pooled in approximate equimolar concentrations for Pacific Biosciences sequencing with each SMRT cell. Amplicon-pooled samples were size selected to filter small fragments. A sequencing library then was prepared and sequenced with the Pacific Biosciences RSII platform. The raw sequencing data were aligned with BLASR 1.3.1 and assembled with ConsenseTools as part of the SMRT Analysis v2.3.0 software suite (Pacific Biosciences) (Chaisson and Tesler 2012). We typically recovered 500 or more traces that were used in the assemblies. Pacific Biosciences RSII sequencing has an estimated indel error rate of 15% (Rhoads and Au 2015). Surveying the collection of FL-L1Hs sequences, we discovered that there was a high rate of single-nucleotide deletions in specific homopolymer tracts (HPT). We used ABI capillary sequencing to perform resequencing of a sampling of FL-L1Hs elements and verified the reoccurring HPT deletion errors. We then used this algorithm to correct HPT errors systematically at reoccurring sites.
Sequenced FL-L1Hs elements were aligned to the preTa consensus sequence (Supplemental Table S3C), and all coordinates are listed relative to this consensus sequence. L1 subfamilies were annotated using previously reported diagnostic positions (Boissinot et al. 2000; Brouha et al. 2003) along with our newly defined diagnostic positions (Fig. 3A). Ambiguous (Ambig) elements do not perfectly match the diagnostic positions of any known L1 subfamily.
Data access
The FL-L1Hs PacBio sequence data generated in this study have been submitted to the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers MZ092004-MZ092701. The 1000 Genomes Project MEI call sets generated in this study have been submitted to NCBI's database of human genomic structural variation (dbVar; https://www.ncbi.nlm.nih.gov/dbvar) under accession number nstd211. The TOPMed Amish, Jackson Heart Study, and GTEx MEI call sets generated in this study have been submitted to the NCBI dbGaP database (https://www.ncbi.nlm.nih.gov/gap/) under accession number phs002463.v1.p1. The UKBB MEI call sets generated in this study have been submitted to the UKBB database (https://www.ukbiobank.ac.uk/) under UKBB application ID 49852. MELTv2.1.5fast and CloudMELT can be found in Supplemental Code S1 and are available at GitHub (https://github.com/Scott-Devine/CloudMELT/releases/tag/v1.0.1). Our FL-L1Hs annotation script can be found in Supplemental Code S2.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Luke J. Tallon, Lisa Sadzewicz, and the Genomics Resource Center at the Institute for Genome Science for PacBio sequencing. We also thank John Moran for helpful discussions. This work was supported by the following National Institutes of Health (NIH) grants: R01 HG002898 (S.E.D.), T32 DK067872 (N.T.C.), T32 CA154274 (G.L.R.), and F31 HG009223 (E.J.G.). This research was conducted using the UK Biobank resource under application number 49852.
Author contributions: J.C., N.T.C., A.A.M., and S.E.D. developed and tested MELTv2.1.5fast and CloudMELT; N.T.C., E.J.G., and S.E.D. designed and performed FL-L1Hs PacBio sequencing experiments; N.T.C., D.M.T., G.L.R., and S.E.D. performed insertional mutagenesis analyses; D.M.T. prepared display items; J.A.P. and C.C.H. provided data and performed analysis; N.T.C. and S.E.D. performed population and subfamily analysis, designed experiments, generated data, prepared display items, and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275323.121.
- Received February 2, 2021.
- Accepted September 16, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








