
Profiling neural editomes reveals a molecular mechanism to regulate RNA editing during development
Abstract
Adenosine (A) to inosine (I) RNA editing contributes to transcript diversity and modulates gene expression in a dynamic, cell type–specific manner. During mammalian brain development, editing of specific adenosines increases, whereas the expression of A-to-I editing enzymes remains unchanged, suggesting molecular mechanisms that mediate spatiotemporal regulation of RNA editing exist. Herein, by using a combination of biochemical and genomic approaches, we uncover a molecular mechanism that regulates RNA editing in a neural- and development-specific manner. Comparing editomes during development led to the identification of neural transcripts that were edited only in one life stage. The stage-specific editing is largely regulated by differential gene expression during neural development. Proper expression of nearly one-third of the neurodevelopmentally regulated genes is dependent on adr-2, the sole A-to-I editing enzyme in C. elegans. However, we also identified a subset of neural transcripts that are edited and expressed throughout development. Despite a neural-specific down-regulation of adr-2 during development, the majority of these sites show increased editing in adult neural cells. Biochemical data suggest that ADR-1, a deaminase-deficient member of the adenosine deaminase acting on RNA (ADAR) family, is competing with ADR-2 for binding to specific transcripts early in development. Our data suggest a model in which during neural development, ADR-2 levels overcome ADR-1 repression, resulting in increased ADR-2 binding and editing of specific transcripts. Together, our findings reveal tissue- and development-specific regulation of RNA editing and identify a molecular mechanism that regulates ADAR substrate recognition and editing efficiency.
RNA modification is a molecular process that regulates gene expression by shaping the transcriptional profiles of cells in a multicellular organism. Individual nucleosides in RNA can be deleted or inserted, can undergo base modifications (which together are historically referred to as “RNA editing” events), or can be chemically modified. More than 150 different types of nucleoside changes have been identified on cellular RNAs to date (Ontiveros et al. 2019). The most prevalent RNA editing event on mRNA is the hydrolytic deamination of adenosine (A) to generate inosine (I). This reaction is catalyzed by the adenosine deaminase that act on RNA (ADAR) family, which specifically target double-stranded (ds) regions of RNA (Nishikura 2010; Walkley and Li 2017). The base-pairing properties of inosine vary from adenosine, and inosine is mostly recognized as guanosine by cellular machinery (Bass and Weintraub 1988; Licht et al. 2019a). A-to-I editing affects gene expression through protein recoding, RNA export, splicing, stability, regulation of the immune system, and heterochromatin formation (Deffit and Hundley 2016; Shevchenko and Morris 2018). Neither is every adenosine edited within a ds region, nor are all the mRNA copies of a single gene edited within every cell during development. The extent of editing varies from 0% to 100% at a single adenosine within a given mRNA. Differential editing can lead to changes in gene regulation. To understand how the gene expression landscape changes during differentiation and growth of an organism, it is critical to understand how the specificity and efficiency of A-to-I RNA editing are regulated.
A-to-I editing is essential for proper neuronal development in mammals (Li and Church 2013; Behm and Öhman 2016). Altered RNA editing levels in central nervous system (CNS) transcripts occur in many neuropathological disorders, including brain tumors and psychiatric and neurodegenerative diseases (Slotkin and Nishikura 2013; Gallo et al. 2017). Editing at a well-studied site in one CNS transcript, glutamate receptor subunit B (GRIA2), causes a glutamine (Q)-to-arginine (R) change in the protein, which reduces calcium influx into cells. Proper Q/R site editing is essential (Higuchi et al. 2000). Furthermore, the level of Q/R site editing is regulated in a tissue- and development-specific manner during development (Nutt and Kamboj 1994; Kawahara et al. 2003; Wahlstedt et al. 2009). Recent genome-wide sequencing studies revealed a similar spatiotemporal regulation of RNA editing across different neural types during mammalian brain development (Ekdahl et al. 2012; Hwang et al. 2016; Zaidan et al. 2018; Lundin et al. 2020). These studies also revealed that spatiotemporal editing was perturbed in neurological conditions, including glioblastoma and spinal cord injury (Hwang et al. 2016).
Similar to mammals, editing is important for proper neuronal development and function in the model organism, Drosophila melanogaster. Flies lacking ADAR are morphologically wild-type but show temperature-sensitive paralysis and uncoordinated locomotion (Palladino et al. 2000). Profiling RNA from cell populations in the adult fly brain revealed the presence of precise, spatial regulation of RNA editing within the developed brain (Sapiro et al. 2019). This study also identified cell type–specific editing events, suggesting RNA editing could contribute to differential functions of neural populations. Together, genome-wide studies have reported tissue- and development-specific editing in mammalian and fly brains; however, the mechanisms that regulate spatiotemporal RNA editing remain unknown.
The recent establishment of cell sorting techniques from different stages of Caenorhabditis elegans has opened a new era of profiling transcriptomes of a number of cell types and tissues during development (Spencer et al. 2014; Washburn and Hundley 2016b; Deffit et al. 2017; Kaletsky et al. 2018). In addition, C. elegans is an excellent model system to study the molecular and cellular mechanisms that regulate RNA editing in vivo, as animals lacking A-to-I editing are viable but show neurological phenotypes, including chemotaxis defects and altered lifespan (Tonkin et al. 2002; Sebastiani et al. 2009; Ganem et al. 2019). The C. elegans genome encodes two proteins with homology with the ADAR family, an editing-deficient family member, ADR-1, and the A-to-I editing enzyme, ADR-2. Previous C. elegans studies identified a few global mechanisms of regulating RNA editing. For example, C. elegans ADBP-1 positively regulates editing by promoting ADR-2 localization to the nucleus (Ohta et al. 2008), and ADR-1 promotes editing by physically interacting with and directing ADR-2 to specific substrates (Rajendren et al. 2018). In addition, recent studies suggest the presence of tissue-specific editing regulation in C. elegans. For example, ADR-2-mediated editing of a reporter transcript is inhibited by ADR-1 in neural cells (Washburn and Hundley 2016b). Further, ADR-2 regulates expression of clec-41, a gene involved in chemotaxis, in a neural-specific manner (Deffit et al. 2017). However, little is known about the mechanisms that regulate tissue-specific RNA editing. Unlike in mammals and flies, developmental editing levels of nematode neural transcripts have not been previously studied.
In this study, a transcriptome-wide approach is taken to identify changes in neural editing during development. Together with biochemical assays, these studies aim to elucidate a molecular mechanism that regulates neurodevelopmental editing.
Results
The C. elegans adult neural editome consists of nearly 600 A-to-I editing sites
To examine the C. elegans adult neural editome, synchronized larval-stage 1 (L1) worms were grown to adulthood for 50 h at 20°C before neural cell isolation. Neural cells were isolated from wild-type and adr-2(-) worms expressing green fluorescent protein (GFP) under the control of a pan-neuronal promoter, rab-3, as previously described (Spencer et al. 2014; Deffit et al. 2017; Kaletsky et al. 2018). Briefly, adult worms were subjected to chemomechanical disruption, and by using fluorescent-activated cell sorting (FACS), GFP-positive neural cells were selected from the live, single-cell population (Supplemental Fig. S1A,B). The percentage of adult neural cells isolated was similar to previously published studies using this technique (Kaletsky et al. 2018). Successful isolation of neural cells from nonneural cells was confirmed using real-time quantitative PCR (qPCR) to measure expression of both the neural-specific gene unc-64 (Saifee et al. 1998) and the muscle-specific gene myo-3 (Supplemental Fig. S1C; Ardizzi and Epstein 1987).
High-throughput sequencing of poly(A) selected RNA from the isolated adult neural cells was performed, and reads were uniquely mapped to the C. elegans reference genome using the Spliced Transcripts Alignment to a Reference (STAR) alignment program (Dobin et al. 2013). Pairwise comparisons of the replicates indicated high reproducibility (Supplemental Fig. S2A). We compared our adult neural transcriptome to the previously published adult C. elegans neural transcriptome, which was performed with a similar technique, except the neural GFP expression was driven by a different pan-neuronal promoter (Kaletsky et al. 2018). Comparison of the transcriptomes using DESeq2 revealed that a majority of genes showed no significant difference in expression (P-adj < 0.05) (Supplemental Fig. S2B; Supplemental Table S1).
A-to-I RNA editing sites were identified de novo from the adult neural RNA-seq data set using the Software for Accurately Identifying Locations Of RNA editing (SAILOR) program (Deffit et al. 2017). Each predicted site is reported with a confidence score that evaluates a number of parameters, including read coverage and percentage of editing at each site. Sites with a confidence score of ≥0.99 were used for downstream analysis. By using these stringent cut-offs, 574 candidate sites were predicted from the wild-type adult neural RNA-seq data set, and 17 sites were predicted for the adr-2(-) data set. As adr-2(-) worms lack A-to-I editing (Tonkin et al. 2002), the sites identified by SAILOR for the adr-2(-) worms are false positives. Subtraction of these false positives led to identification of 570 A-to-I editing sites in the wild-type adult neural editome, which were assigned to 66 different neural transcripts (Supplemental Table S2).
To determine whether this developmental- and tissue-specific approach identified novel editing sites, the adult neural editome was compared with a list of 203,934 editing sites (9444 transcripts) identified from previous C. elegans studies (Washburn et al. 2014; Whipple et al. 2015; Zhao et al. 2015; Deffit et al. 2017; Goldstein et al. 2017; Reich et al. 2018; Ganem et al. 2019). Except one (Deffit et al. 2017), the previous studies used whole-worm lysates to identify editing sites. Many of these studies performed control experiments, in which sites were also computationally predicted for adr-2(-) worms (that lack all A-to-I editing) and any false-positive sites were removed. This list also includes edited sites from all developmental stages. Nearly, 23% of the adult neural editing sites (130 out of 570) had not been previously identified (Fig. 1A), which suggests that these adenosines may be edited only or more highly in adult neurons. Nearly 80% of the novel sites (104 out of 130) mapped to different regions of 53 previously identified edited transcripts, whereas 10 neural transcripts were identified as novel edited mRNAs (Fig. 1A). The genic distribution of A-to-I editing sites indicates that the vast majority (>80%) of adult neural editing sites are located in the 3′ untranslated regions (UTRs) of transcripts (Fig. 1B). In addition, as the UTRs of many C. elegans genes are not well annotated and our previous studies showed that the intergenic regions primarily reside within 2000 bp upstream of or downstream from the annotated genes (Deffit et al. 2017), it is highly possible that some of the editing sites identified in intergenic regions also reside in 3′ UTRs.
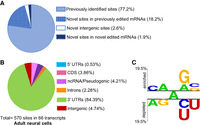
De novo identification of editing sites in adult neurons. (A) The adult neural editing sites identified were cross-referenced with previously reported sites to identify novel editing sites. (B) Genomic distribution of adult neural sites. (CDS) Coding region; (UTR) untranslated region; (ncRNA) noncoding RNA. (C) A Two Sample Logo analysis of editing sites identified in adult neural cells compared with adenosines present in the same dsRNA region. Enriched and depleted nucleotides are shown above and below the axis, respectively. The level of conservation is represented by letter height. Logos were generated using a Student's t-test with P < 0.05 and no Bonferroni correction.
To determine the nearest neighbors of the 570 editing sites identified in adult neural cells, the percentage of occurrence for the 5′ and 3′ neighboring nucleotides were weighted. As the list of 570 sites contains editing sites mapped to different genomic locations, a background list was used to match the genomic distribution of these identified 570 edited sites. To determine whether or not the appearance of particular nucleotides in the nearest-neighbor positions was statistically significant (P < 0.05), a Two Sample Logo analysis (Vacic et al. 2006) was used to compare the edited adenosines to the sequence context of all adenosines in the ds region (Fig. 1C). Our data suggest that in adult neural cells, C/A and G/A are overrepresented whereas A/G and U/C are underrepresented at the 5′ and 3′ sides of editing sites, respectively. The identified 5′ neighboring nucleotides are consistent with previously published mammalian ADAR preferences (Polson and Bass 1994; Lehmann and Bass 2000). However, A is less preferred at the 3′ side by mammalian ADARs. In sum, our transcriptome-wide approach to identify tissue- and developmental-specific editing sites yielded 570 editing sites in adult neural cells, including 130 novel sites mapped to 63 individual transcripts.
The neural editome changes during development
Previous data from our laboratory reported 7361 editing sites in neural cells isolated from L1 worms (Deffit et al. 2017). This number is more than 10-fold higher than the number of adult neural editing sites identified herein. In addition, in contrast to the adult neural editome, nearly half of the previously identified L1 neural editing sites were annotated to intronic regions. It is possible that technical differences in sequencing and/or bioinformatic analysis could contribute to these differences. To rule out differences owing to variations in read length and bioinformatics analysis, the length of the L1 neural RNA-seq reads was shortened to 75 bases (see methods) and then reprocessed through the bioinformatics pipeline with the same parameters used for the adult neural RNA-seq reads. This reanalysis predicted 6213 candidate sites in wild-type and 28 false-positive sites in adr-2(-) L1 neural cells. After subtracting the false positives, 6202 A-to-I editing sites were identified in the L1 neural editome (Supplemental Table S3). Despite the slight reduction in number of L1 edited sites, these data still suggest a 10-fold higher number of editing sites in the C. elegans larval nervous system compared with the adult nervous system.
To test whether the quantity of editing sites between the life stages could be attributed to read coverage differences, the number of uniquely aligned reads were counted using SAMtools at various points in the computational analysis. Although the adult neural RNA-seq data set had twice as many aligned reads, the SAILOR analysis for the adult data set had 3.3 times fewer sequencing reads compared with the L1 data set (Fig. 2A). Before identifying editing sites, SAILOR discards duplicate reads (those that start and end with the same end nucleotide) and reads that contain more than one non A-to-G variant. Thus, the reads used for SAILOR identification had a depth of five reads covering 81.3% and 57.9% of the reference genome in the L1 and adult neural RNA-seq data sets, respectively. To simply test whether the difference in editing site number is caused by the differences in read coverage, the same number of reads (100,000 after removal of duplicated reads) was randomly chosen from both data sets and input into SAILOR. This approach identified 721 and 187 edited sites with a confidence score of ≥0.99 in the L1 and adult neural RNA-seq data sets, respectively. The fourfold larger number of L1 editing sites is consistent with our conclusion that the L1 neural editome is larger than the adult neural editome.
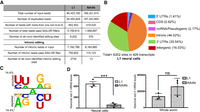
C. elegans neural editome change during development. (A) Comparison of the RNA-seq data sets from L1 and adult neural cells. The number of input reads for each step of the SAILOR bioinformatic pipeline is listed along with the number of de novo identified edited sites. (B) Genomic distribution of the identified L1 neural editing sites was determined using the WormBase annotations (WS275). (C) A Two Sample Logo analysis of editing sites identified in L1 neural cells compared with adenosines present in the same dsRNA region. Enriched and depleted nucleotides are shown above and below the axis, respectively. The level of conservation is represented by letter height. Logos were generated using a Student's t-test with P < 0.05 and no Bonferroni correction. (D) qPCR was used to quantify adr-2 expression in neural cells (left) and in whole-worm lysate (right) for both L1 and adults. The average of three independent replicates is plotted; error bars, SEM. Statistical significance was calculated using a Student's t-test. (***) P < 0.001; (ns) P > 0.05.
In support of developmental differences in the neural editomes and consistent with the initial publication of the L1 analysis, the largest number of L1 neural editing sites (44.0%) localized to introns (Fig. 2B). It is possible that a higher number of intronic reads in the L1 data set could lead to a higher number of intronic editing sites identified. To address this possibility, the number of intronic reads in both RNA-seq data sets was counted using featureCounts. Both RNA-seq data sets contained a similar number of intronic reads (Fig. 2A). SAILOR analyzed 6.5-fold fewer intronic reads for the adult data set but identified 210-fold less intronic sites compared with the L1 data set. In sum, the C. elegans neurodevelopmental editomes differ in both editing site number and distribution, with a significantly higher number and intronic distribution of editing sites in L1 neural cells and a majority of 3′ UTR editing sites in adult neural cells. The nearest-neighbor preferences of the L1 editing sites suggest that U/A and G/A are overrepresented, whereas A/G and U/C are under-represented at the 5′ and 3′ sides, respectively (Fig. 2C). Compared with the preferences of the adult neural edited sites (Fig. 1C), L1 neural edited sites show similar neighbor preferences at the 3′ side, but at the 5′ side U is overrepresented specifically in L1 neural editing sites.
One possibility for the fewer editing sites identified in the adult nervous system could be that adr-2 expression decreases during development. Previous whole-worm analysis suggested similar, and perhaps slightly higher, adr-2 mRNA expression in adult worms compared with L1 worms (Hundley et al. 2008). To determine whether adr-2 expression is altered during neural development, adr-2 mRNA levels were quantified in L1 and adult neural cells. Absolute mRNA levels for adr-2 in neural cells and whole-worm lysates were measured for three independent biological replicates using qPCR. Our data indicate that neural adr-2 mRNA expression decreases during development, whereas global adr-2 mRNA expression remains unchanged (Fig. 2D). Together, these data suggest that the tissue-specific decrease in adr-2 mRNA expression in adult neural cells contributes to the smaller adult neural editome.
Differential expression of edited transcripts contributes to stage-specific neural editomes
The neural-specific decrease in adr-2 mRNA expression likely contributes to the decreased number of editing sites identified in adult neural cells. However, the distribution of editing sites identified within specific regions of transcripts also changes during development. To gain insight into whether editing has altered developmental contributions to neural transcriptome diversity and/or whether altered gene expression is affecting developmental editing site identification, variant detection at the identified sites was performed for both neural RNA-seq data sets using SAMtools (see Methods) (Li et al. 2009). First, a composite list of editing sites in the C. elegans nervous system was generated by merging the L1 and adult editing sites, which yielded a total of 6352 editing sites mapped to 453 unique transcripts. Transcripts were classified as edited if at least one variant (G) read was detected (Fig. 3A; Supplemental Table S4). Of the 6352 editing sites, 4642 and 68 sites were identified as edited only in the L1 or adult neural RNA-seq data sets, respectively (Fig. 3B). Among the 4642 edited sites in L1 neural cells, 1740 sites mapped to transcripts that were edited within a different region in adult neural cells and, thus, were excluded from the list of L1-specific edited transcripts (Fig. 3A,C). Likewise, among the 68 sites edited only in adult neural cells, more than half mapped to transcripts that were edited within a different region in the L1 neural RNA-seq data set (Fig. 3A). Thus, there were only a handful of neural sites (1.1%) edited specifically in adults (Fig. 3B). In contrast, the percentage of neural transcripts edited specifically in early development (73.1%) was significantly higher, and more than three-quarters of these transcripts are edited within introns (Fig. 3C).
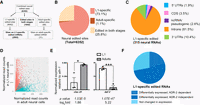
Differential expression of many transcripts during neural development contributes to the stage-specific neural editomes. (A) Workflow for identifying stage-specific edited sites following the quantification of all the neural editing sites. (B) Distribution of stage-specific edited sites. (C) Genomic distribution of L1-specific edited transcripts based on the genomic location of identified edited sites. (CDS) Coding region; (UTR) untranslated region; (ncRNA) noncoding RNA. (D) Dots represent individual genes that are down-regulated (red; 4476, log2fold < −0.5), up-regulated (blue; 5171, log2fold > 0.5), or not significantly different (gray; 7697, P-adj > 0.05) between three biological replicates of wild-type neural L1 and adult RNA-seq data. (E) The average of three independent replicates of qPCR validation of neurodevelopmentally, differentially expressed genes is plotted; error bars, SEM. Statistical significance was calculated by Student's t-test. (*)P < 0.05; (***) P < 0.001. (F) Representation of the L1-specific edited transcripts based on differential expression during neural development. Dark blue represents transcripts differentially expressed in neural development and ADR-2 dependent (dotted) or independent (solid). Light blue represents transcripts that are not differentially expressed in neural development.
The above data suggest that editing impacts transcriptome diversity in the nervous system in a stage-specific manner. However, whether editing of these neural transcripts also leads to differential expression in the two life stages is unknown. To identify genes with altered expression during neural development and to identify the role of editing in any observed expression differences, differential gene expression analysis was performed on three biological replicates of wild-type neural RNA-seq data from both stages using DESeq2 (see Methods). Principle component analysis (PCA) indicates the three biological replicates of each sample cluster best, and the clusters of the two developmental stages were more distinct from one another than any individual samples (Supplemental Fig. S3A). The expression of 9620 genes were significantly altered (P-adj < 0.05) between L1 and adult neural cells isolated from wild-type animals (Fig. 3D; Supplemental Table S5). Compared with the neural L1 expression, slightly more than half of these genes (5171) significantly increased, whereas the other 4476 significantly decreased expression in adult neural cells (Fig. 3D). Independent qPCR validation of one up-regulated gene, clec-41, and one down-regulated gene, daf-2, confirmed the trends observed in the RNA-seq data (Fig. 3E). Gene Ontology (GO) analysis of the genes up-regulated (Supplemental Fig. S3B) and down-regulated (Supplemental Fig. S3B) in adult neural cells was distinct.
To determine whether the differential neural expression during development contributes to the L1-specific neural editome, the stage-specific edited transcripts were overlapped with the differentially expressed neural transcripts. More than two-thirds of the L1-specific edited transcripts (203 of 315) are down-regulated in adult neural cells (Fig. 3F). These data are consistent with previous studies (for review, see Pinto and Levanon 2019) and suggest that the decreased gene expression in adult neural cells affects the overall amount of editing observed.
Previous studies showed that ADR-2 regulates neural-specific gene expression in L1 worms (Deffit et al. 2017). To determine the extent to which ADR-2 contributes to neurodevelopmental gene expression, DESeq2 was performed on L1 and adult neural RNA-seq data sets from adr-2(-) animals (Supplemental Table S6; Supplemental Fig. S3C). The expression of 7088 genes was significantly altered (P-adj < 0.05) between L1 and adult neural cells from adr-2(-) worms (Supplemental Fig. S3C). Compared with L1 expression, nearly two-thirds of genes (4374) increased, whereas the other 2714 decreased, in expression in adult neural cells lacking adr-2(-). Compared with wild-type neural development, there is a large difference in the number of genes with decreased expression in adr-2(-) adult neural cells (4476 to 2714). These data suggest that ADR-2, and perhaps RNA editing, plays an important role in promoting the neural developmental down-regulation of several hundreds of genes.
To identify differentially expressed neural genes that are dependent on ADR-2, the genes with altered expression in wild-type adult neural cells were overlapped with those with altered expression in adr-2(-) adult neural cells (Supplemental Table S7). This analysis identified 1941 genes that were dependent on adr-2 for proper neurodevelopmental gene expression. These genes were enriched for gene ontologies including response to stimulus and determination of adult lifespan (Supplemental Fig. S3D), which is consistent with previous studies showing that worms lacking editing show chemotaxis defects and altered lifespan (Tonkin et al. 2002; Sebastiani et al. 2009; Ganem et al. 2019). It is also possible that these 1941 genes include some genes that are already differentially expressed in L1 neural cells isolated from adr-2(-) animals compared with wild-type L1 neural cells. To address this concern, DESeq2 was also performed between wild-type and adr-2(-) L1 neural cells. This analysis identified five (C39H7.4, rncs-1, asah-1, nep-17, and hpo-15) of the 1941 genes are already down-regulated in the adr-2(-) neural cells at the L1 stage. qPCR of one of these five genes (C39H7.4) confirmed the differential expression (Supplemental Fig. S3E). Three genes (kgb-1, nhr-122, and srw-17) from the remaining 1936 genes were randomly selected, and qPCR confirmed that the gene expression for all three genes was developmentally regulated and ADR-2 dependent (Supplemental Fig. S3F). Thus, providing additional support that ADR-2 is important for regulating neurodevelopmental gene expression for approximately 1900 genes.
To determine if editing could contribute to the ADR-2-dependent neurodevelopmental gene regulation, the list of developmentally regulated neural genes was overlapped with the known stage-specific edited transcripts. Nearly 30% of the differentially expressed and L1-specific edited genes were dependent on ADR-2 for proper gene expression during neural development (Fig. 3F). Together, these data indicate that differential expression contributes to the stage-specific neural editomes, and ADR-2 regulates neurodevelopmental gene expression in both an editing-dependent and an editing-independent manner.
A subset of neural transcripts shows increased editing during development
Although our above analysis indicates that stage-specific editing of many transcripts occurs owing to differential gene expression, nearly one-quarter of edited transcripts showed editing in both life stages (Supplemental Fig. S4A; Supplemental Table S4). Of the 119 transcripts edited in both stages, 10% show editing within a specific dsRNA region in a development-specific manner (Supplemental Fig. S4B). In these 12 transcripts, the intronic edited sites are L1 specific. Although 3′ UTR editing is observed in both stages, the editing level in the 3′ UTR increases in adults. The majority of transcripts (90%) are edited within the same dsRNA region throughout development. In these transcripts, even though the same dsRNA region is edited, both the number of sites edited and the editing level vary between L1 and adult neural cells.
As read coverage influences the ability to accurately quantify editing at a given site, to quantitively examine differences in editing levels during development, a threshold of five reads covering the edited nucleotide and one A-to-G variant in both data sets was required. This stringency reduced the number of edited sites found in both developmental stages to 901 (Supplemental Table S8). Despite the decrease in adr-2 expression during neural development (Fig. 2C), ∼80% of these sites showed increased editing in adult neural cells (Fig. 4A). This is consistent with previous observations of increased A-to-I editing during mammalian brain development and with the fact that ADAR expression does not directly correlate with the changes in editing levels (Wahlstedt et al. 2009; Ekdahl et al. 2012; Shtrichman et al. 2012; Zaidan et al. 2018).

Editing level increases in majority of the sites during neural development. (A) Heatmap representing the percentage of editing in L1 and adult neural cells at the sites (901) with more than five reads in neural RNA-seq from both life stages. (B) The 837 sites mapped to annotated transcripts were categorized into five groups depending on differential editing between L1 and adult neural cells. Bar graph represents the number of editing sites in each group. The number of unique transcripts in each group are listed above each bar.
To begin to understand how neurodevelopmental editing is regulated, the sites with changes in editing were annotated to genomic regions. For the adenosines edited in both life stages, 64 editing sites mapped to unannotated regions and were removed from downstream analysis. The remaining 837 editing sites mapped to 67 transcripts. The editing sites were classified into five groups depending on the percentage of change in editing observed in the two life stages (Fig. 4B). Together, the two classes with increased editing held the majority of sites analyzed, indicating a prevalent higher editing efficiency in adult neural cells.
To begin to mechanistically dissect how increased editing occurs during neural development, we focused on sites that showed a >20% increase in editing in adult neural cells. These editing sites map to 23 unique transcripts (Table 1). In contrast to previously identified editing targets that have multiple (five to 125) editing sites in noncoding regions of the C. elegans transcriptome (Tonkin et al. 2002; Zhao et al. 2015; Deffit et al. 2017; Goldstein et al. 2017), nearly two-thirds (65%) of these transcripts have only one or two specific editing sites, and 70% of these transcripts are edited within the 3′ UTR. In sum, despite decreased adr-2 expression in adult neural cells, increased editing at a majority of sites within neural transcripts that are constitutively expressed during development was observed, thus suggesting the presence of a neural-specific molecular mechanism to regulate RNA editing during development.
Neural transcripts show increased editing during neural development
The inhibitory role of ADR-1 on Y75B8A.8 editing is development-specific
A major regulator of RNA editing in C. elegans is the deaminase-deficient ADAR protein, ADR-1 (Washburn et al. 2014; Rajendren et al. 2018). Initial studies of adr-1(-) worms identified both sites that show increased and decreased editing compared with wild-type worms (Tonkin et al. 2002). In previous studies, ADR-1 was shown to promote ADR-2 binding to transcripts, which leads to increased editing (Washburn et al. 2014; Rajendren et al. 2018). However, ADR-1 has also been shown to inhibit editing of a specific transcript, Y75B8A.8, when transgenically expressed in neurons, and this repression is largely masked in studies of RNA isolated from whole worms (Washburn and Hundley 2016b). The genes identified in this study, which show increased editing in adult neural cells include Y75B8A.8 (Table 1).
From the neural RNA-seq data sets, editing of endogenous Y75B8A.8 increased during neural development (Fig. 5A), suggesting ADR-1 repression of editing is most efficient at the L1 stage. To directly test this hypothesis, developmental editing of the Y75B8A.8 3′ UTR was examined in neural cells in the presence and absence of adr-1. Transgenic worms expressing the Y75B8A.8 3′ UTR under the control of the rab-3 pan-neuronal promoter were synchronized at the L1 and adult stages. After RNA isolation, reverse transcription was performed using a reporter-specific primer. Editing at two specific sites (referred to as sites 227 and 228 owing to the nucleotide numbers within the 3′ UTR) within the Y75B8A.8 3′ UTR was apparent in both stages and genotypes (Fig. 5B). Editing at these sites drastically increased (∼40% in both 227 and 228) in adr-1(-) worms at the L1 stage (Fig. 5C, left), whereas a slight increase (∼20% at 228) in one of the sites was observed in adr-1(-) worms at the adult stage (Fig. 5C, right).
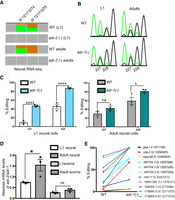
The inhibitory role of adr-1 on editing of neural transcripts is developmentally regulated. (A) Representation of neural RNA-seq reads covering the Y75B8A.8 transcript. Reads from adr-2(-) are negative controls. Green represents adenosine and brown represents guanosine at the marked chromosomal positions. (B) Sanger sequencing chromatograms of cDNA amplified from Y75B8A.8 reporter RNA. Editing sites are listed below the chromatogram. Sites 227 and 228 correspond to chromosomal positions of Chr III: 12,171,074 and 12,171,075. The nucleotides at each position are represented with a different color (green, adenosine; black, guanosine; blue, cytidine; red, thymidine). (C,D) The average of three independent replicates was plotted; error bars, SEM. One-way ANOVA was performed to determine the statistical significance. (****) P < 0.0001; (*) P < 0.05; (ns) P > 0.05. (C) Editing levels of the Y75B8A.8 neural reporter were measured in wild-type and adr-1(-) worms in L1 (left) and adult (right) stages using Sanger sequencing. (D) qPCR was performed to measure the absolute mRNA levels of adr-2 relative to adr-1 in both life stages. (E) Neural RNA-seq data were used to plot the L1 editing levels of representative sites from the group of neural RNAs that showed increased editing in development (P < 0.01, pairwise comparison using two-way ANOVA).
One possibility for the reduced ADR-1 inhibition of editing during neural development is that the relative expression of adr-1 and adr-2 changes during neural development, thus allowing ADR-2 to overcome inhibition by ADR-1. To test for this possibility, the absolute adr-1 mRNA levels in neural cells and whole-worm lysates were measured using qPCR. Our data indicate that neural adr-1 expression decreased during development, whereas adr-1 expression in whole-worm lysates remained unchanged (Supplemental Fig. S5). Absolute mRNA counts for adr-2 (Fig. 2D) were divided by the corresponding life stage of adr-1 mRNA counts, and the observed ratio of adr-2 to adr-1 mRNA expression was found to significantly increase from L1 to adult neural cells (Fig. 5D). In contrast, the adr-2 to adr-1 ratio in whole-worm lysate remained unchanged during development. Consistent with previous studies, adr-1 mRNA levels are higher than those of adr-2 throughout development (Hundley et al. 2008). Thus, these data suggest a tissue-specific regulation of editing during development, in which the levels of an ADAR enzyme (ADR-2) exceed that of a repressive factor (ADR-1) to allow for increased editing during development.
To determine whether the inhibitory role of ADR-1 was specific to Y75B8A.8 or extended to other transcripts that show increased editing in adult neural cells (Table 1), editing levels were quantified for adr-1(-) neural RNA-seq data from both stages (Supplemental Table S9). Editing sites within transcripts that show increased editing during neural development also show increased editing in the absence of adr-1 at the L1 stage (Fig. 5E). This increase is significant across all sites analyzed in Figure 5E (P < 0.01, pairwise comparison). In contrast, a randomly selected group of a similar number of L1 edited sites did not show any significant increase in editing in the absence of adr-1 (Supplemental Fig. S6A). In addition, editing at the majority of the sites showed in Figure 5E did not significantly increase in the absence of adr-1 in the adult stage (Supplemental Fig. S6B). Together, these data suggest that the inhibitory role of adr-1 in neural cells is developmentally regulated. These data are also consistent with previous data indicating an embryo-specific enrichment of editing sites in the absence of adr-1 compared with later (L4) developmental stages (Ganem et al. 2019). Together, these studies suggest that ADR-1 negatively regulates editing primarily during early C. elegans development.
In the absence of adr-1, ADR-2 binding to neural mRNAs increases early in development
Our sequencing data suggest a regulatory mechanism in which adr-2 expression increases relative to adr-1 expression during neural development to result in increased editing of specific neural transcripts. To begin to understand the mechanism of how ADR-1 represses editing of these transcripts during early development, we tested whether ADR-1 binding to transcripts was required for inhibition of editing. Editing of the Y75B8A.8 3′ UTR neural reporter was examined in the presence of either wild-type ADR-1 or an RNA binding–deficient ADR-1 mutant, where mutations (KKxxK to EAxxA) within the first dsRNA binding domain (dsRBD1) abolished ADR-1 binding to mRNA in vivo (Rajendren et al. 2018). Transgenic worms expressing FLAG-tagged wild-type ADR-1 or the ADR-1 RNA binding mutant driven by the endogenous adr-1 promoter along with Y75B8A.8 3′ UTR under the control of the rab-3 pan-neuronal promoter were generated. Synchronized L1 worms were harvested, and total RNA was used to reverse transcribe the neural Y75B8A.8 reporter. By using Sanger sequencing, editing of the neural Y75B8A.8 reporter in the presence of the wild-type ADR-1 transgene (Fig. 6A) was similar to that observed for the neural Y75B8A.8 reporter from wild-type L1 worms (Fig. 5C). However, in the presence of an RNA binding–deficient ADR-1, editing levels significantly increased (Fig. 6B). These data suggest that ADR-1 competes with ADR-2 to bind to Y75B8A.8 in L1 neural cells. In addition, overlap of our previous studies of ADR-1-bound RNAs from whole-worm lysates (Ganem et al. 2019) and the neural transcripts with neurodevelopmentally increased editing identified above indicates that 80% of these transcripts are bound by ADR-1 (Table 1). This suggests that ADR-1 inhibition of editing by binding to neural RNAs could be a widespread mechanism to regulate RNA editing in a tissue- and development-specific manner in C. elegans.
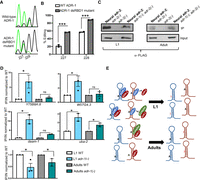
ADR-1 inhibits editing at transcripts that show increased editing in neural development by binding. (A) Sanger sequencing chromatograms of cDNA amplified from Y75B8A.8 reporter RNA isolated from the indicated worms at the L1 stage. (B) Editing in A was quantitatively measured. The average of two independent replicates is plotted; error bars, SEM. Two-way ANOVA was used to determine statistical significance. (***) P < 0.001. (C) Lysates from the indicated worm strains were incubated with FLAG magnetic beads. A portion of the lysates before immunoprecipitation and the IPs was subjected to immunoblotting with a FLAG antibody (Sigma-Aldrich F1804). (D) Bar graph represents the fold enrichment of cDNA present in the IPs divided by cDNA present in the input lysates from the indicated worm strains and normalized to the same ratio in adr-1; adr-2(-) worms. The average of three (Y75B8A.8, WO7G4.3 and lam-2) and two (daam-1 and uba-2) independent replicates were plotted; error bars, SEM. One-way ANOVA was used to determine statistical significance. (*) P < 0.05; (ns) P > 0.05. (E) Proposed model for editing regulation of neural transcripts that show increased editing during development.
To further test the model that ADR-1 represses editing by outcompeting ADR-2 binding to transcripts, we sought to determine whether ADR-2 binding to neural targets is affected by loss of adr-1. To specifically examine ADR-2 binding to neural mRNAs, transgenic worms coexpressing 3X FLAG-tagged adr-2 under control of the rab-3 promoter and the neural Y75B8A.8 reporter were generated in both adr-2(-) and adr-1(-);adr-2(-) backgrounds. Reporter editing indicated the expression of functional ADR-2 in these transgenic lines (Supplemental Fig. S7). To examine ADR-2 binding to neural transcripts, a FLAG RNA immunoprecipitation (RIP) was performed in synchronized L1 and adult worms that were UV crosslinked. Total RNA from IPs and input lysates was used to reverse transcribe specific genes, and qPCR was performed. Equal expression and efficient immunoprecipitation of ADR-2 were observed in the presence and absence of adr-1 for both life stages (Fig. 6C). As expected, FLAG–ADR-2 was not immunoprecipitated from adr-2(-) worms (Fig. 6C). The qPCR data indicate that, on average, sevenfold more Y75B8A.8 neural reporter mRNA was present in ADR-2 IPs from L1 worms lacking adr-1 compared with IPs in the presence of adr-1 (Fig. 6D). In contrast, the enrichment of neural Y75B8A.8 mRNA in ADR-2 IPs from adult worms was similar in the presence and absence of adr-1. To examine the competition of ADR-1 and ADR-2 to bind other transcripts with increased editing during neural development, ADR-2 binding to W07G4.3, daam-1, and uba-2 was examined. Similar to Y75B8A.8, increased ADR-2 binding to all three transcripts was observed in adr-1(-) L1 worms (Fig. 6D), and binding of neural ADR-2 to both W07G4.3 and daam-1 in adult worms was not significantly different in the presence or absence of adr-1. A small (1.7-fold), but significant (P = 0.04) increase in neural ADR-2 binding to uba-2 was detected in adr-1(-) adult worms. In contrast, ADR-2 binding to lam-2, an mRNA in which ADR-2 is known to require ADR-1 for binding (Rajendren et al. 2018), was reduced in both L1 and adult adr-1(-) worms.
In sum, our model suggests that ADR-1 inhibits ADR-2 binding to a specific group of neural transcripts in early development (Fig. 6E). Neural-specific changes in gene expression result in adr-2 increased levels compared with adr-1 in adult worms. The neurodevelopmental gene expression changes allow ADR-2 to bind more efficiently to select neural transcripts, resulting in increased editing. Together, these data reveal a mechanism for neural- and development-specific regulation of RNA editing and provide new insights into molecular mechanisms that regulate ADAR substrate recognition and editing efficiency.
Discussion
In this study, we determined how the effects of editing on transcriptome diversity change during neural development. Fewer editing sites were identified in adult neural cells compared with L1 neural cells, which is in part owing to decreased expression of edited transcripts. Previous studies of developmental editomes had indicated the number of both edited sites and transcripts decreased during C. elegans development (Zhao et al. 2015), but adr-2 mRNA expression slightly increased in adult worms compared to the L1 stage (Hundley et al. 2008). In contrast to those studies from whole worms, we find that adr-2 mRNA expression decreases in neural cells during development, which may contribute to the reduced number of edited transcripts identified in adult neural cells. However, we also found that the distribution of identified sites shifted from intronic regions in L1 neural cells to 3′ UTRs in adult neural cells, suggesting that the effects of RNA editing on neural transcriptome diversity change during development.
In addition, our data indicate that developmental regulation of editing levels occurs for neural transcripts that were edited throughout development. Despite the adr-2 down-regulation, these transcripts showed increased editing in adult neural cells. By performing neural ADR-2 IPs in the presence and absence of adr-1, we showed that ADR-1 competes with ADR-2 to bind these specific neural transcripts at the larval stage. The competition between ADR-1 and ADR-2 to bind RNA leads to decreased editing at the L1 stage. However, this competition is reduced in adult neural cells, which leads to increased editing of these transcripts. Together, our data suggest a model in which ADR-1 acts as a stage- and substrate-specific repressor of editing of specific neural transcripts during C. elegans development. These ADR-1-inhibited transcripts may contain specific secondary structures that are bound by ADR-1 with stronger affinity. One common feature of these targets is the presence of one or a few highly specific editing sites within the 3′ UTR. Bulges and loops within dsRNA, including Alu dsRNA, play an important role in the ability of human ADARs to select specific adenosines to edit both in vitro and across the transcriptome (Lehmann and Bass 1999; Kallman et al. 2003; Kleinberger and Eisenberg 2010). Future studies should focus on a comprehensive effort to identify the RNA cis elements present in neural RNAs inhibited by ADR-1, including secondary microstructures in order to better understand the features that distinguish these neural RNAs from other edited RNAs.
In our study, L1-specific neural edited transcripts are enriched for intronic editing events. Nearly two-thirds of the transcripts edited in introns decrease in expression during development, which may contribute to the reduced editing of intronic regions observed in adult neural cells. Another possibility for the fewer number of intronic editing events could be varying efficiency of RNA splicing in L1 and adult neural cells. Previous studies have shown an interplay between RNA editing and RNA splicing, in which the efficiency of each process affects the outcome of the other process (Solomon et al. 2013; Licht et al. 2016, 2019b). In support of this possibility, we found a significant enrichment for RNA processing genes, including splicing factors rsp-2, rsp-3, swp-1, prp-8, bacs-2, and repo-1, in adult neural cells compared with L1 neural cells (Supplemental Table S5). The intronic edited transcripts may undergo rapid splicing adult neural cells, thus decreasing the number of introns that undergo deamination by ADR-2.
A majority of neural edited sites identified in both developmental stages occur within noncoding regions, which suggests these editing events may not directly influence the function of the gene product. Unlike flies and cephalopods, in which A-to-I editing extensively recodes genetic information (Graveley et al. 2011; St Laurent et al. 2013; Alon et al. 2015; Liscovitch-Brauer et al. 2017), editing in coding regions is rare in C. elegans. However, editing within noncoding regions is known to impact immune and behavioral pathways. For example, neural editing within the clec-41 3′ UTR is important for proper chemotaxis in C. elegans (Deffit et al. 2017). In addition, C. elegans ADARs are involved in silencing long terminal repeat (LTR) retrotransposons and regulating cellular responses to endogenous dsRNA (Reich et al. 2018; Fischer and Ruvkun 2020). Together, these previous studies suggest potential roles of editing within noncoding, repetitive regions of the C. elegans transcriptome.
We also identified adult-specific editing sites that were primarily localized within the coding sequence and 3′ UTR of a few neural transcripts. The functional consequences of editing within coding regions are poorly studied in C. elegans. A recent study reported ADAR-mediated regulation of SLO-2 activity in C. elegans neurons, but no editing of slo-2 transcripts was observed from RNA isolated from whole animals (Niu et al. 2020). We identified editing within the slo-2 coding sequence in adult neural cells. Future studies of whether neural editing of slo-2 transcripts regulates SLO-2 activity will be important to determine if the ability to recode the genome is critical for proper C. elegans nervous system function.
In addition to determining stage-specific editomes, our approach identified transcripts edited throughout C. elegans neural development. Despite the neural-specific down-regulation of the ADR-2 editing enzyme, most neural editing sites identified in these transcripts showed increased editing in adults. This is consistent with previous data in which RNA editing was increased in the adult mammalian brain compared with the neonatal brain, as well as a lack of direct correlation between editing levels and ADAR mRNA expression (Wahlstedt et al. 2009; Zaidan et al. 2018). Together with our data, this indicates the presence of regulatory networks that control the deamination activity of ADARs in brain development from nematodes to mammals. This regulatory network can involve factors that affect localization of ADARs and edited mRNAs, RNA-binding proteins that block binding of ADARs to target RNAs, and proteins that hinder ADARs from finding targets (Deffit and Hundley 2016; Washburn and Hundley 2016a). In C. elegans, a few molecular mechanisms that regulate RNA editing globally have been studied (Ohta et al. 2008; Rajendren et al. 2018). However, mechanisms that regulate tissue- and development-specific RNA editing have not previously been identified.
In analysis of mixed-stage whole-worm extracts, ADR-1 was previously shown to promote ADR-2 association with target mRNAs, which in turn results in increased editing (Rajendren et al. 2018). In that study, Y75B8A.8 was identified as an ADR-2 target where ADR-1 promotes ADR-2 binding to mRNAs. However, editing of the Y75B8A.8 transcript is independent of ADR-1 within the nervous system (Washburn and Hundley 2016b). In contrast to the whole-worm studies, our in vivo binding studies indicate that ADR-2 binding to Y75B8A.8 is inhibited by ADR-1 in L1 neural cells. Here, our data suggest that ADR-1 acts as a repressor at certain neural transcripts early in development, specifically by competing with ADR-2 for mRNA binding. In line with this, previous studies of human cell lines identified three RNA-binding proteins (RPS14, SFRS9, and DDX15) with substrate-specific inhibition of editing, which directly correlates with RNA binding preferences of the regulators (Tariq et al. 2013). Combined with our data, this suggests that ADR-1-inhibited transcripts may have specific cis elements, including sequence motifs and/or secondary structures, that are recognized by ADR-1.
Our data suggest decreased editing in larval neural cells is because of the high expression of adr-1, which binds to and inhibits editing. Our in vitro binding studies show that ADR-1 has a more than 150-fold stronger affinity to dsRNA compared with that of ADR-2 (Supplemental Fig. S8; Rajendren et al. 2018). Neural adr-2 expression relative to adr-1 increases during development, allowing ADR-2 to outcompete ADR-1 for binding to transcripts, resulting in higher editing in adult neural cells. As ADR-2 has the weakest affinity to dsRNA of all tested ADAR family members (Goodman et al. 2012; Rajendren et al. 2018), it is not known whether ADR-2 binds to these targets alone or in complex with a neural RNA binding cofactor. Another interesting observation is that nearly 500-fold higher adr-2 mRNA levels are detected in whole worms compared with neural cells, suggesting a major role of adr-2 in nonneural cells. Future studies of C. elegans ADARs should expand these specific approaches to study editing regulation and ADAR substrate binding in tissues with abundant adr expression.
Methods
Worm strains and maintenance
All worms were maintained under standard laboratory conditions on nematode growth media seeded with Escherichia coli OP50. Bristol strain N2 was used to define the GFP gates in neural cell isolation. The worms used for isolating neural cells (BB76, BB77, and BB78) and for the neural Y75B8A.8 editing assay were previously published (Hundley et al. 2008; Washburn and Hundley 2016b). Transgenic worms were created for this study using standard microinjection techniques and passaged by selecting worms that contained the GFP marker (for details, see Supplemental Material).
Neural cell isolation
Neural cells were isolated from adult worms as previously described (for detailed protocol, see Supplemental Material; Kaletsky et al. 2018) and filtered into sterile FACS tubes. Filtered cells were stained with near IR live/dead fixable dye (Invitrogen) before FACS sorting. The BD FACSAria II sorter was used to sort GFP expressing neural cells from the non-GFP population, and FACSDiva 6.1.1 software was used for analysis (Flow Cytometry Core Facility). Cells were sorted into TRIzol (Invitrogen), snap-frozen, and stored at −80°C.
RNA isolation and qPCR for absolute mRNA levels
Total RNA was extracted using TRIzol and treated with DNase (Thermo Fisher Scientific). RNeasy extraction (Qiagen) was used to further purify the RNA, and cDNA was synthesized with SuperScript III (Invitrogen) using both Oligo(dT) and random hexamer primers (Thermo Fisher Scientific). For the absolute quantification, qPCR standards were made using https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/dna-copy-number-calculator.html. Copy number was calculated for each standard, and the curve was generated using absolute copy number dilutions. qPCR reactions were performed using gene-specific primers (Supplemental Table S10) and SYBR FAST master mix (KAPA) on an Eppendorf Realplex Mastercycler.
Neural RNA sequencing
Total RNA was isolated as described above. Oligo(dT) beads (Invitrogen) were used to enrich for mRNAs, and libraries were generated using a stranded RNA-seq library preparation kit (KAPA) according to the manufacturer's instructions. Briefly, RNA samples were fragmented into 200- to 300-nt pieces and used to synthesize cDNA. This was followed by adapter ligation (KAPA S1 adapter kit) and minimal amplification of the libraries. Libraries were sequenced for SE75 cycles on an Illumina NextSeq 500 instrument at the Center for Genomics and Bioinformatics (CGB-IUB).
Bioinformatics analysis of RNA-seq reads
In brief, reversely stranded SE 75-bp reads were trimmed of adapters and aligned to the C. elegans genome (WS269) using STAR (v2.5.2b) with the following parameters: [outFliterMultimap Nmax 1, outFilterScoreMinOverLread 0.66, outFilterMatchNminOverLread 0.66, outFlterMismatchNmax10, outFilterMismatchNoverLmax 0.3]. Raw RNA-seq reads from isolated L1 neural cells were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) using accession number GSE98869 (Deffit et al. 2017). L1 RNA-seq reads were trimmed for adapters using cutadapt (Martin 2011) and shortened to 75-base-long reads using Trimmomatic-0.39 (Bolger et al. 2014). These reads were aligned with STAR with the same parameters. Uniquely aligned reads were used as inputs to run featureCounts (v.1.5.2) to map the reads to WormBase gene annotations (WS275) using [-s 2] flag. To count the intronic reads in the L1 and adult aligned reads, featureCounts was used with the following flags [-F GTF -a ce11.introns.gtf -o intron.counts.txt].
For differential gene expression, DESeq2 (v1.18.1) (Love et al. 2014) was run with raw read counts obtained from featureCounts. Transcripts that have a significant difference in gene expression (P-adj < 0.05, using Benjamini–Hochberg correction) were considered to be differentially expressed. Previously published raw RNA-seq reads from isolated adult neural cells from were downloaded from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) using accession number PRJNA400796 (Kaletsky et al. 2018) and processed as described.
To identify high-confidence editing sites, uniquely aligned RNA reads for the three biological replicates were merged to increase coverage at edited sites. These merged reads were used as inputs for SAILOR (Deffit et al. 2017). Sites with a confidence of ≥0.99 were chosen for downstream analysis. Annotation of these high-confidence sites was performed with a custom Python script using WormBase WS275 annotations (Supplemental Code S1).
Variant calling using SAMtools
SAILOR-identified sites from L1 and adult neural cells were merged to make a list of all neural editing sites. BAM files (after removing duplicates) were used for variant calling using SAMtools (Li et al. 2009) and a custom Python script (Supplemental Code S2). The C. elegans genome (WS275) was used as a reference to identify variant nucleotides. The numbers of total and variant reads covering the nucleotide were extracted for all the combined neural editing sites. Percent editing was calculated by [(number of variant reads/total number of reads) × 100].
Two Sample Logo analysis
Nucleotide positions of editing sites were identified, and the corresponding nearest-neighbor nucleotides were retrieved using BEDTools (Quinlan and Hall 2010). Five-nucleotide stretches (centered with the edited adenosine) were used to determine the overrepresented and underrepresented nucleotides surrounding the edited sites using Two Sample Logo (http://twosamplelogo.org).
Sanger sequencing of the neural Y75B8A.8 reporter
Gravid adult worms were bleached, and the collected eggs were incubated overnight at 20°C to hatch. A portion of the synchronized L1 worms were stored in TRIzol, and the other portion was plated and cultured for 50 h at 20°C to get adults before storing in TRIzol. RNA was extracted as described above and then reverse transcribed with HH1216 (Supplemental Table S10) using ThermoScript (Invitrogen) followed by PCR with Phusion DNA polymerase (NEB). Negative controls without ThermoScript were conducted for each sample. The region containing editing sites was PCR-amplified, gel-purified, and subjected to Sanger sequencing.
RIP assay
FLAG immunoprecipitation was performed as previously described (Washburn et al. 2014), with minor modifications in lysate concentration used for the different life stages (for details, see Supplemental Material).
Data access
Raw and processed high-throughput sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE151916. Raw Sanger sequencing files can be found in the Supplemental Material for this article.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH; R01 GM130759), the National Science Foundation (award 191750), and a IUSM Ralph W. and Grace M. Showalter award to H.A.H.; an American Heart Association predoctoral fellowship (19PRE34430122) and the Doane and Eunice Dahl Wright Fellowship (IUSM-Bloomington) to S.R.; an NIH National Research Service Award postdoctoral fellowship (F32 GM119257; S.N.D.) and the Indiana Clinical and Translational Sciences Institute (CTSI) funded in part by award number UL1TR002529 from the NIH; and the National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award (Postdoctoral Challenge Award to S.N.D. and Indiana University Medical Student Program for Research and Scholarship [IMPRS] Award to J.T.). Content is solely the authors’ responsibility and does not necessarily represent the official view of the NIH. We thank Christiane Hassel (IUB-Flow Cytometry) for assisting in cell isolation, the Center for Genomics and Bioinformatics (CGB)-IUB for RNA sequencing, and Doug Rusch for assisting with the Two Sample Logo analysis. We thank fellow graduate students Robert Policastro, Kasun Buddika, and Aidan Manning for bioinformatics assistance and Taylor Nicholas for help with some statistical analyses. We thank Dr. Rachel Kaletsky for assistance with adult neural cell isolation and comparison of our adult neural transcriptome with previously published data.
Author contributions: Study conceptualization was by S.R. and H.A.H.; methodology was by S.R., A.D., S.N.D., and P.V.; bioinformatics was by S.R. and J.T.; data curation was by S.R. and A.D.; writing of the original draft was by S.R. and H.A.H.; review and editing were by all the authors; visualization was by S.R., A.D., and J.T.; project administration and supervision were by S.R. and H.A.H.; and funding acquisition was by H.A.H., S.R., S.N.D., and J.T.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.267575.120.
- Received June 17, 2020.
- Accepted November 18, 2020.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








